

Abstract
Herein, we describe a protocol for the isolation of human embryonic stem cells (hESCs)-derived vascular cells at various stages of development. The cells are isolated from 10 to 15-d-old human embryoid bodies (EBs) cultured in suspension. After dissociation, cells are labeled with anti-CD34 or anti-CD31 (PECAM1) antibody and separated from the cell mixture by magnetic-activated cell separation (MACS) or fluorescent-activated cell sorting (FACS). Isolated vascular cells are then cultured in media conditions that support specific differentiation and expansion pathways. The resulting vascular cell populations contain > 80% endothelial-like or smooth muscle-like cells. Assuming typical initial cell adhesion and proliferation rates, the entire procedure can be completed within 1.5 months. Vascular cells isolated and differentiated under the described conditions may constitute a potential cell source for therapeutic application toward repair of ischemic tissues, preparation of tissue-engineered vascular grafts and design of cellular kits for drug screening applications.
INTRODUCTION
Human embryonic stem cells (hESCs) represent an unlimited source of cells instrumental in replacing damaged tissues. In particular, hESCs can differentiate into various subtypes of vascular cells such as endothelial cells (ECs) or smooth muscle cells (SMCs)1–4, and can be potentially applied toward repair of ischemic tissues, engineering artificial blood vessels and heart valves, repair of blood vessels and vascularization of engineered tissues1–3,5,6. Vascularization of engineered tissues is fundamental for tissue survival, structural organization and acceptance after in vivo transplantation. Nonvascularized tissues engineered in vitro are typically too small in size (< 1 mm) to allow for sufficient oxygen diffusion7.
In addition, the mechanisms and molecular cues governing embryogenesis can be further elucidated through the study of in vitro hESC differentiation to ECs and SMCs. Although several reports describe the critical role played by specific growth factors and extracellular matrix components as well as by environmental conditions in the induction of vascular hESC differentiation1,6,8,9, the complete signaling and regulatory mechanisms mediating such differentiation are still poorly understood. Likewise, a pure source of such cells can allow for experimental setups to study endothelial– SMC cross talk and its role in the formation of functional blood vessels during development. Similarly, analysis of vascular differentiation of hESCs may shed light on the pathogenesis of vascular diseases, as well as on the early developmental stages of organs including pancreas, kidney, liver and neural tissue, which are highly reliant on the presence of blood vessels10,11.
Drug screening represents an additional field that can benefit from increased hESC-derived vascular cell accessibility12,13. hESC-derived vascular cells can be isolated to unlimited numbers for high-throughput assays, shortening the timelines for identification of new therapeutics and reducing in vivo testing periods. To date, human tissue samples (biopsies) provide drug discovery protocols with the required vascular cells for research. However, these cells exhibit limited survival times in culture, are limited in number and present significant genetic variability, which affects their applicability in screening technologies12. In contrast, hESC-derived vascular cells may offer valuable advantages over primary human vascular cells in their reproducibility and wide range of available identification and evaluation assays.
Earlier studies focusing on the vascular potential of hESCs6,14 have identified multiple markers specific to progenitor or fully differentiated vascular cells. Some studies describe cells isolation from embryoid bodies (EBs) cultured in suspension for several days1,2. Others induced hESC differentiation to vascular cells in cocultures with mouse embryonic fibroblasts (MEF), OP9, S17 or MS-5 feeder layers15,16.
In this protocol, we describe a methodology to isolate mature ECs (Fig. 1) or vascular progenitor cells (VPCs) that can then be induced to differentiate into endothelial or SMCs (Fig. 2). Specific differentiation to either cell type occurs by spontaneous EB differentiation, cultured in suspension for a variable number of days (Fig. 3 summarizes the method in a flow diagram). This differs from other methods15,16 as it does not require additional cell types (as co-culture or feeder cells) or additional growth factors for EB differentiation (although for VPCs growth factors are required at the last stage of endothelial or SMC differentiation).
Figure 1.
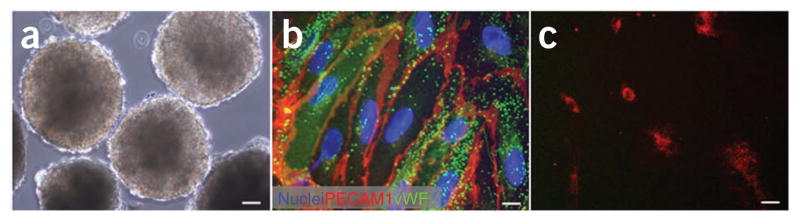
Characterization of hES-derived endothelial cells isolated from 13-d-old EBs by FACS sorting of CD31+ cells. (a) A light microscopy image of EBs on day 13. (b) Immunofluorescence images of hES-derived ECs grown in culture and stained for CD31 and vWF. (c) Dil-labeled ac-LDL uptake by CD31+ cells. Scale bar in a corresponds to 60 μm, in b to 6 μm and in c to 30 μm.
Figure 2.

Immunofluorescent characterization of VPCs and VPC-derived endothelial and smooth muscle cells. Light (a) and fluorescent (b) microscopy images of EBs on day 10 stained for CD34 (b). Scale bars in a and b correspond to 500 and 25 μm, respectively. (c) VPC-derived ECs in light microscopy (left) and stained for CD31 (middle) and vWF (right). (d) VPC-derived SMCs in light microscopy (left) and stained for SM–MHC (middle) and calponin (right). Scale bars in c (left) and d (left) correspond to 50 μm. Scale bars in c (middle and right) and d (middle and right) correspond to 25 μm.
Figure 3.

A flow diagram of VPC and EC-isolation protocol.
The protocol below focuses on the induction of vascular differentiation of hESCs and not on hESC culturing required before this stage. The reader is encouraged to look over protocols for hESC maintenance published earlier in this journal17,18. Typically, we culture hESCs on mitomycin-treated MEF feeder layers, and passage hESCs via mechanical or enzymatic colony disruption.
hESC-derived ECs
Herein we describe a highly efficient method, adapted from a methodology previously reported by the authors2, for the isolation of hESC-derived ECs which make up ~2% of the EBs from which they are purified. hESC-derived ECs are isolated from 13-d-old EBs by fluorescent-activated cell sorting (FACS) separation of CD31+ cells. The ideal time for isolation was chosen by the period in which endothelial-specific genes and proteins are most highly expressed during hESC differentiation. Vascular markers such as CD31, CD34 and vascular endothelial cadherin (VE-cad) peak between days 13 and 15. EBs are then dissociated into single cells and CD31+ cells are isolated by FACS using anti-CD31 antibodies and then expanded in endothelial growth medium. The hESC-derived endothelial cells, isolated as described below, express endothelial markers, including CD31, VE-cad, von Willebrand factor (vWF) and N-cadherin (N-cad), and demonstrate Dil-labelled acetylated low-density lipoprotein (Dil-ac-LDL) uptake (Fig. 1). In addition, after seeding these embryonic ECs on biodegradable synthetic scaffolds, their biofunctionality can be evaluated in severe combined immunodeficient (SCID) mice upon subcutaneous implantation of the cell-embedded scaffold. Immunohistochemical analysis of the retrieved implants demonstrated CD31+ microvessel, which contained mouse blood cells2.
hESC–VPCs
In the protocol below, we also describe the isolation of VPCs based on a recent methodology described by the authors and others1,16. VPCs can differentiate to endothelial1,16 or SMCs1 when using the appropriate media conditions.
Vascular progenitor cells are isolated at earlier stages of EB differentiation (day 10), where CD34 serves as a specific cell marker. VPC SMC-differentiating capacity may serve as an important model in studying early regulatory effects of these cells on initial vascularization stages, with a specific focus on their role in vascular network growth, vascular permeability and blood flow.
Vascular progenitor cells can be isolated by magnetic-activated cell separation (MACS) from 10-d-old EBs, using anti-human CD34 antibodies, as this endothelial/hematopoietic marker is minimally expressed on undifferentiated hESCs but is upregulated in EB (Fig. 2). CD34+ cells can then be subsequently cultured in endothelial growth medium EGM-2 supplemented with vascular endothelial growth factor (VEGF165) to give rise to endothelial-like cells, as demonstrated by cell morphology, gene and protein analyses, in vitro functional assays and in vivo implantation studies1 (see anticipated results). In contrast, CD34+ cells cultured in EGM-2 medium supplemented with platelet-derived growth factor (PDGFBB) give rise to smooth muscle-like cells as confirmed by morphology, gene and protein analyses, functional assays and in vivo studies1 (see ANTICIPATED RESULTS).
MATERIALS
REAGENTS
Cells
hESCs cell lines H9 and H13 (Wicell International Stem Cell Bank, Wicell Research Institute)
Human umbilical vein endothelial cells (HUVECs) (Clonetics, Lonza, cat. no. C2517A)
Human umbilical artery SMCs (HUASMCs) (Clonetics, Lonza, cat. no. CC-2579)
MEF (see REAGENT SETUP)
Growth media and supplements
DMEM (Invitrogen, cat. no. 41965-035)
Knockout DMEM (Invitrogen, cat. no. 10829-018)
DMEM–F12 (Biological Industries, cat. no. 01-170-1A)
Knockout serum replacement (KSR) (Invitrogen, cat. no. 10828-028)
Fetal bovine serum (FBS) (Hyclone, Thermo Fisher Scientific, cat. no. SH30070.03)
L-glutamine (200 mM in 0.85% NaCl) (Invitrogen, cat. no. 25030-081)
-
β-Mercaptoethanol (55 mM in PBSA) (Invitrogen, cat. no. 21985-023)
! CAUTION β-Mercaptoethanol is toxic. Avoid inhalation, ingestion and skin contact.
Nonessential amino acid solution (Invitrogen, cat. no. 11140-050)
Recovery cell culture freezing medium (Invitrogen, cat. no. 12648-010)
Mitomycin C (Sigma-Aldrich, cat. no. M-4287) ! CAUTION Mitomycin C is toxic and carcinogenic. Avoid inhalation, ingestion or contact with skin or mucous membranes. Protect from light.
Trypsin neutralizing solution (TNS) (Clonetics, Lonza, cat. no. CC-5002)
HEPES (Clonetics, Lonza, cat. no. CC-5022)
Penicillin–streptomycin (penstrep) (Biological Industries, cat. no. 03-031-1B)
EGM-2 medium (Lonza, cat. no. CC-3162)
SmGM-2 medium (Lonza, cat. no. CC-3182)
hESC medium (see REAGENT SETUP)
EB medium (see REAGENT SETUP)
EC medium (see REAGENT SETUP)
SMC medium (see REAGENT SETUP)
Enzymes and growth factors
Basic fibroblast growth factor (bFGF) (Peprotech, cat. no. 100-18B) (see REAGENT SETUP)
VEGF165 (10 μg, R&D Systems, cat. no. 293-VE-010) (see REAGENT SETUP)
PDGFBB (1 mg, Peprotech, cat. no. 100-14B) (see REAGENT SETUP)
Collagenase B (Roche Applied Science, cat. no. 11088815001) (see REAGENT SETUP)
Collagenase type IV (Invitrogen, cat. no. 17104-019) (see REAGENT SETUP)
0.05% Trypsin–EDTA (Invitrogen, cat. no. 25300)
Trypsin–EDTA 0.25 mg ml−1 (Clonetics, Lonza, cat. no. CC-5012)
Cell and molecular biology reagents and kits
CD34 cell isolation kit (Myltenyi Biotec, cat. no. 130-046-702)
M-MLV reverse transcriptase (Promega, cat. no. M1705)
Taq DNA polymerase (BIOTAQ) (Bioline, cat. no. BIO-21086)
Ethidium bromide (Sigma-Aldrich, cat. no. E1510) ! CAUTION Ethidium bromide is carcinogenic. Wear gloves and dispose of all contaminated tips, gels and buffers in appropriate containers.
Agarose (IBI-Scientific, cat. no. IB70040)
Trizol (Invitrogen, cat. no. 15596-026) ! CAUTION Trizol is carcinogenic and may irritate eyes, respiratory system and skin. Work under a hood and use gloves.
Primers for RT-PCR (See Table 1)
Paraformaldehyde (PFA) 16% (wt/vol) solution (Electron Microscopy Science, cat. no. 15710) (see REAGENT SETUP)
Formalin solution, neutral buffered, 10% (Sigma-Aldrich, cat. no. HT501128) ! CAUTION PFA or formalin solution is toxic. Work under a hood and use gloves.
Triton X-100 (Sigma-Aldrich, cat. no. 21123) (see REAGENT SETUP)
Antibody diluent (Dako, cat. no. S3022)
4′,6-Diamidino-2-phenylindole (DAPI, Sigma-Aldrich, cat. no. D9542) (see REAGENT SETUP)
Fluoromount-G (Southernbiotech, cat. no. 0100-01)
DNase I (Roche Applied Science, cat. no. 11-284-932) (see REAGENT SETUP)
Propidium iodide (PI) (Sigma-Aldrich, cat. no. P4170) ! CAUTION PI is irritating to the eyes, respiratory system and skin. Wear suitable protective clothing, gloves and eye/face protection.
Dil-ac-LDL (Biomedical Technologies, cat. no. BT-902) (see REAGENT SETUP)
Animal research kit (ARK) (Dako, cat. no. K3954) ! CAUTION ARK contains diaminobenzidine (DAB), which is a suspected carcinogen. Wear suitable protective clothing and gloves.
Reveal buffer (Biocare Medical, cat. no. RV1000)
Background Sniper (Biocare Medical, cat. no. BS966H)
Table 1.
PCR reaction conditions and primer sequences for marker genes of interest.
| Gene transcript | Primer sequences (5′ to 3′, Fw/Rv) | Product (bp) | Cycles | Annealing temperature (°C) | [MgCl2] (mM) |
|---|---|---|---|---|---|
| CD31 | GCTGTTGGTGGAAGGAGTGC GAAGTTGGCTGGAGGTGCTC |
620 | 28 | 55 | 1.5 |
| CD34 | TGAAGCCTAGCCTGTCACCT CGCACAGCTGGAGGTCTTAT |
200 | 30 | 55 | 1.5 |
| KDR–Flk-1 | CTGGCATGGTCTTCTGTGAAGCA AATACCAGTGGATGTGATGGCGG |
790 | 35 | 60 | 1.5 |
| Angiopoietin-1 | GGGGGAGGTTGGACTGTAAT AGGGCACATTTGCACATACA |
362 | 35 | 60 | 1.5 |
| Angiopoietin-2 | GGATCTGGGGAGAGAGGAAC CTCTGCACCGAGTCATCGTA |
535 | 35 | 60 | 1.5 |
| Tie2 | ATCCCATTTGCAAAGCTTCTGGCTGGC TGTGAAGCGTCTCACAGGTCCAGGATG |
512 | 35 | 60 | 1.5 |
| Vascular endothelial (VE)-cad | ACGGGATGACCAAGTACAGC ACACACTTTGGGCTGGTAGG |
596 | 35 | 60 | 1.5 |
| vWF | ATGTTGTGGGAGATGTTTGC GCAGATAAGAGCTCAGCCTT |
656 | 40 | 55 | 1.0 |
| SM–MHC | GGACGACCTGGTTGTTGATT GTAGCTGCTTGATGGCTTCC |
670 | 35 | 60 | 1.5 |
| α-SMA | CCAGCTATGTGAAGAAGAAGAGG GTGATCTCCTTCTGCATTCGGT |
965 | 35 | 60 | 1.5 |
| Caldesmon | AACAACCTGAAAGCCAGGAGG GCTGCTTGTTACGTTTCTGC |
530 | 35 | 60 | 1.5 |
| SMα-22 | CGCGAAGTGCAGTCCAAAATCG GGGCTGGTTCTTCTTCAATGGGG |
928 | 35 | 60 | 1.5 |
| GAPDH | AGCCACATCGCTCAGACACC GTACTCAGCGCCAGCATCG |
302 | 27 | 60 | 1.5 |
PCR conditions: 5 min at 94 °C (hot start), 30–40 cycles (actual number noted in the table), 94 °C for 30 s, annealing temperature (noted in the table) for 30 s and 72 °C for 30 s, followed by a final 7 min extension period at 72 °C. PCR conditions for CD31 and CD34: 15 min at 95 °C, 1 min at 94 °C, annealing temperature (as indicated) for 1 min and 72 °C for 1 min, followed by a final 10 min extension period at 72 °C.
Other reagents and chemicals
Milli-Q water
PBS without Ca2+ and Mg2+ (Invitrogen, cat. no. 14190-250 or Sigma-Aldrich, cat. no. D8537)
EDTA (Sigma-Aldrich, cat. no. 252352)
Nonenzymatic cell dissociation solution (Sigma-Aldrich, cat. no. c5914-100 ml)
Bovine serum albumin (BSA) (Sigma-Aldrich, cat. no. A9418)
Gelatin type A, from porcine (Sigma-Aldrich, cat. no. G-1890) (see REAGENT SETUP)
MACS buffer (see REAGENT SETUP)
Poly-L-lactic acid (PLLA, Polysciences, cat. no. 18582)
Polylactic-glycolic acid (PLGA, Resomer, cat. no. RG 503 H)
Chloroform (Bio-Lab, cat. no. 03080521) ! CAUTION Chloroform is toxic and a suspected carcinogen. Work under the hood and use gloves.
Sodium chloride (Bio-Lab, cat. no. 19030291)
Growth factor-reduced (GFR) BD matrigel matrix (Matrigel, BD Biosciences, cat. no. 354230) (see REAGENT SETUP)
Atropine (Sigma-Aldrich, cat. no. A0132)
Carbachol (Sigma-Aldrich, cat. no. C4382)
Animals
CB17SC-M-F SCID−/− mice, 4-weeks old (Taconic Farms, model no. CB17SC-M) ! CAUT ION Mice must be maintained according to relevant national regulations using protocols and conditions approved by the institutional animal care and use committee.
EQUIPMENT
MACS columns (Myltenyi Biotec, cat. no. 130-042-401)
MACS magnet and support (Myltenyi Biotec, cat. no. 130-042-301)
FACS tubes (BD, cat. no. 352235)
15 ml Falcon tubes (BD, cat. no. 352196)
50 ml Falcon tubes (BD, cat. no. 352070)
24-Well plate (Nunc, cat. no. 142475)
Low-adhesion 10 cm petri dishes (Corning, cat. no. 3262)
Cell strainers (40 μm, BD, cat. no. 352340)
500 ml Medium filtration systems (Corning, cat. no. 431097)
50 ml Medium filtration systems (Corning, cat. no. 430320)
Pipette aid (BD Falcon, cat. no. 357590)
Micropipette set (Gilson, cat. nos. P20, P200 and P1000)
Glass Pasteur pipettes sterilized using autoclave
Plastic disposable pipettes, 1, 5, 10, 25 and 50 ml
Lab-Tek II chamber slides (VWR International, cat. no. 53106-306)
Nalgene ‘Mr. Frosty’ freezing container (Fisher Scientific, cat. no. 15-350-50)
Biosafety cabinet with aspirator for tissue culture
Tissue culture incubator, humidified, 5% CO2/95% air, 37 °C
Inverted light microscope with phase contrast (×4, ×10, ×20 and ×40 objectives)
Confocal or fluorescent microscope
Centrifuge suitable for 15 and 50 ml tubes with 4 °C cooling option
Water bath
Hemocytometer
UV lamp for analysis of agarose–ethidium bromide gels
UV-visible spectrophotometer
PCR thermocycler
Ice machine and ice baskets
Vortex
Flow cytometry cell sorter: FACStar with CellQuest software or FACSAria with FACSDiva software (BD Biosciences)
Flow cytometer analyzer: FACScan, FACSCalibur or LSR-II with CellQuest or FACSDiva software (BD Biosciences)
REAGENT SETUP
hESC line
Grow hESCs on mitomycin C-inactivated MEF as described previously17,18. Briefly, culture hESCs in ESC medium in a 5% CO2 incubator at 37 °C. Change growth medium daily and passage cells using 1 mg ml−1 collagenase every 5–6 d onto a fresh gelatin-coated mitomycin-inactivated MEF plate.
MEF
Prepare MEF as described before19. Briefly, remove embryos (E13) from a pregnant mouse, wash with PBS, mince and trypsinize to a single cell suspension. Grow MEF in DMEM supplemented with 10% (vol/vol) FBS, split a confluent culture to 1:3 ratio and freeze cells that are not needed immediately. For mitomycin-inactivation treatment, incubate MEF with DMEM containing mitomycin C (8 μg ml−1) for 2–3 h, wash three times with PBS, remove cells using trypsin–EDTA and seed 1.6 × 106 cells per 100 mm plate coated with 0.1% gelatin. A day later, MEFs are ready to be used as a feeder layer for undifferentiated hESCs.
hESC medium
DMEM–F12 supplemented with 20% (vol/vol) KSR, 1 mM L-glutamine, 1% (vol/vol) nonessential amino acids, 0.1 mM β-mercapto-ethanol and 4 ng ml−1 bFGF. Store at 4°C up to 2 weeks.
EB medium
Supplement 400 ml knockout DMEM with 100 ml FBS (or KSR), 2.5 ml L-glutamine, 1 ml β-mercaptoethanol, 5 ml nonessential amino acid solution and filter through a 0.2 μm filter. Store at 4°C. The solution can be used up to 1 month.
EC medium
Add 250 μl VEGF solution to 50 ml EGM-2 (final concentration of VEGF is 50 ng ml−1). Store at 4 °C up to 2 weeks.
SMC medium
Add 250 μl PDGFBB solution to 50 ml EGM-2 (final concentration of PDGFBB is 50 ng ml−1). Store at 4 °C up to 2 weeks.
Gelatin solution
For a 0.1% (wt/vol) solution, dissolve 0.5 g gelatin in 500 ml milli-Q water. Sterilize by autoclaving for 30 min. Let the solution cool for 30 min before coating 24-well plates. For a 1% (wt/vol) gelatin solution, dissolve 5 g gelatin in 500 ml milli-Q water and prepare as for a 0.1% solution.
Gelatin-coated 24-well plates
Pipette 1 ml of gelatin solution to each well of a 24-well plate. Incubate at 37 °C for at least 1 h and up to 2 weeks. Aspirate excess gelatin solution before use.
Collagenase type-IV solution
Dissolve 100 mg collagenase in 50 ml DMEM. Filter the solution using a 50 ml filtration system and store at 4 °C. The solution can be used up to 2 weeks.
Collagenase B solution
Dissolve 0.15 U collagenase B per ml DMEM. Sterilize the solution by filtration (0.22 μm filter) and store at 4 °C. The solution can be used up to 2 weeks.
bFGF solution
Dissolve 100 mg BSA in 2 ml PBS to form a 5% (wt/vol) BSA solution. Filter (0.22 μm) the solution. Prepare a 0.1% (wt/vol) BSA solution by adding 0.1 ml of the 5% (wt/vol) BSA solution to 4.9 ml sterile PBS. Add 1 ml of the 0.1% (wt/vol) BSA solution to a vial containing 10 μg bFGF. Aliquot the bFGF–BSA solution into sterile eppendorfs (0.25 ml per vial) and store at −20 °C up to 6 months.
VEGF165 solution
Add 0.1 ml of a 5% (wt/vol) BSA solution to 4.9 ml sterile PBS to yield a 0.1% (wt/vol) BSA solution. Add 1 ml of the 0.1% (wt/vol) BSA solution to a vial containing 10 μg of VEGF165. Aliquot the VEGF solution into sterile eppendorfs (0.250 ml per vial) and store them in the freezer at −20 °C up to 6 months.
PDGFBB solution
Prepare a 4 mM HCl solution by adding 1 ml HCl 1N to 249 ml H2O. Add 300 μl of a 5% (wt/vol) BSA solution to 2.7 ml 4 mM HCl. Filter the solution using a 0.22 μm filtration system. Add 1 ml of the filtered solution to a vial containing 10 μg PDGFBB. Aliquot the PDGFBB solution (0.250 ml aliquots) and store at −20 °C.
MACS buffer
Dissolve 0.5 g BSA in 50 ml PBS. Dissolve 0.0744 g EDTA in 50 ml PBS. Mix the BSA and EDTA solutions and sterilize by filtration (0.22 μm filter). ▲ CRITICAL Prepare fresh solutions on the day of experiment.
FACS buffer
Add 5% (vol/vol) FBS and 1% (vol/vol) penstrep to PBS and filter (0.22 μm filter). Store at 4 °C up to 2 weeks.
DNase solution
Reconstitute lyophilized DNase (100 mg) in 10 ml double distilled, sterile water. Aliquot and store at −20 °C up to 6 months.
3% PFA
Dilute 10 ml 16% (wt/vol) PFA in 40 ml PBS. Aliquot and store at −20 °C.
0.2% Triton X-100 solution
Add 200 μl Triton X-100 to 100 ml PBS and mix thoroughly. Store at room temperature (22–25 °C).
DAPI solution
Dilute DAPI 1:1,000 in PBS as follows: pipette 1 μl DAPI into 1 μl PBS and mix well. Add 5 μl PBS. 1 μl after another, and mix thoroughly. Then gradually add 994 μl PBS. ▲ CRITICAL Prepare the dilution fresh, on ice and in the dark.
Matrigel preparation
Thaw one bottle of Matrigel overnight at 4 °C on ice. Aliquot to sterilized Eppendorf tubes and freeze at −20 °C for up to 6 months. Before use, thaw aliquots overnight on ice. Coat 24-well plates with 0.4 ml per well and incubate for 30 min at 37 °C.
PLLA–PLGA scaffolds
Dissolve PLLA and PLGA (1:1) in chloroform to a 5% (wt/vol) polymer solution. Load 0.24 ml of PLLA–PLGA solution into molds packed with 0.4 g sodium chloride particles. Allow the solvent to evaporate and immerse in distilled water for 8 h. Change water every hour. This immersion allows for salt leaching and formation of an interconnected pore structure. Scaffolds can be stored in a desiccator for several months. Cut the sponges to 0.5 mm × 4 mm × 5 mm. Before transplantation, sterilize the scaffolds overnight in 70% (vol/vol) ethyl alcohol and wash three times in PBS2,20.
EQUIPMENT SETUP
FACS setup
FACSAria equipped with FACSDiva software is used for cell analysis and sorting. Set FACS parameters: adjust forward scatter and side scatter to exclude cell aggregates and cell debris. Set 530-nm band pass filter for FITC detection, and 585 nm for PI detection. For sorting, adjust to low speed using a wide nozzle of 100 μm. Use control samples to determine appropriate settings and to identify the population to be sorted. Prepare the control samples identically to experiment sample (as described in PROCEDURE, Step 3A) aside from the following changes:
For isotype control—use anti-IgG1κ–FITC antibody (1:4 dilution, 10 μl antibody per 1 × 106 cells) in place of anti-CD31–FITC antibody to confirm specific antibody binding and block nonspecific Fc receptor binding; for positive control—use ECs, such as HUVEC, that express CD31 to confirm CD31–FITC antibody-binding capacity; for Negative control—no antibody is placed over samples, to assess the background autofluoresence of cells; and for Cell viability control—use PI (0.5 μg ml−1) staining to detect dead cells. This can help in identifying and excluding dead cells from sorted cells.
PROCEDURE
EB formation and culturing ● TIMING 10–13 d
-
1|
Aspirate hESC medium and detach hESC colonies from MEF feeder layer with collagenase type IV solution. Between 1–2 EB petri dishes (10 cm diameter, total yielding between 2–5 × 106 cells) may be necessary to obtain 0.3–1 × 105 hESC-derived VPCs using their isolation protocol (Step 3B), and between 5–10 EB petri dishes (10 cm diameter, total yielding between 1–2 ×107 cells) may be necessary to obtain 0.3–1 × 105 ECs using their isolation protocol (Step 3A).
▲ CRITICAL STEP Optimal maintenance and genetic stability of hESCs are best obtained between passages 30–50.
▲ CRITICAL STEP The collagenase treatment must be optimized for each batch, as specific enzyme activity may vary.
-
2|
Gently remove hESC colonies from MEF and transfer to a 15 ml Falcon tube (typically, two 10-cm petri dishes of hESCs per Falcon tube). Centrifuge the cell suspension at 200g for 3 min at room temperature. Aspirate the supernatant and resuspend the cells in 10 ml EB medium. Add the cell suspension to a low-adherence 10 cm petri dish and incubate cells in a CO2 incubator. Change EB medium every 2–3 d.
▲ CRITICAL STEP MEF are often removed with the hESC colonies; however, most of the MEF die in the subsequent days of culture.
?TROUBLESHOOTING
Isolation of vascular cells from EBs
-
3|
Proceed with the isolation to specifically isolate hESC–ECs (option A) or hESC–VPCs (option B). The VPC and EC isolation protocol is summarized in a flow diagram in Figure 3.
(A) Isolation of hESC–ECs ● TIMING 4–6 h (i–viii) and 3–5 weeks (ix–xiii)
Transfer 13-d-old EBs from 100 mm petri dish into a 15 ml conical tube and allow EBs to sediment for 5–10 min. Aspirate supernatant and wash EBs with 10 ml PBS. Allow to sediment again for 5–10 min and aspirate supernatant.
-
Add 3 ml of cell dissociation solution to dissociate EBs into single cells. Incubate at 37 °C, 5% CO2 for 15 min, while removing the tube from incubator every 5 min for pipetting. Pipette the cells up and down several times until EBs dissolve and the solution becomes cloudy.
▲ CRITICAL STEP It is important to disaggregate EBs well to single cells as remnant aggregates are removed later by cell straining, thus resulting in cell loss and yield decrease.
? TROUBLESHOOTING
Add 4 ml FACS buffer, pipette cells and pass them through a 40 μm cell strainer to dissociate aggregates. Centrifuge at 300g for 5 min at 4 °C.
Aspirate supernatant, wash cells with 5 ml FACS buffer and count cells using a hemocytometer. Centrifuge at 300g for 5 min at 4 °C.
-
Aspirate supernatant and resuspend pellet with 40 μl FACS buffer supplemented with 10 μl anti-CD31–FITC antibody per 1 × 106 cells. Mix thoroughly using a micropipette. Incubate for 30 min at 4 °C in the dark.
▲ CRITICAL STEP From this step on, keep samples and reagents on ice and protect from light until FACS analysis is carried out.
-
Rinse cells with 10 ml FACS buffer and centrifuge at 300g for 5 min in 4 °C. Aspirate supernatant. Wash again with 5 ml FACS buffer and pass through a 40 μm cell strainer. Centrifuge at 300g for 5 min at 4 °C.
▲ CRITICAL STEP Cell straining is necessary to remove aggregates before FACS analysis. Aggregates may block the FACS flow and stop the sorting process.
Aspirate supernatant and resuspend with 1 ml FACS buffer per 107 cells. Transfer to a FACS conical tube and cover. Keep sample on ice until FACS analysis. FACS analysis should be performed within 2 h.
-
Shortly thereafter, vortex samples before placing the tube in FACS. Use control samples to properly identify the desired sorting population (prepared as described in EQUIPMENT SETUP). Sort CD31-positive cells.
▲ CRITICAL STEP Prepare control samples in parallel to sample preparations. Prepare 1–5 × 105 cells for each control sample (see EQUIPMENT SETUP).
▲ CRITICAL STEP Keep cell conditions as sterile as possible during FACS sorting.
? TROUBLESHOOTING
Expansion of hESC-derived endothelial cells: transfer sorted CD31+ cells into 15 ml conical tubes and centrifuge at 200g for 5 min at room temperature. Aspirate supernatant and resuspend in 1 ml EGM-2 medium containing 1% (vol/vol) penstrep.
Seed CD31+ cells on 1% gelatin-coated 24-well plates at a density of 105 cells per well. Incubate in 5% CO2, 37 °C.
Change medium after 3–4 d of seeding and every other day thereafter. First confluency is expected within 2–3 weeks.
-
Passage cells when they reach 80% confluency in the following manner: wash cells with HEPES solution and then harvest with Trypsin–EDTA solution placed over cells for 2–3 min. Neutralize trypsin with TNS and centrifuge at 200g for 5 min at room temperature. Split cells by a 1:3 dilution.
■ PAUSE POINT Upon passaging, cells can be stored in liquid nitrogen for later use. To prepare for storage, resuspend cells in recovery cell-culture freezing medium (see Materials; typically 1 × 106 cells per 0.5 ml freezing medium), freeze overnight at − 80 °C in a Nalgene freezing container containing propanol and then transfer to liquid nitrogen for long-term storage.
Passage cells to passage 3–4.
(B) Isolation of hESC-derived VPC s ● TIMING 5 h (i–x) and 15–30 d (xi–xiv)
-
Transfer the 10-d-old EBs (see EB formation and culturing, Step 2) from the petri dish to a 15 ml Falcon tube. Let the EBs settle in the tube before aspirating the medium. Add 2–3 ml collagenase B solution and incubate for 2 h in the CO2 incubator, 37 °C.
▲ CRITICAL STEP Regularly agitate tube to maximize the contact and activity of collagenase B with EBs.
Remove the tube from the incubator and centrifuge the cell suspension at 300g for 3 min at room temperature. Aspirate supernatant and wash EBs with 5 ml PBS. Centrifuge cells at 300g for 3 min at room temperature and aspirate supernatant. Add 2 ml of cell dissociation solution and incubate at room temperature for 10 min while agitating regularly to maximize the dissociation effect.
-
Remove the tube from the incubator and pipette colonies up and down using a 1,000 μl pipette.
▲ CRITICAL STEP Improper single-cell isolation from EB colonies results in cell clumps that cannot be passed through the cell strainer in step (iv) below, resulting in a decreased cell yield.
? TROUBLESHOOTING
Add 2 ml 5% FBS (vol/vol, in PBS) to the cell suspension and pass the cells through a 40 μm cell strainer. Centrifuge cells at 300g for 5 min at room temperature and aspirate the supernatant. Resuspend cells in 10 ml 5% FBS (vol/vol, in PBS) and count cells with a hemocytometer.
Centrifuge cells at 300g for 5 min at 4 °C and aspirate the supernatant. Resuspend cells in 300 μl 5% FBS (vol/vol, in PBS) and label cells by adding 100 μl FcR blocking solution and 100 μl of anti-CD34 microbeads (both reagents are included in the CD34 cell-isolation kit; see REAGENTS section). Keep the cell suspension on ice for 30 min.
-
After cell labeling, rinse cells with 10 ml 5% FBS (vol/vol, in PBS) and centrifuge at 300g for 5 min at 4 °C. Aspirate supernatant and repeat this washing step one more time. Finally, resuspend cells (up to 1 × 108) in 500 μl MACS buffer.
▲ CRITICAL STEP Cell clumps can clog the MACS column in step (viii) below. Therefore, cell suspension must be thoroughly pipetted up and down before its application to the MACS column.
Insert LS columns with the column wings facing the front of the MACS separator. Prepare LS columns by rinsing with 3 ml of buffer. Discard effluent and change collection tube.
-
Apply cell suspension to the prepared LS column. Collect unlabeled cells which pass through. Wash LS column three times with MACS buffer, adding buffer each time only after the column reservoir is empty.
? TROUBLESHOOTING
Remove LS column from the separator and place it over a new collection tube. Pipette 5 ml MACS buffer onto the LS column. Immediately flush out fraction with the magnetically labeled cells by firmly applying the plunger supplied with the column.
-
Centrifuge the cell suspension at 300g for 5 min at 4 °C, resuspend the cell pellets in 500 μl MACS buffer and repeat steps (vii–ix) to receive a high purity cell fraction.
▲ CRITICAL STEP After two passages through the MACS columns, the purity of the cells is typically above 80%. However, flow cytometry is recommended for the evaluation of cell purity using an anti-CD34 monoclonal antibody recognizing a region other than that recognized by the beads used in step (v) (e.g., anti-human CD34 antibody clone AC136, commercially available through Miltenyi Biotec).
Differentiation of VPCs into endothelial or SMCs: pipette 1 ml 0.1% gelatin solution to each well of a 24-well plate and incubate at 37 °C for at least 1 h. Aspirate excess gelatin solution before use.
Centrifuge CD34+ cells at 300g for 5 min at room temperature and aspirate supernatant. Resuspend cells in EC medium and plate (3 × 104 cells per well) in the gelatin-coated wells containing EC medium for endothelial differentiation or SMC medium for SMC differentiation.
-
Change the medium 3–4 d after cell seeding and then daily thereafter.
▲ CRITICAL STEP Cell confluency is typically achieved within 8–15 d, although time-to-confluency is highly dependent on the degree of adhesion and the growth capacities of each cell type (EC versus SMC). If the cell adhesion yield is low, cells may become apoptotic.
? TROUBLESHOOTING
-
Passage cells when they reach 80% confluency. Wash cells with PBS and subsequently harvest them with a 0.05% Trypsin–EDTA solution for 2–3 min. Block further trypsin activity by washing the harvested cells with TNS.
■ PAUSE POINT Upon passaging, cells can be stored in liquid nitrogen for later use. To prepare for storage, resuspend cells in recovery cell culture freezing medium (see MATERIALS section; typically 1 × 106 cells per 0.5 ml freezing medium), freeze overnight at − 80 °C in a Nalgene freezing container containing propanol and then transfer to liquid nitrogen for long-term storage.
Cell characterization
-
4|
Characterize the differentiated cells at passages 3–4 using the following methods of analysis: immunostaining (option A), gene expression (option B), flow cytometry (option C), ac-LDL uptake (option D), Matrigel assay (option E), contraction assay (option F) and/or in vivo transplantation (option G).
(A) Immunostaining ● TIMING 3–4 h
Transfer the differentiated cells (as described in Steps 3A(xii) and 3B(xiv)) suspended in EC or SMC medium (depending on the cell type) to gelatin-coated chamber slides and allow for cell attachment overnight. Wash slides twice with PBS and fix cells with 4% PFA (wt/vol) for 30 min at room temperature.
Wash twice with PBS and then permeabilize cells with 0.2% Triton X-100 (vol/vol) prepared in PBS, for 30 min.
Aspirate the permeabilization solution, wash twice with PBS and block nonspecific antigen sites with 3% BSA (wt/vol) prepared in PBS for 30 min.
Aspirate blocking solution and incubate cells with relevant antibodies for 1 h at room temperature. For endothelial cells, incubate cells with the following anti-human primary antibodies: CD31, CD34, vWF, VE-cad, N-cad, vinculin or FITC-phalloidin for actin staining. Use an isotype-matched IgG control for each experiment. Use the following anti-human primary antibodies for the characterization of SMCs: α-SMA, SM–MHC or calponin. Dilutions of all antibodies are carried out with antibody diluent solution (see REAGENTS SETUP section) and are summarized in Table 2.
Wash cells twice with PBS.
Incubate cells with secondary antibody (dilutions as indicated in Table 2) for 30 min, in the dark.
Aspirate the antibody solution, wash twice with PBS and then stain cell nuclei with DAPI solution (see REAGENT SETUP) for 15 min.
Aspirate the DAPI solution, wash twice with PBS and remove the wells according to the instructions of the chamber slide manufacturer.
-
Add fluorescent mounting medium (Fluoromount G) and place a cover slip over the slide. Let the slide dry overnight in the dark.
■ PAUSE POINT Slides can be stored in a dark place at room temperature for up to 1 year.
-
View the immunostaining with a fluorescence or confocal microscope.
? TROUBLESHOOTING
Table 2.
Antibody sources and working concentrations.
| Dilution | Supplier | Cat. no. | |
|---|---|---|---|
| Primary antibody | |||
| Mouse monoclonal anti-human CD31 | 1:20 | Dako | M 0823 |
| Mouse monoclonal anti-human CD34 | 1:20 | Dako | M 7165 (clone QBEnd/10) |
| Mouse monoclonal anti-human CD34 | 1:1,000 | Lab Vision | MS-363-P1ABX (clone QBEnd/10) |
| Mouse monoclonal anti-human vWF | 1:20 | Dako | M 0616 |
| Rabbit polyclonal anti-human vWF | 1:200 | Dako | A0082 |
| Mouse monoclonal anti-human alpha smooth muscle actin (α-SMA) | 1:50 | Dako | M 0851 |
| Mouse monoclonal anti-human smooth muscle myosin heavy chain (SM–MHC) | 1:50 | Dako | M 3558 |
| Mouse monoclonal anti-human calponin | 1:50 | Dako | M 3556 |
| Negative control mouse IgG2a | 1:50 | Dako | X 0943 |
| Negative control mouse IgG1 | 1:20 | Dako | X 0931 |
| Mouse monoclonal anti-vascular endothelial (VE)-cadherin | 1:50 | Santa-Cruz | sc-9989 (F-8) |
| Mouse monoclonal anti-vinculin | 1:400 | Sigma | V9131 |
| Rabbit anti-N-cadherin | 1:50 | Santa-Cruz | sc-7939 |
| FITC-phalloidin | 1:500 | Sigma | P5282 |
| Secondary antibody | |||
| Sheep anti-mouse IgG–Cy3 | 1:50 | Sigma | C 2181 |
| Rabbit anti-mouse IgG–FITC | 1:20 | Dako | F 0261 |
| Goat anti-mouse IgG–Cy3 | 1:100 | Jackson ImmunoResearch | 115-166-072 |
| AlexaFluor-488 goat anti-rabbit IgG | 1:1,000 | Molecular Probes | A-11034 |
| Fluorecense-conjugated antibodies for fluorescent-activated cell sorting (FACS) | |||
| Mouse anti-human CD31–FITC | 1:4 | BD Pharmingen | 555445 |
| Mouse anti-human IgG1κ–FITC isotype | 1:4 | BD Pharmingen | 555748 |
| Control mouse anti-human CD34–FITC | 1:11 | Miltenyi Biotech | 130-081-001 |
| Mouse anti-human KDR–Flk1-PE | Ready to use | R&D Systems | (clone AC136) FAB357P |
| Mouse anti-human IgG1-PE isotype control | Ready to use | R&D Systems | (clone 89106) IC002P |
(B) Gene expression analyses ● TIMING 2 d
Extract total RNA from 1 × 106 EC or SMCs, using Trizol according to the manufacturer’s protocol.
-
Quantify total RNA by a UV spectrophotometer.
■ PAUSE POINT RNA can be stored at − 80 °C until use.
-
Carry out a reverse transcription (RT) reaction with 1 μg extracted RNA using the M-MLV RT enzyme kit, according to manufacturer’s instructions.
■ PAUSE POINT cDNA can be stored at − 80 °C until use.
-
Perform PCRs with BIOTAQ DNA polymerase using 1 μl of RT product per reaction. Primer sequences, reaction conditions and optimal cycle numbers are summarized in Table 1. To ensure semiquantitative results of the RT-PCR assays, the number of PCR cycles for each set of primers must be verified to be within the linear range of the amplification.
In addition, all RNA samples must be adjusted to yield equal amplification of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an internal standard.
Separate amplified products on 2% agarose gels prepared with ethidium bromide (0.4 μg ml− 1). Illuminate the gel with UV light to observe the bands.
(C) Flow cytometry analyses ● TIMING 3 h
Remove cells from plate using trypsin and transfer into a 15 ml conical tube. Centrifuge at 200g for 5 min at room temperature. Aspirate supernatant and wash cells with 5 ml PBS.
Count cells using a hemocytometer and calculate the number of cells required for all samples, where 1–5 × 105 cells are suggested for each sample. Centrifuge at 300g for 5 min, 4 °C. Wash again with 5 ml FACS buffer and centrifuge at 300g for 5 min, 4 °C.
-
Aspirate supernatant and incubate cells with antibodies diluted as summarized in Table 2. Stain cells with anti-CD31–FITC, anti-CD34–FITC, anti-Flk1–VEGFR-2-PE (purchased ready to use, 10 μl per 105 cells) or with an isotype-matched IgG control antibody (see Table 2). Mix thoroughly using a micropipette. Incubate for 30 min at 4 °C in the dark.
▲ CRITICAL STEP From this step on, keep samples and reagents on ice and protect from light until FACS analysis is carried out.
-
Wash samples with 5 ml FACS buffer and centrifuge at 300g for 5 min, 4 °C. Aspirate supernatant. Wash again with 5 ml FACS buffer and pass through a 40 μm cell strainer. Centrifuge at 300g for 5 min in 4 °C.
▲ CRITICAL STEP Cell strainer is necessary to remove cell clumps before using FACS.
Aspirate supernatant and resuspend cells in 300 μl FACS buffer. Add PI (0.5 μg ml− 1) for detection of dead cells. Transfer to a FACS conical tube and cover. Keep samples on ice until FACS analysis is carried out within 2 h.
Shortly vortex FACS tubes before analysis. Use HUVEC as a positive control sample and an EC or SMC cell sample not treated with the labeling antibody as negative control (see EQUIPMENT SETUP). Exclude dead cells by gating out PI-positive cells.
(D) ac-LDL uptake ● TIMING 5 h
Incubate cells with 10 μg ml−1 Dil-ac-LDL for 4 h at 37 °C.
Wash cells three times with PBS.
Fix cells with 3% PFA (wt/vol) for 30 min. Wash twice with PBS.
View ac-LDL uptake under a fluorescent microscope and compare that of HUVEC and CD31− cells treated in a similar manner.
(E) Matrigel assay ● TIMING 1–3 d
Seed cells on Matrigel-coated 24-well plates to a concentration of 1 × 105 cells per well in 300 μl of their respective culture media. Incubate 1 h at 37 °C and then add 1 ml of fresh medium.
Incubate for 1–3 d at 37 °C.
Observe cord-like structures using a phase-contrast microscope. Cord formation can also be observed by electron microscopy.
(F) Contraction assay ● TIMING 1 d
Seed VPC-derived SMCs on a gelatin-coated 24-well plate at a concentration of 7 × 104 cells per well in 1 ml of their culture media. In a separate experimental plate use HUASMCs as control.
Wash cells, quantify the cell area using a light microscope and induce cell contraction by addition of 1 × 10− 5 M carbachol diluted in DMEM medium for 30 min. In a separate experiment, induce cell relaxation by addition of 1 × 10− 4 M atropine prepared in DMEM for 1 h. Quantify cell area using a light microscope, and then induce cell contraction by incubating cells with 1 × 10− 5 M carbachol for 30 min.
Calculate cell contraction by the difference of cell area between time zero and time 30 min. Use bright-field images (magnifications ×10 or ×20) for this purpose.
(G) Transplantation into SC ID mice ● TIMING 10–31 d
Remove cells from plate using trypsin and transfer to a 15 ml conical tube. Count cells using a hemocytometer and centrifuge at 200g for 5 min at room temperature. Aspirate supernatant.
Resuspend 1 × 106 cells per scaffold in a 50 μl culture medium and Matrigel mix (1:1) and allow to absorb into the PLLA–PLGA polymer sponge. Incubate at 37 °C for 30 min. Transplant the scaffolds subcutaneously (s.c.) in the dorsal region of 4-week-old SCID mice. Alternatively, for VPC-derived ECs and SMCs, resuspend 0.5 × 106 cells in 20 μl of cell medium and mix with 350 μl Matrigel on ice. Inject subcutaneously the cell suspension in the dorsal region of 4-week-old nude mice.
Sacrifice mice from 7 to 28 d after transplantation and retrieve implants.
Fix scaffolds overnight in 10% buffered formalin (vol/vol) at 4 °C.
-
Embed in paraffin and cut to 5 μm sections for immunohistological staining.
■ PAUSE POINT Slides can be stored at 4 °C for 1–3 years.
Deparaffinize sections and incubate with Reveal buffer at 95 °C for 20 min to allow for antigen recovery. Cool slides for 20 min at RT and wash twice with PBS.
Block sections with Sniper for 5 min at RT.
-
Stain sections using the ARK kit reagents and according to the kit instructions. Shortly thereafter, incubate slides for 2 h with prepared biotinylated mouse primary antibodies against human CD31 (Dako), vWF (Dako, M0616) and CD34 (Lab Vision) (see Table 2). Then, incubate sections with streptavidin peroxidase and complete the staining with DAB Chromogen, which results in a brown-colored precipitate at the antigen site (all reagents are included in the kit).
■ PAUSE POINT Stained sections can be stored at room temperature for 1–3 years.
View sections using a phase-contrast microscope.
● TIMING
Steps 1 and 2, EB formation and culturing: 10–13 d
Step 3A, Isolation of hESC–ECs: 4–6 h (i–viii) and 3–5 weeks (ix–xiii)
Step 3B, Isolation of hESC-derived VPCs: 5 h (i–x) and 15–30 d (xi–xiv)
Step 4A, Immunostaining: 3–4 h
Step 4B, Gene expression analyses: 2 d
Step 4C, Flow cytometry analyses: 3 h
Step 4D, ac-LDL uptake: 5 h
Step 4E, Matrigel assay: 1–3 d
Step 4F, Contraction assay: 1 d
Step 4G, Transplantation into SCID mice: 10–31 d
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 3.
Table 3.
Troubleshooting table.
| Step | Problem | Possible reason and solution |
|---|---|---|
| 2 | Embryoid body (EB) medium becomes yellow during EB growth, indicating increase of acidity level in medium EB attachment to the dish |
This happens because of the consumption of oxygen by the growing EBs, resulting in high levels of secreted CO2, which then leads to a pH reduction. Split EBs into two plates to decrease number of cells in each plate and replace EB medium with fresh medium Scrape EBs off the dish using a cell scraper |
| 3A(ii) and 3B(iii) | Inefficient cell dissociation | Prolong the treatment time with cell dissociation solution. The cell suspension can be incubator; force dissociation by thorough incubated for 1–2 additional min in the CO2 pipetting |
| 3A(ii) | Formation of DNA aggregates during EB dissociation | Often because of DNA release from dead cells. Add DNase solution (10 μl–1 ml) to dissolve DNA aggregates |
| 3A(viii) | Low yield of CD31+ cells after sorting |
|
| 3B(viii) | Column occlusion | Cell aggregates may block the magnetic-activated cell separation (MACS) columns. Transfer the column from the magnet to a 50 ml falcon tube. Flush out all cells by firmly applying the plunger supplied with the column. Add 2 ml of 5% FBS (vol/vol, in PBS) to the cell suspension and centrifuge the cells at 200g for 3 min at 4 °C. Aspirate the supernatant and thoroughly resuspend the cells in 500 μl of MACS buffer |
| 3B(xiii) | Cell contamination | Cells with atypical morphology that attach within the first day. Remove the cells with the pipette tip. If unsuccessful, check the proliferation rate of the cells over time. If they show a low proliferation rate, it is unlikely that they will interfere with the differentiation program of CD34+ cells |
| 4A(x) | No cell staining | Confirm both antibody activity/binding capacities and staining protocol by using primary endothelial (human umbilical vein endothelial cells (HUVECs)) and smooth muscle cells (human umbilical artery smooth muscle cells (HUASMCs)) as positive controls. These cells express typical endothelial and smooth muscle cell markers |
| 4A–C | Low expression of vascular markers | The vascular potential of hESCs may differ between cell lines. More specifically, our experience has shown that differentiation of CD34+ cells to the endothelial lineage is less efficient in H13 when compared with the H9 cell line |
ANTICIPATED RESULTS
This protocol presents a simple method for the isolation of hES-derived ECs, with a typical yield of 2% CD31-positive cells of the EBs dissociated on day 13 of their culturing. Further culturing of the isolated CD31-positive cells shows high purity of nearly 80% CD31-expressing cells2 when assessed after several passages. The remaining 20% are fibroblast-like cells, as determined by their typical elongated morphology and reactivity to anti-SMA antibodies using immunohistochemical staining. A total of 5 × 104 CD31+ cells plated and cultured for three passages yield ~1 × 106 EC cells. The isolated ECs demonstrate phenotypic endothelial characteristics as determined by flow cytometry, immunostaining, LDL incorporation and formation of blood vessel-like structures both in vitro and in vivo2. FACS analysis of passage six CD31+ isolated cells detected CD34 and Flk1 expression levels similar to those expressed in HUVEC2. In addition, immunostaining of these isolated cells showed expression of both CD31 and vWF (Fig. 1b), as well as standard EC organization of the VE-cad, N-cad and vinculin junction molecules2. CD31+ cells isolated in this manner also demonstrated the endothelial biofunctional characteristics by efficiently incorporating ac-LDL as detected by fluorescence microscopy after incubation with Dil-ac-LDL (Fig. 1c). Furthermore, these CD31+ ECs formed blood vessel-like structures both in vitro by Matrigel and in vivo in PLLA–PLGA scaffolds transplanted in SCID mice. These vessels anastomosed with the mouse vasculature2.
Vascular progenitor cells represent 10% of the overall EB cell population cultured in suspension for 10 d. Upon MACS isolation, > 90% of the isolated VPCs express CD34 antigen. At this stage, CD34+ cells co-express high levels of CD31 (~55%), α-SMA (~45%) and SSEA4 (~43%), moderate levels of KDR–Flk-1 (~16%) and low levels of the hematopoietic marker CD45 (~1%)1. The differentiation of 3.5 × 104 CD34+ cells cultured for three passages in EC medium yields ~1 × 106 ECs, whereas the same initial number of CD34+ cells grown in SMC medium for the same number of passages, yields ~4 × 106 cells. VPC-derived ECs are characterized by cobblestone-like cell morphology and express typical endothelial markers at gene or protein levels, such as CD31, CD34, KDR–Flk-1, VE-cad and vWF (Fig. 2), and incorporate ac-LDL. In addition, these cells do not express the SM-MHC, SMα-22 or angiopoietin-1 SMC markers. In contrast, VPC-derived SMCs are characterized by a spindle-like shaped morphology, express the α-SMA, SM-MHC, calponin, caldesmon and SMα-22 SMC markers (Fig. 2), and have the ability to contract and relax in response to addition of common pharmacological agents such as carbachol and atropine. However, in some cases VPC-derived SMCs express some endothelial markers including angiopoietin-2 and Tie2 (ref. 1) (see below). Subcutaneous transplantation of VPC-derived ECs alone or together with VPC-derived SMCs, in the dorsal region of nude mice, led to microvessel formation. Most of these microvessels were patent with empty lumens, whereas a small percentage (~5–6%) contained mouse RBCs1. α-SMA+ cells formed small, independent tubules or surrounded human microvessels1.
Although the ECs and SMCs obtained from this differentiation and expansion protocol may not be identical to mature endothelial and SMCs, they share many features with mature vascular cells. Their differences may stem from their embryonic source or incomplete stages of differentiation. For example, the endothelial-derived cells isolated at day 13–15 from EBs and expanded in vitro for six passages express similar levels of CD34 and Flk-1 to those detected in HUVEC, but not similar levels of CD31 (78% versus 98% in HUVEC)2. In addition, VPC-derived SMCs are not completely differentiated as demonstrated by low expression of CD31 (~5%) and CD34 (~1%) markers while also expressing Tie2 and angiopoietin-2 markers characteristic of ECs1.
When comparing the two methods of EC isolations, both CD31+ -based and CD34+ -based isolations yielded EC populations with similar characteristics, as demonstrated by their typical endothelial cell morphology as well as expression of typical endothelial markers such as CD31, CD34, KDR–Flk-1, VE-cad and vWF (Figs. 1 and 2). Flow cytometry analyses revealed differences in the expression levels of some membranal endothelial markers: 98% of the ECs expended from CD34+ isolations expressed CD31 versus 78% from the CD31+ isolation, 65% of the ECs expended from CD34+ -isolated cells express CD34 versus 16% expressing CD34 in CD31+ -isolated cultures, and only 12% of the CD34+ -derived ECs expressed Flk1, whereas 19% of the CD31+ -isolated ECs expressed this protein. ECs originating from CD34+ isolations expressed similar levels of CD31 (98%) to those detected in HUVEC, although ECs derived from CD31+ isolations expressed similar levels of CD34 (~16%) and Flk-1 (~19%) to those detected in HUVEC1,2.
Effective incorporation of ac-LDL and formation of blood vessel-like structures both in vitro by Matrigel and in vivo by transplantation into immunodeficient mice1,2 were used to determine EC functionality of both CD31+ and CD34+ -derived populations.
Despite similarities in their external markers, the two population groups were isolated at different yields, owing to their ratios in the initial EB cell population. Although CD34+ cells represent 10% of the total EB cell population on 10-d culture, CD31+ cells represent only 2% of the cell population in 13-d old EBs. Therefore, five-fold more EBs would be required to reach the same number of isolated ECs when using the CD31+ -based isolation method when compared with the number required using CD34+ -based isolation. However, in the VPC protocol, VEGF is needed to differentiate the VPC to ECs.
Acknowledgments
We would like to acknowledge the financial support of NIH grants HL060435 and DE013023 (R.L.), Marie-Curie Reintegration Grants (S.L. and L.S.F.), MIT-Portugal program (L.S.F.), Crioestaminal/Associação Viver a Ciência (L.S.F.) and FCT (PTDC/SAU-BEB/098468/2008) (L.S.F.). TPK was supported by the Swiss National Science Foundation (grant number PBELP3-127902).
Footnotes
AUTHOR CONTRIBUTIONS All authors wrote this paper.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/.
References
- 1.Ferreira LS, et al. Vascular progenitor cells isolated from human embryonic stem cells give rise to endothelial and smooth muscle like cells and form vascular networks in vivo. Circ Res. 2007;101:286–294. doi: 10.1161/CIRCRESAHA.107.150201. [DOI] [PubMed] [Google Scholar]
- 2.Levenberg S, Golub JS, Amit M, Itskovitz-Eldor J, Langer R. Endothelial cells derived from human embryonic stem cells. Proc Natl Acad Sci USA. 2002;99:4391–4396. doi: 10.1073/pnas.032074999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levenberg S. Engineering blood vessels from stem cells: recent advances and applications. Curr Opin Biotechnol. 2005;16:516–523. doi: 10.1016/j.copbio.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 4.Levenberg S, et al. Differentiation of human embryonic stem cells on three-dimensional polymer scaffolds. Proc Natl Acad Sci USA. 2003;100:12741–12746. doi: 10.1073/pnas.1735463100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levenberg S, et al. Engineering vascularized skeletal muscle tissue. Nat Biotechnol. 2005;23:879–884. doi: 10.1038/nbt1109. [DOI] [PubMed] [Google Scholar]
- 6.Levenberg S, Zoldan J, Basevitch Y, Langer R. Endothelial potential of human embryonic stem cells. Blood. 2007;110:806–814. doi: 10.1182/blood-2006-08-019190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 8.Ferreira LS, et al. Bioactive hydrogel scaffolds for controllable vascular differentiation of human embryonic stem cells. Biomaterials. 2007;28:2706–2717. doi: 10.1016/j.biomaterials.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coultas L, Chawengsaksophak K, Rossant J. Endothelial cells and VEGF in vascular development. Nature. 2005;438:937–945. doi: 10.1038/nature04479. [DOI] [PubMed] [Google Scholar]
- 10.Lammert E, Cleaver O, Melton D. Induction of pancreatic differentiation by signals from blood vessels. Science. 2001;294:564–567. doi: 10.1126/science.1064344. [DOI] [PubMed] [Google Scholar]
- 11.Shen Q, et al. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science. 2004;304:1338–1340. doi: 10.1126/science.1095505. [DOI] [PubMed] [Google Scholar]
- 12.Pouton CW, Haynes JM. Embryonic stem cells as a source of models for drug discovery. Nat Rev Drug Discov. 2007;6:605–616. doi: 10.1038/nrd2194. [DOI] [PubMed] [Google Scholar]
- 13.Sartipy P, Bjorquist P, Strehl R, Hyllner J. The application of human embryonic stem cell technologies to drug discovery. Drug Discov Today. 2007;12:688–699. doi: 10.1016/j.drudis.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 14.Bai H, Wang ZZ. Directing human embryonic stem cells to generate vascular progenitor cells. Gene Ther. 2008;15:89–95. doi: 10.1038/sj.gt.3303005. [DOI] [PubMed] [Google Scholar]
- 15.Vodyanik MA, Bork JA, Thomson JA. Slukvin, II human embryonic stem cell-derived CD34+ cells: efficient production in the coculture with OP9 stromal cells and analysis of lymphohematopoietic potential. Blood. 2005;105:617–626. doi: 10.1182/blood-2004-04-1649. [DOI] [PubMed] [Google Scholar]
- 16.Wang ZZ, et al. Endothelial cells derived from human embryonic stem cells form durable blood vessels in vivo. Nat Biotechnol. 2007;25:317–318. doi: 10.1038/nbt1287. [DOI] [PubMed] [Google Scholar]
- 17.Lerou PH, et al. Derivation and maintenance of human embryonic stem cells from poor-quality in vitro fertilization embryos. Nat Protoc. 2008;3:923–933. doi: 10.1038/nprot.2008.60. [DOI] [PubMed] [Google Scholar]
- 18.Braam SR, et al. Feeder-free culture of human embryonic stem cells in conditioned medium for efficient genetic modification. Nat Protoc. 2008;3:1435–1443. doi: 10.1038/nprot.2008.140. [DOI] [PubMed] [Google Scholar]
- 19.AEM Isolation and propagation of mouse embryonic fibroblasts and preparation of mouse embryonic feeder layer cells. Curr Protoc Stem Cell Biol. 2007;Chapter 1 doi: 10.1002/9780470151808.sc01c03s3. [DOI] [PubMed] [Google Scholar]
- 20.Mooney DJ, et al. Long-term engraftment of hepatocytes transplanted on biodegradable polymer sponges. J Biomed Mater Res. 1997;37:413–420. doi: 10.1002/(sici)1097-4636(19971205)37:3<413::aid-jbm12>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]