

Abstract
Multicomponent reactions allow for more bond-forming events per synthetic operation, enabling more step and time economical conversion of simple starting materials to complex and thus value-added targets. These processes invariably require that reactivity be relayed from intermediate to intermediate over several mechanistic steps until a termination event produces the final product. Here we report a multicomponent process in which a novel 1,2,3-butatriene equivalent (TMSBO: TMSCH2C≡CCH2OH) engages chemospecifically as a two-carbon alkyne component in a metal-catalyzed [5+2] cycloaddition with a vinylcyclopropane to produce an intermediate cycloadduct. Under the reaction conditions, this intermediate undergoes a remarkably rapid 1,4-Peterson elimination, producing a reactive four-carbon diene intermediate that is readily intercepted in either a metal-catalyzed or thermal [4+2] cycloaddition. TMSBO thus serves as an yne-to-diene transmissive reagent coupling two powerful and convergent cycloadditions - the homologous Diels-Alder and Diels-Alder cycloadditions - through a vinylogous Peterson elimination, and enabling flexible access to diverse polycycles.
A pre-eminent goal of synthesis is to produce targeted structural complexity and thus functional value with step and time economy in a green if not ideal fashion.1–3 Multicomponent reactions provide a powerful means to achieve this end as they allow for the multiply convergent assembly of complex targets from two or more simple, often commercially available, fragments. Multicomponent processes rely on relaying or regenerating reactivity such that the “product” of each step is a reactive “starting material” for a subsequent bond-forming step until a termination event occurs.4–10 While in many multicomponent processes a single type of reactive intermediate (e.g., cation, anion, radical, etc.) is regenerated with each step, the propagation of reactivity can also involve a change in reactive species or reagents. For example, in considering approaches to a new family of selective kinase inhibitors (Figure 1, II), a subject of much current clinical interest,11 it occurred to us that access to designed, structurally simplified staurosporine-like 5-6-7 polycyclic targets incorporating the critical activity determining functionality of the natural product lead could be realized in one operation by using 1,2,3-butatriene (V) as a reactivity regenerating reagent (Figure 1).12–15 According to this plan, butatriene would function as a two-carbon ene component in an initiating rhodium-catalyzed [5+2] cycloaddition with a vinylcyclopropane (VCP), during which it would be transformed into a reactive four-carbon diene IV for a subsequent Diels-Alder or related cycloaddition. While the importance of butatriene equivalents has been recognized, equivalency is achieved only through 2 or more reactions.16–19 We describe herein a highly effective, regioselective butatriene (cumulene) equivalent that allows for the realization of this single-flask multicomponent process.
Figure 1.

Retrosynthetic analysis of staurosporine analogues based on butatriene-enabled cycloadditions. With the goal of step-economically generating a library of simplified staurosporine analogues with selective kinase inhibitory function (FOS: function-oriented synthesis), we envisaged a 1,2,3-butatriene (V) reacting first in a [5+2] cycloaddition to provide a 1,3-diene ready for a subsequent [4+2] cycloaddition.
Butatriene itself has found only limited use in synthesis partly due to difficulties with its preparation, safe handling, physical properties (gas at room temperature) and propensity to readily polymerize even at low temperatures (−40 °C).20–23 Its reactions with various reagents under a variety of reaction conditions often provide polymeric materials. These problems extend as well to many other low molecular weight cumulenes, restricting their synthetic utility. Control of chemoselectivity in the reactions of such trienes poses an additional problem in their synthetic use arising from the differing steric and electronic features of the component π-systems. 4-(Trimethylsilyl)but-2-yn-1-ol (TMSBO 2, Figure 2) represents a potential butatriene equivalent that could circumvent all of these problems. It is an easily handled and prepared24–29 liquid at room temperature and would be expected to engage an ynophilic partner exclusively at its 2,3-π bond. The resultant product would be poised for a relatively under-exploited vinylogous Peterson elimination,30–37 thereby producing in situ a reactive diene for a subsequent [4+2] cycloaddition (or other diene-initiated reaction). Overall, TMSBO would serve as an yne-to-diene transmissive reagent coupling two powerful and convergent cycloadditions – the homologous Diels-Alder and the Diels-Alder cycloadditions – through a vinylogous Peterson elimination, all effected in one synthetic operation (Figure 2).
Figure 2.

General depiction of the [5+2] cycloaddition/vinylogous Peterson elimination/[4+2] cycloaddition cascade and mechanistic possibilities. The 1,2,3-butatriene equivalent, TMSBO, reacts under rhodium catalysis to give an intermediate cycloadduct. Under the reaction conditions, we observed the spontaneous deprotection of the enol ether and an unexpectedly facile vinylogous Peterson elimination, giving directly the 1,3-diene we had envisaged in our retrosynthetic analysis. The Peterson elimination presumably occurs by either a Lewis acid promoted elimination or a zwitterionic cycloreversion. In the presence of a suitable dienophile, a one-flask conversion to polycyclic products can be achieved.
Results and Discussion
To test whether TMSBO 2 (Figure 2) could function as a butatriene equivalent in a [5+2] cycloaddition, its reaction with commercially available VCP 1 in the presence of [(naph)Rh(COD)]SbF638 (2 mol%) was initially investigated. The reaction proceeded smoothly at room temperature, producing after 6 hours a single product in high yield. Significantly, this product proved not to be the expected cycloadduct VII but rather the diene 3 resulting from both a [5+2] cycloaddition and vinylogous Peterson elimination. The expected [5+2] cycloadduct was not isolated or detected by TLC analysis during the reaction, suggesting that it undergoes facile elimination at a rate comparable to its rate of formation. An alternative mechanistic possibility in which TMSBO is first converted to butatriene is rendered unlikely as a control experiment indicates that TMSBO is unchanged after multiple days under the reaction conditions in the absence of VCP 1. To further probe the mechanism of this transformation, the reaction of the methoxy derivative of TMSBO, TMSCH2C≡CCH2OMe (TMSBOMe), was explored. Interestingly, in this case, NMR studies suggest that not only does the cycloaddition reaction proceed less rapidly, but additionally, the rate of elimination to form the diene product is also slowed. The facility of the elimination with TMSBO and the contrasting behavior of TMSBOMe suggests a steric effect in the Lewis acid activation stage of the 1,4-elimination VIII or an unexplored cycloreversion of a zwitterion formed through bond formation between the oxygen and silicon atoms IX (Figure 2). Mechanistic studies on these various pathways are in progress and will be reported in due course.
The above results establish that TMSBO is a superb substrate for [5+2] cycloadditions and serves as a highly effective butatriene equivalent. These findings set the stage for numerous alkyne-to-diene based reactions that would engage a third component, including Diels-Alder cycloadditions and metal-catalyzed variants, to produce polycyclic products. Given the alkynophilic nature of the [5+2] cycloadditions, we next sought to determine whether the initial [5+2]/elimination process could be conducted in the presence of an alkene Diels-Alder dienophile. This proved to be highly successful. As illustrated in Table 1, a variety of alkene dienophiles can be mixed with TMSBO 2 and VCP 1 in the presence of the Rh(I) catalyst to give directly the product of the [5+2] cycloaddition/elimination/[4+2] cycloaddition sequence. Maleate, fumarate and acrylate esters did not interfere with the catalyst, initial cycloaddition and subsequent elimination and could thus be added at the outset of the process although heating was required for completion of the final cycloaddition. In contrast, 4-phenyl-1,2,4-triazoline-3,5-dione did complex the catalyst, interfering with the initial cycloaddition. However, by delaying its addition until after the initial cycloaddition and elimination, the three-component, one-flask process can be achieved in good yield.
Table 1.
[5+2]/vinylogous Peterson/[4+2] adducts using VCP 1 and TMSBO 2
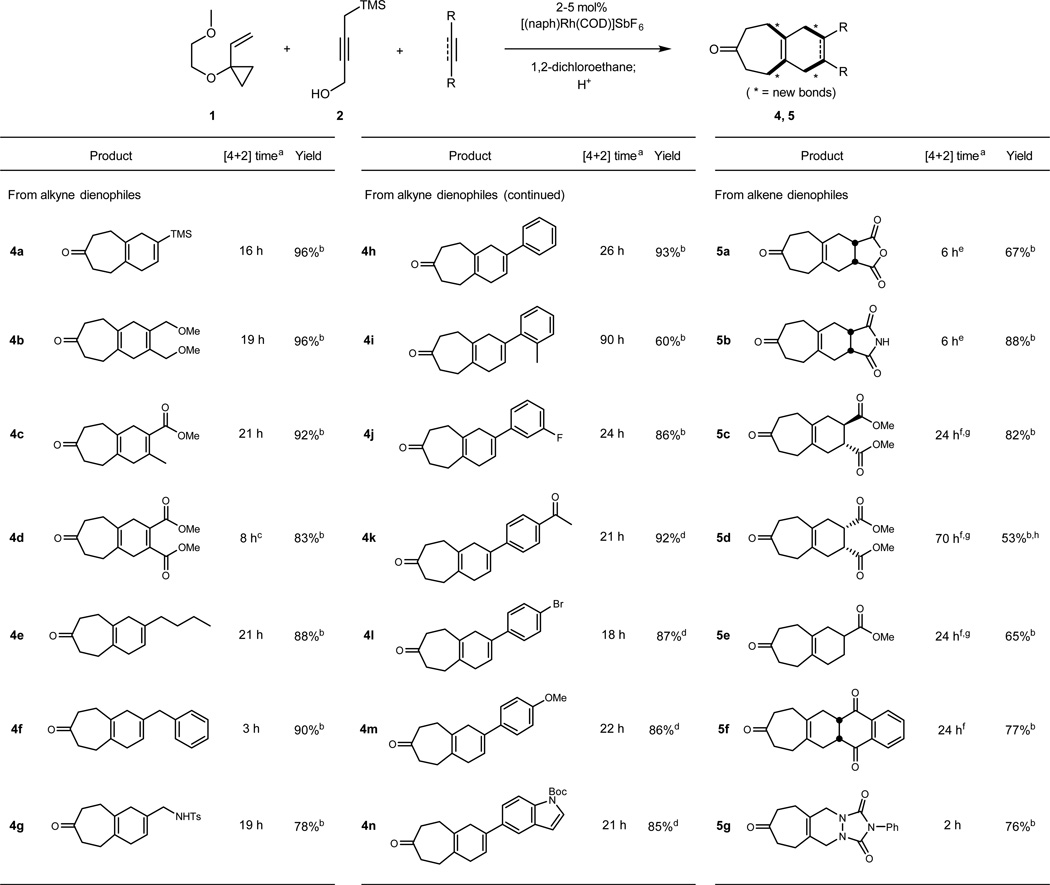 |
Reactions were conducted at room temperature unless otherwise noted.
Unless otherwise noted, this time is the time required for only the [4+2] portion of the reaction cascade
2 mol% [(naph)Rh(COD)]SbF6
The [4+2] cycloaddition was effected at 70 °C
5 mol% [(naph)Rh(COD)]SbF6
This time reflects the total time for both the [5+2] and [4+2] processes
Reaction run at 0.330 M
The [4+2] cycloaddition was effected at 100 °C
Product is a 1.8:1 mixture of cis:trans esters starting from pure dimethyl maleate
In addition to alkenes, alkynes – abundantly available synthetic building blocks – can be used as a third component in the process. For this purpose, addition of the second alkyne is delayed until after the initial [5+2] and elimination processes are complete. Significantly, both Diels-Alder [4+2] cycloadditions and transition metal-catalyzed formal [4+2] cycloadditions are possible. The former provide three component products in high yield (entries 5a–5g, 8, 10). Alkynes that do not react through a conventional Diels-Alder cycloaddition because of a poor HOMO-LUMO gap can be induced to react through an alternative, multistep metal-catalyzed [4+2] cycloaddition. In this case, the catalyst used in the [5+2] cycloaddition is subsequently employed as a catalyst in the second [4+2] cycloaddition. For example, addition of VCP 1 and TMSBO to a solution of the catalyst followed by addition of 3-phenylpropyne gives the three component product 4f in 90% yield. That the second cycloaddition is Rh(I) catalyzed is supported by a control experiment in which diene 3 and 3-phenylpropyne are allowed to react in the absence of catalyst. No product is formed even after 2 days. A wide range of electron-rich, electron poor, internal and terminal alkynes are accommodated in this sequence using either the thermal or metal-catalyzed [4+2] cycloaddition option (Table 1, entries 4a–4n).
In connection with our original interest in kinase inhibitors, we have also found that one can readily produce the corresponding arenyl core ring through oxidation of the initially formed cyclohexadienyl products or alternatively by starting with a higher oxidation level dienophile and effecting a double elimination after the cascade (Table 2). Finally, while our initial focus was on the exploration of the viability of this one operation multicomponent process, we have also found that it can be extended to variations in both the starting VCP and the reactivity transmissive reagent (Table 3). Thus, the substituted VCP 9 readily enters into the sequence producing in one operation the polycycle 10. Similarly, a methyl substituted butatriene equivalent 11 also reacts readily to form products 12a and 12b, providing yet a further site for diversification.
Table 2.
Oxidative aromatization of reaction products
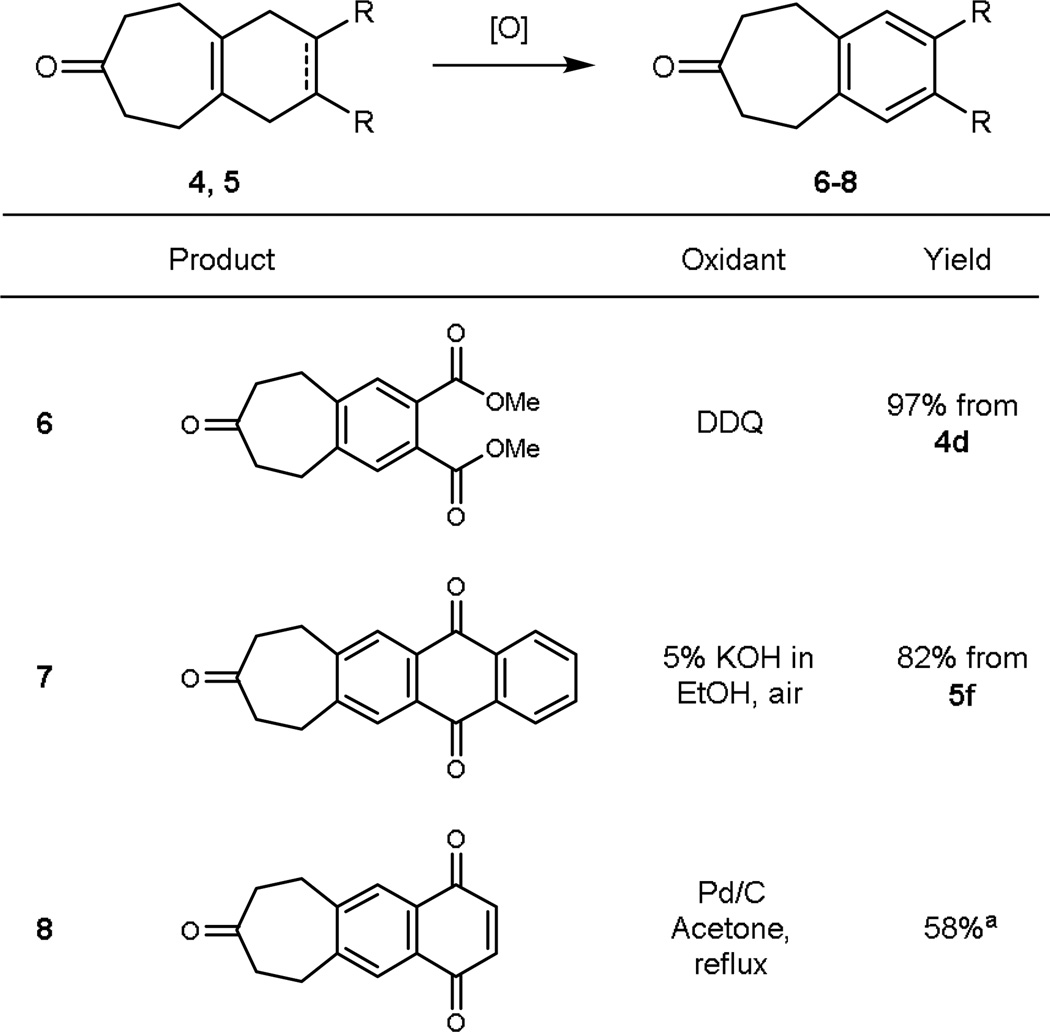 |
The initial cycloadduct oxidizes readily and was thus converted directly to compound 8
Table 3.
Additionally functionalized [5+2]/vinylogous Peterson/[4+2] products
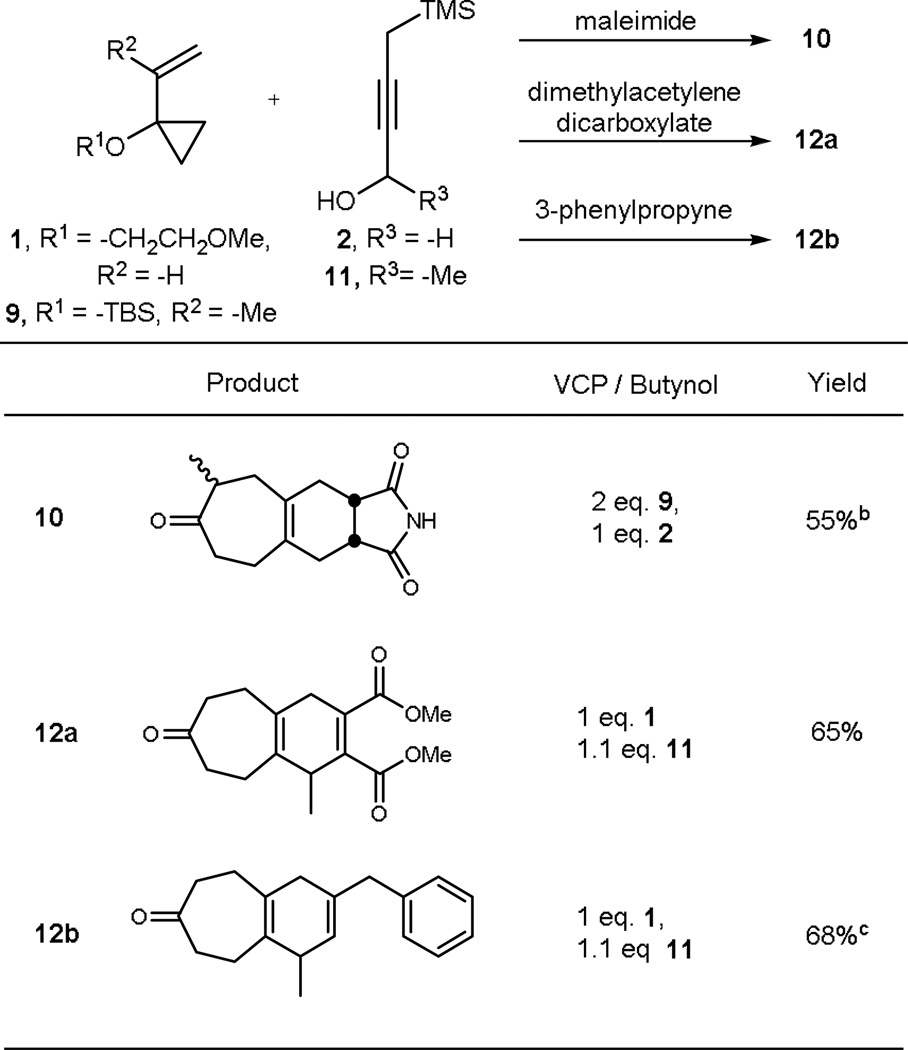 |
Reactions run at room temperature with 5 mol% [(naph)Rh(COD)]SbF6
1:1 mixture of diastereomers
4.2:1 mixture of regioisomers (major isomer shown)
Alkynes and dienes are among the most common and useful building blocks in organic synthesis. In this study, we have shown how the versatile two-carbon π-reactivity of a suitably functionalized alkyne can be transformed through a metal-catalyzed [5+2] cycloaddition and vinylogous Peterson elimination into a cisoid diene capable of engaging a range of dienophiles in a conventional Diels-Alder or a metal-catalyzed [4+2] cycloaddition. Drawing on the benefit of 4 carbon-carbon bond forming events in one operation, the overall process translates three simple and readily available building blocks into complex, polycyclic, added-value products. Both activated and unactivated dienophiles can be used in the second cycloaddition allowing flexible access to highly diversified polycycles. In the case of unactivated alkynes, the catalyst used to effect the first ([5+2]) cycloaddition is also used to effect the second ([4+2]) cycloaddition and possibly figures in facilitation of the intervening Peterson elimination. For conventional dienophiles, the second cycloaddition taps into the tremendous breadth and versatility of the Diels-Alder reaction. More generally, the key dual purpose, dual-function reagent (TMSBO) and its congeners that enable these back-to-back complexity-increasing cycloadditions should prove useful in serially coupling other cycloadditions including, for example, serial [4+2]/[4+2] cycloadditions, [3+2]/[4+2] cycloadditions, [2+2]/[4+4] cycloadditions and others. Studies on these processes, on this strategy for enhancing step economy through multi-purpose designed reagents and on the use of the serial [5+2]/[4+2] cycloadditions to generate kinase inhibitor libraries are in progress.
Methods
General Alkyne Dienophile Method
To an oven-dried or flame-dried vial was added 1.1 equivalents of TMSBO 2 as a neat liquid followed by 1,2-dichloroethane (DCE)(0.162 M with respect to VCP). To this solution is added 1 equivalent of VCP 1 as a neat liquid, and then 2–5 mol% [(naph)Rh(COD)]SbF6 in one portion. The reaction was stirred for 6 hours or until consumption of 1 could be observed by TLC. The second alkyne is then added and the reaction monitored by TLC for consumption of the intermediate diene. The reaction is quenched with dilute acid and the resulting products purified via column chromatography. Results for these substrates are summarized in Table 1.
General Alkene Dienophile Method
To an oven-dried vial was added 2 mol% [(naph)Rh(COD)]SbF6, followed by 1,2-dichloroethane (0.165 or 0.330 M with respect to VCP as specified in Table 1). To this solution is added 1 equivalent of VCP 1 as a neat liquid, 1.1 equivalents of TMSBO 2 as a neat liquid, and then 1.3 equivalents of the corresponding alkene dienophile (an exception was made for 4-phenyl-1,2,4-triazoline-3,5-dione as noted above). The reaction was stirred for 6 hours or until consumption of 1 could be observed by TLC. Depending on the reactivity of the dienophile, the reaction was either quenched, allowed to continue stirring at room temperature, or heated to 100 °C as noted in Table 1. The reaction was then monitored by TLC for consumption of the intermediate diene. The reaction is quenched with dilute acid and the resulting products purified via column chromatography. Results for these substrates are summarized in Table 1.
Supplementary Material
Acknowledgements
This research was supported by the National Science Foundation (CHE1265956) and the National Institutes of Health (CA031841). Additional funding was provided by the National Science Foundation Graduate Research Fellowship (R.V.Q.), Abbott Laboratories Stanford Graduate Fellowship (M.S.J.), Kanazawa University (F.I.), and the German Academic Exchange Service (M.P.).
Footnotes
Contributions
P.A.W. conceived the study. F.I. and M.P. performed the initial syntheses of butynols and evaluated initial viability of [5+2] and [5+2]/[4+2] reactions. D.N.F., M.S.J., and R.V.Q. determined the substrate profile and performed the synthesis and characterization of all reported compounds. M.S.J., R.V.Q., and P.A.W. wrote the paper. All authors commented on the manuscript.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Wender PA, et al. Toward the ideal synthesis. New transition metal-catalyzed reactions inspired by novel medicinal leads. Pure Appl. Chem. 2002;74:25–31. [Google Scholar]
- 2.Wender PA, Miller BL. Synthesis at the molecular frontier. Nature. 2009;460:197–201. doi: 10.1038/460197a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wender PA, Miller BL. In: Organic Synthesis: Theory and Applications. Hudlicky T, editor. Greenwich, CT: JAI Press; 1993. pp. 27–66. [Google Scholar]
- 4.Tietze LF, Modi A. Multicomponent domino reactions for the synthesis of biologically active natural products and drugs. Med. Res. Rev. 2000;20:304–322. doi: 10.1002/1098-1128(200007)20:4<304::aid-med3>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 5.Clavier H, Pellissier H. Recent Developments in Enantioselective Metal-Catalyzed Domino Reactions. Adv. Synth. Catal. 2012;354:3347–3403. [Google Scholar]
- 6.Slobbe P, Ruijter E, Orru RVA. Recent applications of multicomponent reactions in medicinal chemistry. Med. Chem. Commun. 2012;3:1189–1218. [Google Scholar]
- 7.Kang T, Kim W-Y, Yoon Y, Kim BG, Lee H-Y. Tandem Cycloaddition Reactions of Allenyl Diazo Compounds Forming Triquinanes via Trimethylenemethane Diyls. J. Am. Chem. Soc. 2011;133:18050–18053. doi: 10.1021/ja207591e. [DOI] [PubMed] [Google Scholar]
- 8.Van der Heijden G, Ruijter E, Orru R. Efficiency, Diversity, and Complexity with Multicomponent Reactions. Synlett. 2013;24:666–685. [Google Scholar]
- 9.Hopf H, Sherburn MS. Dendralenes Branch Out: Cross-Conjugated Oligoenes Allow the Rapid Generation of Molecular Complexity. Angew. Chem. Int. Ed. 2012;51:2298–2338. doi: 10.1002/anie.201102987. [DOI] [PubMed] [Google Scholar]
- 10.Souweha MS, Enright GD, Fallis AG. Vinigrol: A Compact, Diene-Transmissive Diels–Alder Strategy to the Tricyclic Core. Org. Lett. 2007;9:5163–5166. doi: 10.1021/ol702202b. [DOI] [PubMed] [Google Scholar]
- 11.Gani OABSM, Engh RA. Protein kinase inhibition of clinically important staurosporine analogues. Nat. Prod. Rep. 2010;27:489–498. doi: 10.1039/b923848b. [DOI] [PubMed] [Google Scholar]
- 12.Awuah E, Capretta A. Development of Methods for the Synthesis of Libraries of Substituted Maleimides and α,β-Unsaturated-γ-butyrolactams. J. Org. Chem. 2011;76:3122–3130. doi: 10.1021/jo1025805. [DOI] [PubMed] [Google Scholar]
- 13.Tanramluk D, Schreyer A, Pitt WR, Blundell TL. On the origins of enzyme inhibitor selectivity and promiscuity: a case study of protein kinase binding to staurosporine. Chem. Biol. Drug Des. 2009;74:16–24. doi: 10.1111/j.1747-0285.2009.00832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chae HJ, et al. Molecular mechanism of staurosporine-induced apoptosis in osteoblasts. Pharmacol. Res. Off. J. Ital. Pharmacol. Soc. 2000;42:373–381. doi: 10.1006/phrs.2000.0700. [DOI] [PubMed] [Google Scholar]
- 15.Omura S, et al. A new alkaloid AM-2282 OF Streptomyces origin. Taxonomy, fermentation, isolation and preliminary characterization. J. Antibiot. (Tokyo) 1977;30:275–282. doi: 10.7164/antibiotics.30.275. [DOI] [PubMed] [Google Scholar]
- 16.Angus RO, Johnson RP. Butatriene cycloaddition equivalent approach to the multiple linear homologation of six-membered rings and the synthesis of benzocyclobutenes. J. Org. Chem. 1983;48:273–276. [Google Scholar]
- 17.Hojo M, Tomita K, Hirohara Y, Hosomi A. New access to Di-exo-methylenecyclobutanes via [2 + 2] cycloaddition of 3-methylthio-4-trimethylsilyl-1,2-butadiene with alkenes mediated by a Lewis acid. Tetrahedron Lett. 1993;34:8123–8126. [Google Scholar]
- 18.Hojo M, Murakami C, Nakamura S, Hosomi A. Allenylmethylsilane Derivative as a Synthetic Equivalent of 1,2,3-Butatriene: Synthesis and Reactions of Di-exo-methylenecyclobutanes and -cyclobutenes. Chem. Lett. 1998;27:331–332. [Google Scholar]
- 19.Hojo M, Tomita K, Hosomi A. Synthesis and reactions of methyl (trimethylsilylmethyl)-acetylenecarboxylate. A general method for the generation of di-exo-methyleneisoxazolines and novel access to fused isoxazoles. Tetrahedron Lett. 1993;34:485–488. [Google Scholar]
- 20.Leroyer L, Maraval V, Chauvin R. Synthesis of the Butatriene C4 Function: Methodology and Applications. Chem. Rev. 2012;112:1310–1343. doi: 10.1021/cr200239h. [DOI] [PubMed] [Google Scholar]
- 21.Schubert WM, Liddicoet TH, Lanka WA. THE SYNTHESIS OF BUTATRIENE. J. Am. Chem. Soc. 1952;74:569–569. [Google Scholar]
- 22.Chow H-F, Cao X-P, Leung M. Facile synthesis of alkyl and aryl substituted 1,2,3-butatrienes. J. Chem. Soc. Chem. Commun. 1994:2121–2122. [Google Scholar]
- 23.Wang KK, Liu B, Lu Y. Facile Synthesis of [3]Cumulenes via 1,4-Elimination of Hydroxytrimethylsilane from 4-(Trimethylsilyl)-2-butyn-1-ols. J. Org. Chem. 1995;60:1885–1887. [Google Scholar]
- 24.Malkov AV, et al. A Novel Bifunctional Allyldisilane as a Triple Allylation Reagent in the Stereoselective Synthesis of Trisubstituted Tetrahydrofurans. Chem. – Eur. J. 2011;17:7162–7166. doi: 10.1002/chem.201100513. [DOI] [PubMed] [Google Scholar]
- 25.Jervis PJ, Kariuki BM, Cox LR. Stereoselective Synthesis of 2,4,5-Trisubstituted Tetrahydropyrans Using an Intramolecular Allylation Strategy. Org. Lett. 2006;8:4649–4652. doi: 10.1021/ol061957v. [DOI] [PubMed] [Google Scholar]
- 26.Chiu SK, Peterson PE. The preparation of propargyltrimethylsilanes. Tetrahedron Lett. 1980;21:4047–4050. [Google Scholar]
- 27.Tong R, et al. Total Syntheses of Durgamone, Nakorone, and Abudinol B via Biomimetic Oxa- and Carbacyclizations. J. Am. Chem. Soc. 2007;129:1050–1051. doi: 10.1021/ja068826+. [DOI] [PubMed] [Google Scholar]
- 28.Ambasht S, Chiu SK, Peterson PE, Queen J. Preparation of Trimethylsilylmethyl Derivatives Using Phase Transfer Methods. Synthesis. 1980;1980:318–320. [Google Scholar]
- 29.Wein AN, Tong R, McDonald FE. An economical synthesis of 4-trimethylsilyl-2-butyn-1-ol. Org. Synth. 2011;88:296. [Google Scholar]
- 30.Fleming I, Morgan IT, Sarkar AK. The stereochemistry of the vinylogous Peterson elimination. J. Chem. Soc. [Perkin 1] 1998:2749–2764. [Google Scholar]
- 31.Harmata M, Bohnert GJ. A 4+3 Cycloaddition Approach to the Synthesis of (±)-Sterpurene. Org. Lett. 2003;5:59–61. doi: 10.1021/ol027176l. [DOI] [PubMed] [Google Scholar]
- 32.Angell R, Parsons PJ, Naylor A, Tyrrell E. An Extremely Mild Method for the Construction of E -1,3-Dienes. Synlett. 1992;1992:599–600. [Google Scholar]
- 33.Takano S, Otaki S, Ogasawara K. A Facile Synthesis of the Substituted Tetrahydronapthalenes by the Benzo-Peterson Reaction. Heterocycles. 1985;23:2811. [Google Scholar]
- 34.Takano S, Sato N, Otaki S, Ogasawara K. The Total Synthesis of (±)-Sikkimotoxin via the Benzo-Peterson Reaction. Heterocycles. 1987;25:69. [Google Scholar]
- 35.Takano S, Otaki S, Ogasawara K. Stereocontrolled synthesis of (±)-deoxypodophyllotoxin via the benzyl equivalent of the Peterson reaction. J. Chem. Soc. Chem. Commun. 1985:485–487. [Google Scholar]
- 36.Angoh AG, Clive DLJ. A synthetic equivalent for the butadienyl carbonium ion: use of 4-(trimethylsilyl)but-2-ynal for preparation of 1,3-dienes and macroexpansion of cyclic ketones. J. Chem. Soc. Chem. Commun. 1984:534–536. [Google Scholar]
- 37.Ahmed M, Atkinson CE, Barrett AGM, Malagu K, Procopiou PA. Synthesis of C-19-Functionalized 1α-Hydroxyvitamin D2 Analogues via Ring-Closing Metathesis. Org. Lett. 2003;5:669–672. doi: 10.1021/ol027406w. [DOI] [PubMed] [Google Scholar]
- 38.Wender PA, Williams TJ. [(arene)Rh(cod)]+ Complexes as Catalysts for [5+2] Cycloaddition Reactions. Angew. Chem. Int. Ed. 2002;41:4550–4553. doi: 10.1002/1521-3773(20021202)41:23<4550::AID-ANIE4550>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.