

Background: The impact of EBV variation on T cell immunity is unclear.
Results: We determined the structures of EBV epitopes bound to HLA-B*35:01/08 and examined the associated T cell responses.
Conclusion: Viral mutation and HLA polymorphism interplay causes changes in epitope conformation.
Significance: We provide a basis for the impact of EBV strain variation on cellular immunity in humans.
Keywords: Immunology, Major Histocompatibility Complex (MHC), Structural Biology, T Cell Receptor (TCR), X-ray Crystallography
Abstract
Mutations within T cell epitopes represent a common mechanism of viral escape from the host protective immune response. The diverse T cell repertoire and the extensive human leukocyte antigen (HLA) polymorphism across populations is the evolutionary response to viral mutation. However, the molecular basis underpinning the interplay between HLA polymorphism, the T cell repertoire, and viral escape is unclear. Here we investigate the T cell response to a HLA-B*35:01- and HLA-B*35:08-restricted 407HPVGEADYFEY417 epitope from Epstein-Barr virus and naturally occurring variants at positions 4 and 5 thereof. Each viral variant differently impacted on the epitope's flexibility and conformation when bound to HLA-B*35:08 or HLA-B*35:01. We provide a molecular basis for understanding how the single residue polymorphism that discriminates between HLA-B*35:01/08 profoundly impacts on T cell receptor recognition. Surprisingly, one viral variant (P5-Glu to P5-Asp) effectively changed restriction preference from HLA-B*35:01 to HLA-B*35:08. Collectively, our study portrays the interplay between the T cell response, viral escape, and HLA polymorphism, whereby HLA polymorphism enables altered presentation of epitopes from different strains of Epstein-Barr virus.
Introduction
The human leukocyte antigen (HLA)7 complex is the most polymorphic region of the human genome, with >10,500 HLA alleles documented presently. The maintenance of this diversity reflects the need at a population level to present a wide array of pathogen-derived antigens (Ags) to enable sufficient and protective immunity by the host (1). Each HLA allomorph can bind a large repertoire of peptides (p), with peptide specificity being determined via six pockets (A-F) within the Ag binding cleft of the molecule (2). Typically, two HLA pockets represent the main peptide anchors, with neighboring HLA residues also impacting on peptide binding. For example, within the HLA-B*35 family, the B- and F-pockets preferentially bind P2-Pro and PΩ-Phe, respectively, with residues lining the D/E pockets (3, 4), including position 156, also impacting on peptide binding (5–7). HLA polymorphism can also exert more subtle effects on the bound peptide repertoire, such as influencing peptide loading and extent of tapasin dependence, and/or altering epitope conformation and flexibility (8, 9). However, the full extent of the impact of HLA polymorphism on the responding T cell repertoire remains to be elucidated.
The T cell repertoire comprises a vast array of αβ T cell receptors (TCRs) to cope with the challenge of the myriad of potential peptide HLA landscapes (10, 11). Despite this diversity, T cells are for the most part genetically restricted to interacting with foreign peptide bound to an individual's self-HLA molecules (12). For example, the HLA restriction paradigm states that T cells from an HLA-B*35:08+/HLA-B*35:01− individual should not cross-react with HLA-B*35:01+ antigen-presenting cells despite HLA-B*35:08 and HLA-B*35:01 differing by only one residue at position 156 (HLA-B*35:08, Arg; HLA-B*35:01, Leu).
In the periphery self-HLA molecules bound to processed viral antigens are sensed as foreign, thereby leading to T cell activation and lysis of the virally infected cell. However, to escape immune surveillance, viruses have deployed a wide range of strategies, including interfering with the Ag-processing machinery and mutating T cell epitopes (13, 14). Mutations within major histocompatibility complex (MHC)-bound peptides can potentially impact on MHC binding or TCR recognition, which can subsequently lead to immune escape. However, the interplay characterizing the molecular arms race between viral escape, HLA polymorphism, and the ensuing adaptive immune response is not fully understood.
To understand the impact of HLA polymorphism on anti-viral immunity, we have investigated the protective immune response to Epstein-Barr virus (EBV), a ubiquitous human pathogen. EBV is controlled by a robust cellular immune response that is directed against a defined number of EBV antigens. There are two distinct strains of EBV, type 1 and type 2, that are primarily divergent within the genes EBNA-2 and EBNA-3 (15, 16). In addition, variation in EBNA-1, BZLF1, and LMP1 has been identified, which has led to a greater diversity of known epitope mutations (17). Indeed, it has been suggested that variation in the latent nuclear protein EBNA-1 is linked to the development of EBV-associated diseases (18). One EBNA-1 epitope that contains known mutations is HPVG (HPVGEADYFEY), which has been identified in EBV strains infecting Caucasians (19, 20). The EBV strains infecting Chinese individuals (21, 22) encode a variation at position 5 of this epitope, with an Asp replacing the Glu (HPVG-D5), whereas in some type 1 EBV isolates (19, 23) a glutamine is encoded at position 5 (HPVG-Q5). In addition, the Ag876 EBV strain (type 2 EBV) is prevalent in Africa and encodes a Gly to Ala variation at position 4 of the HPVG epitope (HPVG-A4) (24).
The HPVG epitope is restricted to two closely related allomorphs, HLA-B*35:01 and HLA- B*35:08. The cytotoxic T cell (CTL) response toward this epitope is biased with preferential use of the TRBV9 gene, which pairs with two different TCR α-chains (TRAV20 or TRAV29) based on the HLA restriction element used (25). Previously we investigated the CTL response to the HPVG determinant in HLA-B*35:01+ individuals and the mode of recognition by a prototypic TRBV9+ public TCR, termed TK3. Although the TK3 clone could recognize both HLA-B*35:01 and HLA-B*35:08 presenting the HPVG peptide, it exhibited much higher reactivity toward HLA-B*35:01 (20). However, the molecular basis of the TK3 HLA allomorph specificity was unclear. As the TK3 TCR directly contacted P5-Glu of the HPVG epitope, we asked whether the different EBV strains harboring mutations surrounding P5 could impact on HLA-B*35:01-restricted recognition by the TK3 TCR. Here we report the cellular, biophysical, and structural analysis comparing the TK3 clone response in the context of the two HLA allomorphs presenting the HPVG epitope and three naturally occurring EBV strain variants.
EXPERIMENTAL PROCEDURES
Expression, Refolding, and Purification of Recombinant TCR and pHLA
The production of the TK3 TCR, HLA-B*35 allomorphs in complex with HPVG and its variants, were previously described (20). Briefly, individual components of the TCR and HLA molecules were expressed in BL21 Escherichia coli cells as inclusion bodies and solubilized in 8 m urea buffer containing 10 mm Tris, pH 8, 0.5 mm Na-EDTA, and 1 mm dithiothreitol. TCR and pHLA proteins were then refolded separately in refolding buffer containing 3 or 5 m urea, 100 mm Tris-HCl, pH 8, 2 mm Na-EDTA, 400 mm l-Arg-HCl, 0.5 mm oxidized glutathione, and 5 mm reduced glutathione. The refolded solution was dialyzed twice against 10 mm Tris-HCl, pH 8, overnight to allow sufficient buffer exchange. The dialyzed samples were then purified through diethylaminoethanol cellulose, size-exclusion, and anion exchange chromatography columns. The quality and purity of the proteins were analyzed via SDS-PAGE gels.
Surface Plasmon Resonance Measurement and Analysis
All surface plasmon resonance experiments were conducted at 25 °C on the BIAcore 3000 instrument using HBS buffer (10 mm HEPES, pH 7.4, 150 mm NaCl, and 0.005% surfactant P20) with 1% BSA to prevent nonspecific binding. The human TCR-specific monoclonal antibody 12H8 (26) was coupled to research grade CM5 chips with standard amine coupling. The experiment was conducted as previously described (27), with a concentration range of 0.78–200 μm pHLA complexes. BIAevaluation Version 3.1 was used for data analysis with the 1:1 Langmuir binding model.
Thermal Melting Experiment
Thermal stability experiments of HLA-B*35:01 or HLA-B*35:08 bound to the HPVG epitope and its variants were carried out using a real time PCR instrument (Corbett RotorGene 300). Purified pHLA samples were concentrated to 5 and 10 μm in TBS buffer (10 mm Tris-HCl, pH 8, and 150 mm NaCl), and the experiment was conducted as previously described (28).
Crystallization of pHLA and TK3 TCR-pHLA Complexes
Using hanging drop vapor diffusion techniques, large rod-shape crystals of pHLA were obtained at 20 °C with a 5 mg/ml protein concentration mixing with reservoir solutions containing 0.1 m trisodium citrate, pH 5.6, 16% PEG 4K, and 0.2 m ammonium acetate at 1:1 ratio. Plate-like crystals of TK3 TCR-pHLA complexes were obtained at 6 mg/ml protein concentration in a reservoir solution consisting of 0.2 m LiSO4, 0.1 m trisodium citrate, pH 5.6, and 18% PEG 3350.
Data Collection and Structure Determination
All crystals were soaked in the reservoir solutions with 30% PEG concentration before being flash-frozen in liquid nitrogen. Datasets were collected at the Australian synchrotron (Clayton) with an ADSC-Quantum 210 CCD on MX1 and an ADSC-Quantum 315r CCD on MX2 detector at 100 K. All data were processed and scaled with the XDS program (29) and Scala from the CCP4 suite program, respectively (30). The structures were solved using the molecular replacement program, PHASER (31), with the published structure of HLA-B*35:01 (PDB code 2FYY) (25) and TK3 TCR-HLA-B*35:01 (PDB code 3MV7) (20) minus the peptide as models. Structural refinements were performed with PHENIX (32) and BUSTER (33) programs, and manual building of the models were carried out with Coot (34). The TCR was numbered according to the IMGT unique numbering system (35) whereby the CDR1 loops start at residue number 27, the CDR2 loops start at number 56, and the CDR3 loops start at residue number 105. The final models have been validated using the Protein Data Base validation web site, and the final refinement statistics are summarized in Tables 2 and 3. Coordinates have been submitted to the PDB database: HLA-B*35:08-HPVG-A4 (PDB code 4PRB), HLA-B*35:08-HPVG-D5 (PDB code 4PRD), HLA-B*35:08-HPVG-Q5 (PDB code 4PRE), HLA-B*35:01-HPVG-A4 (PDB code 4PRN), HLA-B*35:01-HPVG-D5 (PDB code 4PR5), HLA-B*35:01-HPVG-Q5 (PDB code 4PRA), TK3 TCR-HLA-B*35:08-HPVG (PDB code 4PRI), TK3 TCR-HLA-B*35:08-HPVG-D5 (PDB code 4PRH) and TK3 TCR-HLA-B*35:01-HPVG-Q5 (PDB code 4PRP). All molecular graphic representations were created using PyMOL (36).
TABLE 2.
Data collection and refinement statistics of pHLA structures
| Structures | B*35:01-HPVG-A4 | B*35:01-HPVG-D5 | B*35:01-HPVG-Q5 |
|---|---|---|---|
| Resolution (Å) | 1.65 (1.75-1.65) | 1.8 (1.9-1.8) | 1.85 (1.95-1.85) |
| Space group | P212121 | P212121 | P212121 |
| Unit cell parameters (Å) | 50.7, 81.3, 110.4 | 50.6, 81.2, 109.1 | 50.7, 81.4, 109.5 |
| Temperature (K) | 100 | 100 | 100 |
| Total number of observations | 400,579 (61,312) | 311,047 (46,089) | 289,411 (42,022) |
| Number of unique reflections | 55,695 (8,882) | 42,487 (6,249) | 39,526 (5,701) |
| Completeness (%) | 100 (100) | 100 (100) | 100 (99.9) |
| Rmergea (%) | 5.8 (40.5) | 6.6 (44.7) | 7.6 (42.7) |
| I/σI | 20.7 (4.53) | 26.2 (4.76) | 21.6 (5.1) |
| Multiplicity | 7.2 (6.9) | 7.3 (7.3) | 7.3 (7.3) |
| Rfactorb (%) | 18.4 | 17.8 | 18.9 |
| Rfreeb (%) | 22.1 | 21.3 | 22.4 |
| r.m.s.d. bond lengths (Å) | 0.012 | 0.010 | 0.008 |
| r.m.s.d. bond angles (°) | 1.234 | 1.097 | 1.074 |
| Ramachandran plot (%) | |||
| Favored/allowed | 99.4 | 99.1 | 99.4 |
| Generously allowed | 0.6 | 0.9 | 0.6 |
| Disallowed | 0.0 | 0.0 | 0.0 |
| Structures | B*35:08-HPVG-A4 | B*35:08-HPVG-D5 | B*35:08-HPVG-Q5 |
|---|---|---|---|
| Resolution (Å) | 1.75 (1.85-1.75) | 1.75 (1.85-1.75) | 1.65 (1.75-1.65) |
| Space group | P212121 | P212121 | P212121 |
| Unit-cell parameters (Å) | 50.8, 82.1, 110.5 | 50.9, 82.0, 110.4 | 50.9, 81.7, 110.8 |
| Temperature (K) | 100 | 100 | 100 |
| Total number of observations | 344,591 (51,279) | 345,635 (51,872) | 403,652 (61,175) |
| Number of unique reflections | 47,355 (7,124) | 47,357 (7,124) | 55,751 (8,726) |
| Completeness (%) | 100 (100) | 99.9 (99.7) | 98.9 (96.8) |
| Rmergea (%) | 5.5 (48.9) | 5.4 (32.0) | 5.1 (35.2) |
| I/σI | 25.6 (4.29) | 24.1 (6.5) | 26.9 (5.9) |
| Multiplicity | 7.3 (7.3) | 7.3 (7.3) | 7.2 (7.0) |
| Rfactorb (%) | 20.0 | 19.6 | 17.7 |
| Rfreeb (%) | 23.3 | 23.4 | 20.3 |
| r.m.s.d. bond lengths (Å) | 0.016 | 0.012 | 0.014 |
| r.m.s.d. bond angles (°) | 1.201 | 1.172 | 1.255 |
| Ramachandran plot (%) | |||
| Favored/allowed | 99.1 | 99.4 | 99.4 |
| Generously allowed | 0.9 | 0.6 | 0.6 |
| Disallowed | 0.0 | 0.0 | 0.0 |
a Rmerge = Σ|Ihkl − 〈Ihkl〉|/ΣIhkl.
b Rfactor = Σhkl‖Fo| − |Fc‖/Σhkl|Fo| for all data except ≈5%, which were used for Rfree calculation. Values in parentheses are for the highest resolution shell.
TABLE 3.
Data collection and refinement statistics of the TK3 TCR-pHLA complexes
| Structures | TK3-B*35:08-HPVG | TK3-B*35:08-HPVG-D5 | TK3-B*35:01-HPVG-Q5 |
|---|---|---|---|
| Resolution (Å) | 41.7-2.4 (2.5-2.4) | 50-2.5 (2.6-2.5) | 47.3-2.5 (2.64-2.5) |
| Space group | P1 | P1 | P1 |
| Unit cell parameter (Å) | 44.58, 62.37, 100.45 | 44.83, 62.08, 98.33 | 45.14, 62.60, 97.81 |
| Unit cell parameter (°) | 98.17, 94.59, 109.11 | 92.69, 101.92, 108.21 | 92.04, 102.53, 109.70 |
| Temperature (K) | 100 | 100 | 100 |
| Total number of observations | 142,189 (16,985) | 125,672 (14,351) | 125,365 (18,871) |
| Number of unique reflections | 73,467 (8,684) | 32,833 (3,671) | 32,843 (4,832) |
| Completeness (%) | 93.6 (96.2) | 96.9 (98.2) | 97.2 (98.0) |
| Rmergea (%) | 7.0 (32.3) | 7.5 (44.5) | 8.2 (49.0) |
| I/σI | 10.77 (2.69) | 11.79 (3.27) | 11.9 (3.7) |
| Multiplicity | 1.9 (1.9) | 3.8 (3.9) | 3.8 (3.9) |
| Rfactorb (%) | 21.3 | 22.5 | 18.0 |
| Rfreeb (%) | 26.1 | 26.5 | 22.8 |
| r.m.s.d. bond lengths (Å) | 0.011 | 0.010 | 0.010 |
| r.m.s.d. bond angles (°) | 1.17 | 1.13 | 1.07 |
| Ramachandran plot (%) | |||
| Favored/allowed | 98.9 | 99.2 | 99.0 |
| Generously allowed | 0.7 | 0.6 | 0.6 |
| Disallowed | 0.4 | 0.2 | 0.4 |
a Rmerge = Σ|Ihkl − 〈 Ihkl 〉|/ΣIhkl.
b Rfactor = Σhkl‖Fo| − |Fc‖/Σhkl|Fo| for all data except ≈5%, which were used for Rfree calculation. Values in parentheses are for the highest resolution shell.
Cellular Assays
The TK3 CTLs clone was tested in duplicate for cytotoxicity in standard 5-h chromium release assays. In brief, CTLs were assayed against 51Cr-labeled lymphoblastoid cell line targets (E:T ratios = 4:1, 2:1, 1:1, 0.5:1) or an EBV-negative phytohemagglutinin-stimulated T cell line that was pretreated with various concentrations of synthetic peptide or left untreated (E:T ratio = 1:1). Toxicity testing of all peptides was performed before use by adding peptide to 51Cr-labeled phytohemagglutinin blasts in the absence of CTL effectors. The mean spontaneous lysis for target cells in the culture medium was always <20%, and the variation from the mean specific lysis was <10%.
The ability of TK3 CTLs to secrete IFN-γ, TNF, granzyme A, granzyme B, MIP-1β, and FasL in response to HPVG and its variants was assessed using a CBA Flex Set (BD Biosciences). First, the T2 cell line, transfected with HLA-B*35:01 or T2-HLA-B*35:08, was incubated individually for 1 h with various concentrations of HPVG, HPVG-A4, HPVG-D5, and HPVG-Q5. After washing these peptide-coated stimulator cells to remove unbound peptide, they were combined with TK3 cells at a 1:20 stimulator-to-responder ratio. The cells were incubated at 37 °C for 20 h before supernatants were removed for CBA analysis according to the manufacturer's protocol. Briefly, CBA capture bead pools were incubated with culture supernatants for 1 h at room temperature, after which a pool of phycoerythrin-conjugated detection reagents was added and incubated for 2 h at room temperature. The capture bead pools were then washed and analyzed on a FACSCanto II (BD Biosciences). Analyte production was calculated by comparing test samples to standard curves using FCAP Array Software (Soft Flow Hungary, Pécs, Hungary).
RESULTS
HLA Polymorphism Affects Epitope Recognition by the TK3 T Cell Clone
The TK3 T cell clone, isolated from an HLA-B*35:01+ EBV seropositive individual (25), preferentially lysed HLA-B*35:01+ EBV-infected target cells presenting the HPVG epitope over HLA-B*35:08+ targets. This was despite these two allomorphs differing by only one residue at position 156 (Fig. 1A). We next asked whether the three HPVG variants (HPVG-A4, HPVG-Q5, HPVG-D5) were equivalently recognized by the TK3 clone when presented by HLA-B*35:01 or HLA-B*35:08.
FIGURE 1.
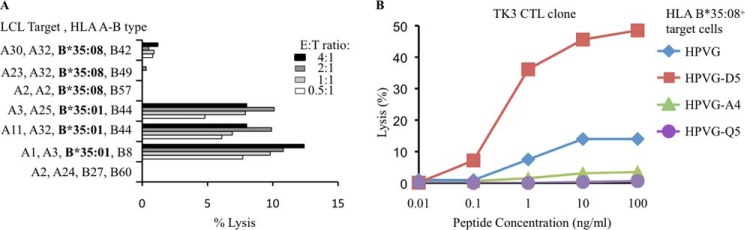
TK3 clone reactivity. A, preferential recognition of HLA-B*35:01+ lymphoblastoid cell line (LCL) cells by the TK3 CTL clone compared with HLA-B*35:08+ lymphoblastoid cells presenting the HPVG peptide. B, recognition of HPVG and three EBV variants of the epitopes by the TK3 clone using a cytotoxicity assay with HLA-B*35:08+-phytohemagglutinin blasts as target cells.
As previously observed, the TK3 CTL clone recognized the HPVG-A4 variant comparably to the wild type (WT) HPVG epitope in the context of self-HLA-B*35:01, whereas the HPVG-Q5 and HPVG-D5 variants showed markedly diminished TK3 recognition (20). Next, we performed a cytotoxicity assay using HLA-B*35:08+ target cells in the presence of the HPVG peptide and its three variants. Surprisingly, the effects of some of the EBV variants on TK3 activation in the context of HLA-B*35:08 were markedly different compared with the presentation by HLA-B*35:01 (Fig. 1B). Namely, the HPVG-A4 and HPVG-Q5 variants, which bound to HLA-B*35:08, poorly stimulated the TK3 clone compared with the WT HPVG epitope. Moreover, although the HPVG-D5 epitope markedly diminished TK3 activation when presented by HLA-B*35:01, this variant markedly improved TK3 CTL recognition when presented by HLA-B*35:08 (Fig. 1B).
This data suggested that HLA polymorphism changes the immunogenicity hierarchy of the TK3 CTL clone toward the HPVG variant epitopes, with the WT HPVG epitope being preferentially recognized by HLA-B*35:01 target cells, whereas the HPVG-D5 variant was preferentially recognized in the context of HLA-B*35:08.
Difference in Cytokine Production Due to EBV Polymorphism
To further assess the impact of HLA polymorphism and Ag variation on TK3 CTL activation and polyfunctionality, we used a bead-based multianalyte flow immunoassay (37). This assay enabled the simultaneous detection of the cytokines IL-2, TNF, IFN-γ, and MIP-1β in addition to FasL and granzymes A and B. These analytes were all detected in culture supernatants after TK3 CTL activation, and as expected, this clone was best stimulated by HPVG presented by HLA-B*35:01 (Fig. 2). Although the levels of released analytes were moderately lower in the presence of the HPVG-A4 peptide (Fig. 2), the absolute cytokine/granzyme A levels (Fig. 2) enabled efficient killing of the HLA-B*35:01 target cells presenting the HPVG-A4 peptide (20). However, the concentrations of these analytes produced by TK3 in the presence of the HPVG-D5 and HPVG-Q5 variants and HLA-B*35:01+-target cells were markedly diminished. In contrast, the HPVG-D5 peptide presented by the HLA-B*35:08+ target cells was able to stimulate the TK3 clone at a greater level than the WT HPVG epitope (Fig. 2). As such, the EBV variant HPVG-D5 has a variable impact on T cell recognition depending on whether it is presented by HLA-B*35:01 (negative impact) or HLA-B*35:08 (positive impact). Collectively, the analyte production (Fig. 2) and cytotoxicity data (Fig. 1) indicate that, although HPVG represents the most immunogenic peptide when presented by HLA-B*35:01, the variant HPVG-D5 is the most immunogenic epitope when presented by HLA-B*35:08 for the TK3 clonotype.
FIGURE 2.

Analyte production by TK3. Analytes released by TK3 after stimulation with HPVG peptide or one of its three variants, presented by T2-HLA-B*35:01 (solid lines) or T2-HLA-B*35:08 APCs (dashed lines) are shown. The culture supernatants of stimulated TK3 cells were assessed by CBA and analyte production in response to HPVG (blue), HPVG-A4 (green), HPVG-D5 (red), and HPVG-Q5 (purple) stimulation at a range of peptide concentrations is shown.
pHLA Complex Stability and HPVG Polymorphism
To understand the effect of EBV polymorphism on HPVG epitope presentation, we questioned whether mutation within this epitope could impact the stability of the HLA-B*35:01 and HLA-B*35:08 complexes. Although the HPVG peptide exhibited greater flexibility when bound to HLA-B*35:01 than HLA-B*35:08 (25), this did not affect pHLA stability (Tm was 67.3 °C for HLA-B*35:01-HPVG and 64.3 °C for HLA-B*35:08-HPVG). Although the HLA-B*35:08-HPVG-Q5 complex showed reduced stability (Tm of 56.9 °C), the HPVG-A4 and HPVG-D5 variants did not significantly affect the Tm for either HLA-B*35:08 or HLA-B*35:01 (Table 1). Thus, the impact of the EBV variants on TK3 CTL clone activation is not attributable to differential impact on pHLA stability.
TABLE 1.
Thermal stability measurement of HLA-B*35:01 or HLA-B*35:08 in complex with the HPVG peptide and its variants
Tm is the thermal melt point of each protein, representing the temperature required to unfold 50% of the sample.
| Proteins | Tm |
|---|---|
| °C | |
| B*35:01-HPVG | 67.3 ± 0.3 |
| B*35:01-HPVG-A4 | 68.6 ± 0.2 |
| B*35:01-HPVG-D5 | 68.0 ± 0.4 |
| B*35:01-HPVG-Q5 | 65.0 ± 0.6 |
| B*35:08-HPVG | 64.3 ± 1.4 |
| B*35:08-HPVG-A4 | 67.3 ± 0.9 |
| B*35:08-HPVG-D5 | 61.0 ± 1.7 |
| B*35:08-HPVG-Q5 | 56.9 ± 0.7 |
TK3 TCR Binding Affinity
To better understand the basis of the HLA restriction and epitope specificity of TK3, we undertook surface plasmon resonance-based measurements of the TK3 TCR and HLA-B*35:08 bound to HPVG and its variant epitopes. The affinity (Kd) of the TK3 TCR for the HLA-B*35:01-HPVG complex was 2.2 μm (20), whereas its affinity for HLA-B*35:08-HPVG was lower, at 93.2 μm (Fig. 3A). The affinities of the TK3 TCR for HPVG-A4 and HPVG-Q5 bound to HLA-B*35:08 were ∼100 μm (Fig. 3, B and C), whereas for HLA-B*35:08-HPVG-D5 the Kd was 25.4 μm with a kon of 1.087 ± 0.302 × 104/ m−1 s−1 and koff of 0.389 ± 0.047/s (Fig. 3D). Thus, consistent with the functional assays, the TK3 TCR preferentially recognizes the HPVG-D5 variant over the HPVG epitope when bound to HLA-B*35:08.
FIGURE 3.

Surface plasmon resonance sensograms of the TK3 TCR with HLA-B*35:08 bound to HPVG and it variants. A range of concentrations (0.78 to 200 μm) of HLA-B*35:08 in complex with either HPVG (A), HPVG-A4 (B), HPVG-Q5 (C), or HPVG-D5 (D) was used for surface plasmon resonance response analysis with the TK3 TCR. The experiments have been conducted in duplicate, and the values represent the means ± S.E. RU, response units.
HLA Structural Changes Induced by HPVG Variants
To determine the structural basis for the differential immunogenicity profiles of the HPVG variants, we solved the high resolution crystal structures of HPVG-A4, HPVG-D5, and HPVG-Q5 complexed with HLA-B*35:01 and HLA-B*35:08 (Table 2) and compared them to the respective HLA-B*35:01/08-HPVG complexes.
As shown previously, the HPVG peptide bound to HLA-B*35:08 was fully resolved (Fig. 4A) and indicated a rigid peptide, whereas the central part of the peptide was mobile when bound to HLA-B*35:01 (Fig. 4B) (25). P4-Gly of the HPVG epitope pointed toward solvent, and thus the HPVG-A4 variant was readily accommodated within HLA-B*35:08 and HLA-B*35:01 (Fig. 4, C and D). Both structures adopt a similar conformation, comparable to the HLA-B*35:08-HPVG structure (root mean square deviation (r.m.s.d.) < 0.25 Å). Curiously, the HPVG-A4 variant was better resolved in the Ag binding cleft in comparison to the HPVG peptide when presented by HLA-B*35:01.
FIGURE 4.

Binary structures of pHLA complexes. Structures of four HPVG epitopes (stick format) bound to HLA-B*35:08 (pale blue) or HLA-B*35:01 (pale pink) (25). The HPVG peptide is in cyan and pink in panels A and B, HPVG-A4 is in dark blue and magenta in panels C and D, the HPVG-D5 is in blue and purple in panels E and F, and the HPVG-Q5 is in pale blue and pale pink in panels G and H bound to HLA-B*35:08 and HLA-B*35:01, respectively. Arg-97 and the polymorphic residue 156 (Arg in HLA-B*35:08 and Leu in HLA-B*35:01) of the antigen binding cleft are represented in stick and colored accordingly to the HLA. The variations in the HPVG peptide are highlighted in yellow at positions 4 and 5. The red dashed lines represent the salt bridge between the P5 or P7 of the peptide with the Arg-97 and Arg-156.
P5-Glu of the HPVG epitope was found to act as a secondary anchor residue in HLA-B*35:08 (Fig. 4A). The HLA-B*35:08-HPVG-D5 complex was similar to HLA-B*35:08-HPVG, displaying an r.m.s.d. of 0.13 Å for the Ag binding cleft and 0.29 Å for the peptide (Fig. 4, A and E). The differences between these two pHLA complexes lay primarily at the P5-Asp replacement. The shorter P5-Asp side chain was further away from Arg-97 than the corresponding P5-Glu (Fig. 4E), and as a result the HPVG-D5 epitope showed greater flexibility (poor electron density) within HLA-B*35:08 compared with the WT HPVG epitope due to the lack of salt-bridge with the Arg-97. The structure of HPVG-D5 bound to HLA-B*35:01 was more surprising (Fig. 4F), as it adopted a different conformation compared with the HLA-B*35:08-HPVG-D5 and caused rearrangements within the HLA-B*35:01 cleft itself. The HPVG-D5 conformation was constrained within the HLA-B*35:01 Ag binding cleft, whereby the central region of HPVG peptide (P5-P8) formed one helical turn and was stabilized by intrapeptide contacts. The helical turn enabled the P5-Asp to salt-bridge with Arg-97, the conformation of which was also impacted by the insertion of P9-Phe into the Ag binding cleft. Moreover, P8-Tyr also inserted its aromatic ring between the helical turn of the peptide and the HLA α2-helix. Accordingly, within HLA-B*35:01, structural rearrangements compensate for the shorter side chain of the P5-Asp to improve peptide-HLA interactions and internal peptide constraints.
The HPVG-Q5 peptide did not markedly affect the overall antigen binding cleft conformation of either HLA-B*35:01 (r.m.s.d. of 0.11 Å) or HLA-B*35:08 (r.m.s.d. of 0.22 Å). However, the P5-Gln mutation caused a complete rearrangement of the peptide in both HLA allomorphs (Fig. 4, G and H). Namely, the P5-Gln flipped outward and became solvent-exposed and thus no longer contacted Arg-97 at the base of the Ag binding cleft. To compensate, P7-Asp reoriented from its surface-exposed position within the HLA-B*35:08-HPVG complex to act as a secondary anchor and salt bridge with Arg-97 (Fig. 4G). Although the HPVG-Q5 structure was similar between the two HLA molecules (r.m.s.d. of 0.29 Å), conformational differences were observed around position 156. Namely, the Leu-156 of HLA-B*35:01 allowed the P7-Asp to sit 1 Å deeper in the binding cleft compared with the HLA-B*35:08 structure, in which the large Arg-156 occupied this region (Fig. 4, G and H). Together, our data show that the HPVG variants exist in dramatically different structural states when bound to the highly homologous allomorphs HLA-B*35:01 and HLA-B*35:08.
The TK3 TCR “Untangles” the HPVG-Q5 Epitope
Although the HPVG-Q5 variant exhibited a unique peptide conformation when presented by HLA-B*35:01, the TK3 TCR nevertheless exhibited an affinity of 52 μm toward this epitope (20). To understand how the TK3 TCR could accommodate these differences, we solved the structure of the TK3 TCR in complex with HLA-B*35:01-HPVG-Q5 (Fig. 5 and Table 3).
FIGURE 5.

Ternary structures of TK3 TCR-pHLA complexes. The top panels show an overview of the TK3 TCR in complex with HLA-B*35:01-HPVG (A), HLA-B*35:01-HPVG-Q5 (B), HLA-B*35:08-HPVG (C), or HLA-B*35:08-HPVG-D5 (D). The TK3 TCR is represented in schematics with the α-chain in pale pink, the β-chain in pale blue, and the CDR loops colored in yellow, magenta, and marine for the CDR1, -2 and -3α and in orange, red, and green for the CDR1, -2, and -3β, respectively. The HLA and β2m are represented in schematics and colored in white (HLA-B*35:01 and β2m) or gray (HLA-B*35:08). The peptides are represented in stick format and colored in pale purple for HPVG, green for HPVG-D5, and brown for the HPVG-Q5. The bottom panels show the footprint of the TK3 TCR on the surface of the pHLA complexes, namely HLA-B*35:01-HPVG (A), HLA-B*35:01-HPVG-Q5 (B), HLA-B*35:08-HPVG (C), or HLA-B*35:08-HPVG-D5 (D). The atomic contacts are colored accordingly to the CDR loop and follow the same color schemes the top panel. The black spheres represent the center of mass of the Vα and Vβ domains of the TK3 TCR.
The TK3 TCR docked onto HLA-B*35:01-HPVG-Q5 comparably to the HLA-B*35:01-HPVG complex (overall r.m.s.d of 0.29 Å; Fig. 5, A and B). However, the total buried surface area was moderately smaller (∼1960 Å2) in the HPVG-Q5 complex compared with the HPVG complex (∼2050 Å2), and this also correlated with a reduced number of contacts in the HPVG-Q5 ternary complex (154 contacts compared with 193 contact observed in TK3-HLA-B*35:01-HPVG complex). Despite the striking structural differences observed between the two peptides in the HLA-B*35:01 cleft, upon TK3 TCR binding both peptides adopted the same conformation (r.m.s.d. of 0.08 Å; Fig. 6A). Thus, the TK3 TCR untangled one helical turn within the central region of HPVG-Q5 peptide, in the cleft of HLA-B*35:01, which molded the peptide into a structure that mimicked the TK3-ligated HPVG conformation. This peptide untangling correlated with a 27-fold slower on-rate for TK3 TCR when recognizing HLA-B*35:01-HPVG-Q5 (Kon = 0.15 × 104/ms) compared with HLA-B*35:01-HPVG (Kon = 4.0 × 104/ms) (20). Thus, an induced form of molecular mimicry enabled the TK3 TCR to recognize HPVG variants that adopt differing conformations in the binary pHLA-B*35:01 complexes.
FIGURE 6.

Peptide flexibility enhances the TK3 TCR recognition. A, superposition of the TK3 TCR-HLA-B*35:01-HPVG-Q5 complex (yellow) with the TK3 TCR-HLA-B*35:01-HPVG complex (pink), with the TCR and HLA in schematic format, and the peptide in stick representation. B, superposition of the HLA-B*35:08-HPVG free (orange) and bound to the TK3 TCR (green). The peptide is represented in schematic format with the Cα of the P8-Tyr residue show as a sphere. C, superposition of the TK3 TCR in complex with the HLA-B*35:01-HPVG (pink) and HLA-B*35:08-HPVG (green), with the TCR and HLA in schematic representation and the peptide in stick representation. D, superposition of the HLA-B*35:08-HPVG (green) with HLA-B*35:08-HPVG-D5 (blue) in complex with the TK3 TCR. The red dash lines represent the hydrogen bonds formed between Arg-97, Asp-114, and the P5-Glu. E, superposition of the HLA-B*35:08-HPVG-D5 free (red) and bound to the TK3 TCR (blue). The spheres represent the Cα atom of the P8-Tyr from the HPVG-D65 peptide, the red dashed lines represent the hydrogen bonds formed by Arg-97 with Asp-114 and Tyr-74 in the TK3 TCR-HLA-B*35:08-HPVG-D5 structure. F, superposition of the HLA-B*35:08-HPVG-D5 (blue) and HLA-B*35:01-HPVG (pink) in complex with the TK3 TCR.
The TK3 TCR-HLA-B*35:08-HPVG Ternary Complex
The TRBV9+ TK3 TCR preferentially recognized the HLA-B*35:01-HPVG complex over the HLA-B*35:08-HPVG complex. To understand the basis of this finely tuned HLA-restricted response, we determined the structure of the TK3 TCR-HLA-B*35:08-HPVG complex. This complex was crystallized in the same space group as the TK3 TCR-HLA-B*35:01-HPVG complex and determined to 2.4 Å resolution (Table 3). The TK3 TCR docked diagonally (66°) onto the HLA-B*35:08-HPVG complex and aligned similarly to that of the cognate TK3 TCR-HLA-B*35:01-HPVG complex (α1-α2 domains r.m.s.d. value of 0.24 Å) (Fig. 5C). The total buried surface area of the TK3 TCR-HLA-B*35:08-HPVG interface is 2040 Å2, which was composed of 150 van der Waals (vdw) contacts, 14 H-bonds (Hb), and 1 salt bridge (sb) interactions, and exhibited fewer contacts than the TK3 TCR-HLA-B*35:01-HPVG complex (buried surface area of 2050 Å2, 172 vdw, 18 Hb, 3 sb).
The structures of the HLA-B*35:08-HPVG free and in complex with the TK3 TCR provided an opportunity to analyze the conformational changes of the HPVG epitope upon complexation. The central region of the HPVG epitope was disordered in the HLA-B*35:01 binary complex yet fully resolved and more rigid in the HLA-B*35:08 complex. Comparison of the HLA-B*35:08-HPVG structures in its free and TK3 TCR-bound forms revealed that the HPVG peptide changed conformation markedly upon TCR ligation. In the binary HLA-B*35:08-HPVG complex, the HPVG peptide leaned toward the α1-helix of HLA-B*35:08, and upon TK3 TCR binding, the central region (P7-Asp to P9-Phe) of the peptide was pushed toward the α2-helix, with a maximal displacement of 3.5 Å of the P8-Tyr Cα atom (Fig. 6B). The CDR3α loop of the TK3 TCR pushed the P7-Asp to avoid steric clashes, allowing the P8-Tyr aromatic ring to be enveloped by the three CDRβ loops. A notable feature of the TK3 TCR-HLA-B*35:08-HPVG complex is that the peptide adopted the same conformation as that observed in the TK3 TCR-HLA-B*35:01-HPVG complex (r.m.s.d. of 0.18 Å) (Fig. 6C) despite marked differences observed in their binary complexes (Fig. 4, A and B). The TK3 TCR-induced structural changes observed in the HPVG peptide when bound by the HLA-B*35:08 are consistent with the lower affinity and reduced activity of the TK3 TCR toward the HLA-B*35:08-HPVG complex (Figs. 1A and 2) in comparison to the HLA-B*35:01-HPVG complex (20). Alternatively, the conformational flexibility of the HPVG peptide in HLA-B*35:01 may be the driving force for the high reactivity toward the TK3 TCR. Accordingly, our data suggest that the micro-polymorphism between HLA-B*35:01/08 can play a pivotal role in fine-tuning the dynamics of antigen flexibility, which subsequently impacts on TK3 TCR recognition and ultimately immunogenicity.
A HPVG Variant Changes HLA Restriction
Although the TK3 TCR affinity was low for HLA-B*35:08 presenting the WT HPVG peptide from the most frequent Caucasian EBV strain (Kd = 93.2 μm), it was improved by almost 4-fold with the HPVG-D5 variant from the Chinese EBV strain (Kd = 25.4 μm) (Fig. 3). To understand the mechanism driving the higher affinity of the TK3 TCR toward HPVG-D5, we solved the structure of the TK3 TCR-HLA-B*35:08-HPVG-D5 complex (Fig. 5D). The overall structure was similar to the TK3 TCR-HLA-B*35:08-HPVG ternary complexes. Namely, the TK3 TCR adopted a diagonal docking mode onto HLA-B*35:08-HPVG-D5 (66°), with a buried surface area of 2020 Å2. The main differences between the two ternary complexes were centered on the P5 position of the peptide (Fig. 6D). In HLA-B*35:08-HPVG, Arg-156 is part of a polar network that includes Arg-97 and P5-Glu (Fig. 4A), whereas in HLA-B*35:08-HPVG-D5, this polar network was weakened due to the shorter side chain of P5-Asp (Fig. 4E). Upon TK3 TCR binding, the HPVG-D5 was pushed toward the α2-helix of HLA-B*35:08 by the CDR3α loop (Fig. 6E) in a manner similar to the TK3 TCR-induced HPVG peptide movement. In comparison to the P5-Glu residue, the shorter P5-Asp residue was positioned 1 Å deeper into the cleft upon TK3 TCR ligation, which caused Arg-97 to shift away from the peptide and interact with the Asp-114 and Tyr-74, as seen in HLA-B*35:01-HPVG complex (Fig. 6F). However, the longer P5-Glu residue does not allow these conformational readjustments in HLA-B*35:08 upon TK3 engagement. Accordingly, the HLA-B*35:08-HPVG-D5 complex appeared intrinsically more malleable than the HLA-B*35:08-HPVG complex, which resulted in improved recognition by the TK3 TCR.
DISCUSSION
Viral mutations are well known to subvert the antiviral T cell-mediated response by impacting HLA binding, the Ag presentation machinery, and T cell receptor recognition. Here we show how naturally occurring EBV variants affect the flexibility and conformation of a T cell epitope, which subsequently directly impacts on TCR recognition. We also show how a single HLA micropolymorphism within the HLA-B*35 family alters the conformation, and hence immunogenicity, of the WT viral epitope and variants. Moreover, our findings demonstrate the functional benefit of maintaining HLA micropolymorphism, as one viral variant effectively caused the switching of the HLA restriction element.
The HPVG epitope is restricted to HLA-B*35:01 and HLA-B*35:08, two HLA allomorphs that differ by one residue at position 156, with this polymorphism also dictating the nature of the responding T cell repertoire (25). Namely, in HLA-B*35:01+ individuals the CTL response is characterized by biased TRAV20-TRBV9 usage, whereas in HLA-B*35:08+ individuals it is underpinned by TRAV29-TRBV9 usage (20, 25). Previously, we characterized the molecular basis of recognition by the prototypical TCR TK3 that responds to the HPVG epitope from HLA-B*35:01+ individuals and also showed how TCR polymorphism within the TRBV9 gene impacted this protective immune response (20). We also demonstrated how the viral mutants HPVG-A4, HPVG-D5, and HPVG-Q5 impacted this HLA-B*35:01-restricted response.
In this study we aimed to address the basis of the finely balanced HLA-B*35:01/08 restriction. The single residue polymorphism (HLA-B*35:01, Leu-156; HLA-B*35:08, Arg-156) is located at the base of the Ag binding cleft, so a priori it was unclear how such a buried polymorphism could impact TCR recognition in this system. Previously, how HLA-B*35:01/08 polymorphism impacts on the conformation and immunogenicity of 10-mer, 11-mer (6, 38), and 13-mer EBV determinants have been determined (39). More broadly, buried polymorphisms within the HLA-B*44 (40–42) and HLA-B*57 families (43) can impact on epitope flexibility and TCR recognition in antiviral responses and, in the context of HLA-B*44, in an alloreactive T cell response (42). Here we show that the HPVG epitope is directly impacted by this polymorphic residue, with the magnitude of the effect being dependent on the nature of the naturally occurring viral variants of the HPVG epitope. The impact of these viral mutants was dramatic and unpredictable. For example, the HPVG-Q5 variant caused a large reorientation of the epitope within HLA-B*35:01/08 such that the P5-Gln residue pointed out of the cleft, whereas the P5-Glu residue represented a middle anchor residue in HLA-B*35:01. Despite this difference, the TK3 TCR was still able to ligate to the HLA-B*35:01-HPVG-Q5 epitope, albeit more weakly. The TK3 TCR was able to achieve this recognition by flattening the HPVG-Q5 determinant in a manner analogous to how TCRs have been shown to “bulldoze” bulged antigenic determinants (38). Here, the HPVG-Q5 peptide adopted the same conformation as the HPVG determinant in its TCR-ligated state, thereby indicating that the TK3 TCR can accommodate differing HPVG landscapes via an induced-fit mechanism.
The TK3 TCR was able to cope well with the HPVG-A4 variant when presented by HLA-B*35:01, whereas the HPVG-D5 variant was very poorly recognized. Surprisingly, however, the HPVG-D5 variant was recognized better by TK3 when presented by HLA-B*35:08 than the WT-HPVG epitope. The ability of the TK3 clone to preferentially recognize the HPVG-D5 determinant is due to the greater capacity of this epitope to enable conformational changes within HLA-B*35:08 upon TCR ligation. It is likely that the T cell repertoire in HLA-B*35:01+ individuals, harboring the EBV strain containing the HPVG-D5 variant, will differ from the classical TK3 gene usage (TRAV20/TRBV9) to maintain optimal viral suppression. Our findings highlight the sensitive and unpredictable interplay between T cell repertoire usage, HLA polymorphism, and viral escape.
Acknowledgment
We thank the staff at the Australian Synchrotron for assistance with data collection.
This work was supported by the Australian Research Council and National Health and Medical Research Council of Australia Grants APP1067374 and APP1021620.
- HLA
- human leukocyte antigen
- pHLA
- peptide-human leukocyte antigen complex
- CDR
- complementarity-determining region
- r.m.s.d.
- root mean square deviation
- TCR
- T cell receptors
- MHC
- major histocompatibility complex
- EBV
- Epstein-Barr virus
- HPVG
- HPVGEADYFEY
- CTL
- cytotoxic T cell
- Ag
- antigen.
REFERENCES
- 1. Parham P. (1996) Pictures of MHC restriction. Nature 384, 109–110 [DOI] [PubMed] [Google Scholar]
- 2. Rammensee H. G., Falk K., Rötzschke O. (1993) Peptides naturally presented by MHC class I molecules. Annu. Rev. Immunol. 11, 213–244 [DOI] [PubMed] [Google Scholar]
- 3. Falk K., Rötzschke O., Grahovac B., Schendel D., Stevanović S., Jung G., Rammensee H. G. (1993) Peptide motifs of HLA-B35 and -B37 molecules. Immunogenetics 38, 161–162 [DOI] [PubMed] [Google Scholar]
- 4. Smith K. J., Reid S. W., Stuart D. I., McMichael A. J., Jones E. Y., Bell J. I. (1996) An altered position of the α2 helix of MHC class I is revealed by the crystal structure of HLA-B*3501. Immunity 4, 203–213 [DOI] [PubMed] [Google Scholar]
- 5. Burrows J. M., Wynn K. K., Tynan F. E., Archbold J., Miles J. J., Bell M. J., Brennan R. M., Walker S., McCluskey J., Rossjohn J., Khanna R., Burrows S. R. (2007) The impact of HLA-B micropolymorphism outside primary peptide anchor pockets on the CTL response to CMV. Eur. J. Immunol. 37, 946–953 [DOI] [PubMed] [Google Scholar]
- 6. Tynan F. E., Elhassen D., Purcell A. W., Burrows J. M., Borg N. A., Miles J. J., Williamson N. A., Green K. J., Tellam J., Kjer-Nielsen L., McCluskey J., Rossjohn J., Burrows S. R. (2005) The immunogenicity of a viral cytotoxic T cell epitope is controlled by its MHC-bound conformation. J. Exp. Med. 202, 1249–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tynan F. E., Borg N. A., Miles J. J., Beddoe T., El-Hassen D., Silins S. L., van Zuylen W. J., Purcell A. W., Kjer-Nielsen L., McCluskey J., Burrows S. R., Rossjohn J. (2005) High resolution structures of highly bulged viral epitopes bound to major histocompatibility complex class I. Implications for T-cell receptor engagement and T-cell immunodominance. J. Biol. Chem. 280, 23900–23909 [DOI] [PubMed] [Google Scholar]
- 8. Zernich D., Purcell A. W., Macdonald W. A., Kjer-Nielsen L., Ely L. K., Laham N., Crockford T., Mifsud N. A., Bharadwaj M., Chang L., Tait B. D., Holdsworth R., Brooks A. G., Bottomley S. P., Beddoe T., Peh C. A., Rossjohn J., McCluskey J. (2004) Natural HLA class I polymorphism controls the pathway of antigen presentation and susceptibility to viral evasion. J. Exp. Med. 200, 13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hülsmeyer M., Hillig R. C., Volz A., Rühl M., Schröder W., Saenger W., Ziegler A., Uchanska-Ziegler B. (2002) HLA-B27 subtypes differentially associated with disease exhibit subtle structural alterations. J. Biol. Chem. 277, 47844–47853 [DOI] [PubMed] [Google Scholar]
- 10. Davis M. M., Bjorkman P. J. (1988) T-cell antigen receptor genes and T-cell recognition. Nature 334, 395–402 [DOI] [PubMed] [Google Scholar]
- 11. Turner S. J., Doherty P. C., McCluskey J., Rossjohn J. (2006) Structural determinants of T-cell receptor bias in immunity. Nat. Rev. Immunol. 6, 883–894 [DOI] [PubMed] [Google Scholar]
- 12. Doherty P. C., Zinkernagel R. M. (1975) Enhanced immunological surveillance in mice heterozygous at the H-2 gene complex. Nature 256, 50–52 [DOI] [PubMed] [Google Scholar]
- 13. Früh K., Ahn K., Djaballah H., Sempé P., van Endert P. M., Tampé R., Peterson P. A., Yang Y. (1995) A viral inhibitor of peptide transporters for antigen presentation. Nature 375, 415–418 [DOI] [PubMed] [Google Scholar]
- 14. Ladell K., Hashimoto M., Iglesias M. C., Wilmann P. G., McLaren J. E., Gras S., Chikata T., Kuse N., Fastenackels S., Gostick E., Bridgeman J. S., Venturi V., Arkoub Z. A., Agut H., van Bockel D. J., Almeida J. R., Douek D. C., Meyer L., Venet A., Takiguchi M., Rossjohn J., Price D. A., Appay V. (2013) A molecular basis for the control of preimmune escape variants by HIV-specific CD8+ T cells. Immunity 38, 425–436 [DOI] [PubMed] [Google Scholar]
- 15. Dambaugh T., Hennessy K., Chamnankit L., Kieff E. (1984) U2 region of Epstein-Barr virus DNA may encode Epstein-Barr nuclear antigen 2. Proc. Natl. Acad. Sci. U.S.A. 81, 7632–7636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sample J., Young L., Martin B., Chatman T., Kieff E., Rickinson A. (1990) Epstein-Barr virus types 1 and 2 differ in their EBNA-3A, EBNA-3B, and EBNA-3C genes. J. Virol. 64, 4084–4092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sitki-Green D., Edwards R. H., Webster-Cyriaque J., Raab-Traub N. (2002) Identification of Epstein-Barr virus strain variants in hairy leukoplakia and peripheral blood by use of a heteroduplex tracking assay. J. Virol. 76, 9645–9656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang J. T., Sheeng T. S., Su I. J., Chen J. Y., Chen M. R. (2003) EBNA-1 sequence variations reflect active EBV replication and disease status or quiescent latency in lymphocytes. J. Med. Virol. 69, 417–425 [DOI] [PubMed] [Google Scholar]
- 19. Bell M. J., Brennan R., Miles J. J., Moss D. J., Burrows J. M., Burrows S. R. (2008) Widespread sequence variation in Epstein-Barr virus nuclear antigen 1 influences the antiviral T cell response. J. Infect. Dis. 197, 1594–1597 [DOI] [PubMed] [Google Scholar]
- 20. Gras S., Chen Z., Miles J. J., Liu Y. C., Bell M. J., Sullivan L. C., Kjer-Nielsen L., Brennan R. M., Burrows J. M., Neller M. A., Khanna R., Purcell A. W., Brooks A. G., McCluskey J., Rossjohn J., Burrows S. R. (2010) Allelic polymorphism in the T cell receptor and its impact on immune responses. J. Exp. Med. 207, 1555–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang W. Y., Chien Y. C., Jan J. S., Chueh C. M., Lin J. C. (2002) Consistent sequence variation of Epstein-Barr virus nuclear antigen 1 in primary tumor and peripheral blood cells of patients with nasopharyngeal carcinoma. Clin. Cancer Res. 8, 2586–2590 [PubMed] [Google Scholar]
- 22. Zhang X. S., Wang H. H., Hu L. F., Li A., Zhang R. H., Mai H. Q., Xia J. C., Chen L. Z., Zeng Y. X. (2004) V-val subtype of Epstein-Barr virus nuclear antigen 1 preferentially exists in biopsies of nasopharyngeal carcinoma. Cancer Lett. 211, 11–18 [DOI] [PubMed] [Google Scholar]
- 23. Snudden D. K., Smith P. R., Lai D., Ng M. H., Griffin B. E. (1995) Alterations in the structure of the EBV nuclear antigen, EBNA1, in epithelial cell tumours. Oncogene 10, 1545–1552 [PubMed] [Google Scholar]
- 24. Dolan A., Addison C., Gatherer D., Davison A. J., McGeoch D. J. (2006) The genome of Epstein-Barr virus type 2 strain AG876. Virology 350, 164–170 [DOI] [PubMed] [Google Scholar]
- 25. Miles J. J., Borg N. A., Brennan R. M., Tynan F. E., Kjer-Nielsen L., Silins S. L., Bell M. J., Burrows J. M., McCluskey J., Rossjohn J., Burrows S. R. (2006) TCRα genes direct MHC restriction in the potent human T cell response to a class I-bound viral epitope. J. Immunol. 177, 6804–6814 [DOI] [PubMed] [Google Scholar]
- 26. Borg N. A., Ely L. K., Beddoe T., Macdonald W. A., Reid H. H., Clements C. S., Purcell A. W., Kjer-Nielsen L., Miles J. J., Burrows S. R., McCluskey J., Rossjohn J. (2005) The CDR3 regions of an immunodominant T cell receptor dictate the “energetic landscape” of peptide-MHC recognition. Nat. Immunol. 6, 171–180 [DOI] [PubMed] [Google Scholar]
- 27. Gras S., Burrows S. R., Kjer-Nielsen L., Clements C. S., Liu Y. C., Sullivan L. C., Bell M. J., Brooks A. G., Purcell A. W., McCluskey J., Rossjohn J. (2009) The shaping of T cell receptor recognition by self-tolerance. Immunity 30, 193–203 [DOI] [PubMed] [Google Scholar]
- 28. Gras S., Wilmann P. G., Chen Z., Halim H., Liu Y. C., Kjer-Nielsen L., Purcell A. W., Burrows S. R., McCluskey J., Rossjohn J. (2012) A structural basis for varied abTCR usage against an immunodominant EBV antigen restricted to a HLA-B8 molecule. J. Immunol. 188, 311–321 [DOI] [PubMed] [Google Scholar]
- 29. Kabsch W. (2010) Xds. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 31. McCoy A. J. (2007) Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63, 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Smart O. S., Womack T. O., Flensburg C., Keller P., Paciorek W., Sharff A., Vonrhein C., Bricogne G. (2012) Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr. D Biol. Crystallogr. 68, 368–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lefranc M.-P., Giudicelli V., Kaas Q., Duprat E., Jabado-Michaloud J., Scaviner D., Ginestoux C., Clément O., Chaume D., Lefranc G. (2005) IMGT, the international ImMunoGeneTics information system(R). Nucleic Acids Res. 33, D593–D597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. DeLano W. L. (2002) The PyMOL Molecular Graphics System [Google Scholar]
- 37. Varro R., Chen R., Sepulveda H., Apgar J. (2007) Bead-based multianalyte flow immunoassays: the cytometric bead array system. Methods Mol. Biol. 378, 125–152 [DOI] [PubMed] [Google Scholar]
- 38. Tynan F. E., Reid H. H., Kjer-Nielsen L., Miles J. J., Wilce M. C., Kostenko L., Borg N. A., Williamson N. A., Beddoe T., Purcell A. W., Burrows S. R., McCluskey J., Rossjohn J. (2007) A T cell receptor flattens a bulged antigenic peptide presented by a major histocompatibility complex class I molecule. Nat. Immunol. 8, 268–276 [DOI] [PubMed] [Google Scholar]
- 39. Tynan F. E., Burrows S. R., Buckle A. M., Clements C. S., Borg N. A., Miles J. J., Beddoe T., Whisstock J. C., Wilce M. C., Silins S. L., Burrows J. M., Kjer-Nielsen L., Kostenko L., Purcell A. W., McCluskey J., Rossjohn J. (2005) T cell receptor recognition of a “super-bulged” major histocompatibility complex class I-bound peptide. Nat. Immunol. 6, 1114–1122 [DOI] [PubMed] [Google Scholar]
- 40. Macdonald W. A., Purcell A. W., Mifsud N. A., Ely L. K., Williams D. S., Chang L., Gorman J. J., Clements C. S., Kjer-Nielsen L., Koelle D. M., Burrows S. R., Tait B. D., Holdsworth R., Brooks A. G., Lovrecz G. O., Lu L., Rossjohn J., McCluskey J. (2003) A naturally selected dimorphism within the HLA-B44 supertype alters class I structure, peptide repertoire, and T cell recognition. J. Exp. Med. 198, 679–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Archbold J. K., Macdonald W. A., Gras S., Ely L. K., Miles J. J., Bell M. J., Brennan R. M., Beddoe T., Wilce M. C., Clements C. S., Purcell A. W., McCluskey J., Burrows S. R., Rossjohn J. (2009) Natural micropolymorphism in human leukocyte antigens provides a basis for genetic control of antigen recognition. J. Exp. Med. 206, 209–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Macdonald W. A., Chen Z., Gras S., Archbold J. K., Tynan F. E., Clements C. S., Bharadwaj M., Kjer-Nielsen L., Saunders P. M., Wilce M. C., Crawford F., Stadinsky B., Jackson D., Brooks A. G., Purcell A. W., Kappler J. W., Burrows S. R., Rossjohn J., McCluskey J. (2009) T cell allorecognition via molecular mimicry. Immunity 31, 897–908 [DOI] [PubMed] [Google Scholar]
- 43. Stewart-Jones G. B., Simpson P., van der Merwe P. A., Easterbrook P., McMichael A. J., Rowland-Jones S. L., Jones E. Y., Gillespie G. M. (2012) Structural features underlying T-cell receptor sensitivity to concealed MHC class I micropolymorphisms. Proc. Natl. Acad. Sci. U.S.A. 109, E3483–E3492 [DOI] [PMC free article] [PubMed] [Google Scholar]