

Background: The O-acetylation of peptidoglycan in Gram-negative bacteria is catalyzed by PatB.
Results: PatB can utilize a variety of acetyl donors for transfer to polymerized glycans containing N-acetyl groups and muroglycans containing tri- and tetrapeptide stems.
Conclusion: PatB has broad specificity for acetyl donors but narrow specificity for acceptor glycans.
Significance: This information will help with the identification of possible leads to novel classes of antibiotics.
Keywords: Bacterial Pathogenesis, Carbohydrate Biosynthesis, Cell Wall, Enzyme Kinetics, Peptidoglycan, Chitooligosaccharides, O-Acetylation, O-Acetyltransferase, Substrate Specificity
Abstract
The O-acetylation of the essential cell wall polymer peptidoglycan is a major virulence factor identified in many bacteria, both Gram-positive and Gram-negative, including Staphylococcus aureus, Bacillus anthracis, Neisseria gonorrhoeae, and Neisseria meningitidis. With Gram-negative bacteria, the translocation of acetyl groups from the cytoplasm is performed by an integral membrane protein, PatA, for its transfer to peptidoglycan by O-acetyltransferase PatB, whereas a single bimodal membrane protein, OatA, appears to catalyze both reactions of the process in Gram-positive bacteria. Only phenotypic evidence existed in support of these pathways because no in vitro biochemical assay was available for their analysis, which reflected the complexities of investigating integral membrane proteins that act on a totally insoluble and heterogeneous substrate, such as peptidoglycan. In this study, we present the first biochemical and kinetic analysis of a peptidoglycan O-acetyltransferase using PatB from N. gonorrhoeae as the model system. The enzyme has specificity for muropeptides that possess tri- and tetrapeptide stems on muramyl residues. With chitooligosaccharides as substrates, rates of reaction increase with increasing degrees of polymerization to 5/6. This information will be valuable for the identification and development of peptidoglycan O-acetyltransferase inhibitors that could represent potential leads to novel classes of antibiotics.
Introduction
Bacterial cell walls and, in particular, their essential component peptidoglycan (PG)2 have been a popular and successful target for antibacterial therapy ever since penicillin was introduced to clinicians in the 1940s. However, resistance to the β-lactams and other clinically introduced antibiotics that followed has necessitated the search for new antibacterial targets. One such target may be the enzymes involved in the metabolism of PG O-acetylation.
The O-acetylation of PG is a major virulence factor identified in many bacteria, both Gram-positive and Gram-negative. The list includes important human pathogens, such as Staphylococcus aureus, Bacillus anthracis, all species of Enterococcus, species of Campylobacter, Listeria monocytogenes, Neisseria gonorrhoeae, and Neisseria meningitidis (reviewed in Ref. 1). This modification to PG occurs predominantly at the C-6 hydroxyl group of N-acetylmuramyl (MurNAc) residues, and it is particularly significant because it sterically hinders the activity of muramidases (lysozymes), thereby providing protection from the defensive activity of innate immune systems. Moreover, it totally precludes the function of the lytic transglycosylases, endogenous bacterial enzymes essential for cell growth and division, PG turnover, and the insertion of wall-spanning structures, such as flagella and secretory systems (reviewed in Refs. 1 and 2). The lytic transglycosylases are also muralytic enzymes with the same specificity as lysozyme, but they are not hydrolases. Rather, they perform substrate-assisted catalysis, utilizing the C-6 hydroxyl group of MurNAc residues to cleave PG with the concomitant formation of 1,6-anhydromuramic acid reaction products (3). Consequently, the O-acetylation of PG has been proposed to control the autolytic activity of the lytic transglycosylases at the substrate level (1, 2). This PG modification may also serve as a defense mechanism against lytic enzymes produced by bacteriophage or Type VI secretion effectors, although this remains to be investigated.
Recent reports have demonstrated that the C-6 hydroxyl of N-acetylglucosaminyl (GlcNAc) residues in the PG of Lactobacillus species also are acetylated (4), and this may extend to species of Bacillus (5). With Lactobacillus plantarum, the O-acetylation of GlcNAc appears to control its major autolysin Acm2, an N-acetyl-glucosaminidase (which has specificity for the GlcNAc-MurNAc glycosidic linkage instead of the lysozyme-specific linkage of MurNAc-GlcNAc) (1, 4).
The O-acetylation of PG was discovered over 50 years ago by two independent groups (6, 7), but the processes for this modification were only recently described. Based on genomic studies, two distinct pathways were proposed that appeared to be unique to Gram-positive and Gram-negative bacteria, respectively (8), and subsequent experimental evidence has been obtained to support both. Thus, a two-component system is proposed for Gram-negative bacteria involving the translocation of acetyl groups from the cytoplasm (acetyl-CoA is the presumed source) to the periplasm by an integral membrane protein, PatA (PG O-acetyltransferase A) and then their transfer to PG by a peripheral membrane O-acetyltransferase, PatB (9, 10). With Gram-positive bacteria, a single bimodal membrane protein, Oat (O-acetylpeptidoglycan transferase), appears to catalyze both reactions of the process (11, 12). Interestingly, B. anthracis and strains of Bacillus cereus encode the genes for both systems and in multiple copies (5). To date, however, only phenotypic evidence exists in support of these pathways in any bacterium because no in vitro biochemical assay was available for their analysis. Moreover, none of these enzymes have been demonstrated to modify their native substrate in vitro. This situation reflects the complexities of investigating membrane-associated proteins that act on a totally insoluble and heterogeneous substrate such as PG.
Based on sequence alignment studies, the patB gene of N. gonorrhoeae was originally predicted to encode an SGNH/GDSL family esterase with similarity to the CAZy family CE-3 O-acetylxylan esterases (13) and Ape1 (O-acetyl-PG esterase 1) (14), the latter being shown experimentally to function as a serine esterase (15). Indeed, PatB shares 20.6% amino acid identity and 58.5% similarity with Ape1, including consensus motifs involving a potential catalytic triad of residues expected of a serine esterase. Whereas PatB was found to possess weak esterase activity, phenotypic assays demonstrated its true function to be a PG O-acetyltransferase (9). However, the lack of an appropriate in vitro assay has precluded any understanding of its substrate specificity and biochemical properties. To compound this, the tertiary structure of any PG O-acetyltransferase is not known. Consequently, lacking both a biochemical assay and structural information, still nothing was known about the enzymes despite increased interest in them. In this regard, an inhibitor of Ape1 was demonstrated recently to serve as an antibacterial with specificity against both Gram-positive and Gram-negative bacteria that produce O-acetylated PG (16), demonstrating the sensitivity of those bacteria to its alteration. Furthermore, OatA from species of Staphylococcus has been correlated with pathogenicity, whereas it is required for full virulence in Listeria monocytogenes (17, 18). Given this, together with the role PG O-acetylation has in controlling the autolytic activity of the lytic transglycosylases (2, 9), it would be expected that blocking this reaction through inhibition of PatB would be even more deleterious to bacterial cells.
We recently reported the first in vitro assay for PG O-acetyltransferases that exploits their ability to utilize both chromogenic acetylesterase substrates as acetyl donors and chitooligosaccharides as acceptors (19). In this study, we present the first biochemical analysis of a PG O-acetyltransferase using PatB from N. gonorrhoeae as the model.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents
Acrylamide and glycerol were purchased from Fisher, whereas isopropyl β-d-1-thiogalactopyranoside was from Roche Applied Science, and chitooligosaccharides were products of Toronto Research Chemicals or Carbosynth (Berkshire, UK). All growth media were from Difco. Ni2+-nitrilotriacetic acid (Ni2+-NTA)-agarose was supplied by Qiagen (Valencia, CA), Source Q was purchased from GE Healthcare, graphitized carbon solid phase extraction columns (Carbograph SPE) were products of Grace Canada, Inc. (Ajax, Canada), and Hypercarb porous graphitized carbon columns were supplied by Thermo Electron Corp. (Rockford, IL). Mouse anti-His6 antibody was obtained from Santa Cruz Biotechnology, Inc. Unless otherwise stated, all other chemicals and reagents were purchased from Sigma-Aldrich.
Isolation of Soluble Muropeptides
PG from Escherichia coli was purified as described previously (1). An evenly dispersed 1.0 mg·ml−1 suspension of this PG (generated by sonication) in 50 mm sodium phosphate buffer, pH 6.2, containing 0.2% NaN3 and 5 mm MgCl2 was incubated at 37 °C for 24 h with both hen egg white lysozyme and E. coli PBPs 4 and 7 (kindly provided by K. Young, University of Arkansas School of Medicine) to a final concentration of 100 μg·ml−1 and 10 μg·ml−1, respectively (PBPs 4 and 7 were included to enhance the production of soluble, uncross-linked muropeptides). Insoluble material was collected by centrifugation at 20,000 × g, and the solubilized muropeptides present in the supernatant were fractionated by solid phase extraction as described below. The eluted muropeptides were dried in vacuo at 30 °C and stored at −20 °C prior to use. The muramic acid content of respective samples was determined by high pH anion exchange chromatography as described previously (9).
Bacterial Strains and Culture Conditions
The strains of bacteria used in this study and their genotypes are presented in Table 1. N. gonorrhoeae FA1090 was grown for 24 h at 35 °C on GC medium base supplemented with Kellogg's defined supplement (20, 21) in a humid, 5% CO2 environment, as described previously (9). All plasmids constructed were screened and maintained in E. coli DH5α. When production of high levels of protein was required, E. coli BL21*-λDE3 cells were always freshly transformed with the desired expression plasmid (Table 1) and grown in Super Broth (5 g of sodium chloride, 20 g of yeast extract, and 32 g of tryptone) at 37 °C with agitation. E. coli growth cultures were supplemented with chloramphenicol (35 mg·ml−1), ampicillin (100 mg·ml−1), and kanamycin (50 mg·ml−1) when required.
TABLE 1.
Strains and plasmids used in this study
| Strain/Plasmid | Description | Source/Reference |
|---|---|---|
| Strains | ||
| E. coli BL21*λ (DE3) | F− ompT hsdSB (rB−mB−) gal dcm rne131 (DE3) | Novagen |
| E. coli DH5α | fhuA2 lac(del)U169 phoA glnV44 Φ80′ lacZ(del)M15 gyrA96 recA1 relA1 endA1 thi-1 hsdR17 | Invitrogen |
| E. coli Top10 | mcrA, Δ(mrr-hsdRMS-mcrBC), Phi80lacZ(del)M15, ΔlacX74, deoR, recA1, araD139, Δ(ara-leu)7697, galU, galK, rpsL(SmR), endA1, nupG | Invitrogen |
| Plasmids | ||
| pACPM22 | pBADHis-A derivative encoding PatB truncated by its N-terminal 36 amino acid residues with a C-terminal His6 tag; CmR | Ref. 9 |
| pACPM30 | pBAD His-A derivative encoding PatB truncated by its N-terminal 77 amino acid residues; AmpR | This study |
| pACPM33 | pET-SUMO derivative encoding PatB truncated by its N-terminal 36 amino acid residues with an N-terminal His6-SUMO tag; AmpR | This study |
Engineering of PatB
Due to low yields and protein instability obtained with prior constructs (9), two new forms of PatB were generated. The first involved a further truncation of the enzyme by another 41 N-terminal amino acid residues. Thus, the gene encoding PatBΔ36 was subcloned from pACPM22 (9) by PCR amplification using the primers 3′-ATCGCTCGAGCTGTCCGGCGAAACGC-5′ (forward) and 3′-CAGTAAGCTTTCATGGCTGTGTACTTGAGGTTG-5′ (reverse). The resulting PCR product was then digested using appropriate restriction enzymes and ligated into a similarly digested pBADHis-A plasmid with T4 DNA ligase, yielding pACPM30. The protein product of this construct, PatBΔ77, lacked its 77 N-terminal amino acids.
The second construct, encoding PatBSUMO, was also generated by subcloning from pACPM22. In this case, PCR amplification involved the forward primer 3′-TACTGGCAGCAGACCTACCAC-5′ and the same reverse primer as described above and ligation directly into the Champion pET-SUMO vector according to the manufacturer's specifications, yielding pACPM33. The protein product from this construct lacked its 36-amino acid, periplasmic-localizing signal sequence while possessing an N-terminal fusion of the His6-SUMO.
Production and Purification of PatB
E. coli BL21*-λDE3 was transformed with pACPM30 or pACPM33 for overproduction of the recombinant forms of PatB described in this study. Cells were grown in 1 liter of Super Broth at 37 °C to an A600 nm of 0.6, and expression of genes was induced with isopropyl β-d-1-thiogalactopyranoside (1 mm, final concentration) for 3 h. Cells were harvested by centrifugation (6,000 × g, 15 min, 4 °C), and they were resuspended in 30 ml of 20 mm sodium phosphate buffer, pH 8.0, containing 500 mm NaCl and 10 mm imidazole (lysis buffer) and subjected to lysis by four successive passages through an EmusliFlex C-3 homogenizer (AVESTIN, Inc., Ottawa, Canada) at 15,000 p.s.i. The lysate was clarified by centrifugation (6,000 × g, 15 min, 4 °C), and the PatB forms were isolated by affinity chromatography on Ni2+-NTA-agarose. The resin (500 μl) was washed with the lysis buffer, and bound proteins were eluted in lysis buffer containing 250 mm imidazole. Recovered proteins were dialyzed at 4 °C exhaustively against 20 mm sodium phosphate buffer, pH 8.0, containing 100 mm NaCl and 50 mm l-arginine and then against the same buffer lacking the NaCl, followed by the same buffer but lacking the NaCl and arginine (this sequence of buffer changes was found to be essential to retain PatB in solution; regardless, all buffer changes to a lower ionic strength led to the precipitation of a fraction of the protein).
PatB was further purified by anion exchange chromatography on Source Q. Samples were applied to the resin, previously equilibrated in 20 mm sodium phosphate buffer, pH 8.0, at a flow rate of 1 ml·min−1, and the enzyme was recovered by application of a linear gradient of 0–1.0 m NaCl over 50 min. PatB eluted in ∼100 mm NaCl. The purified protein was maintained at 4 °C and used immediately.
Determination of Reaction Conditions for Optimal PatB Activity
The dependence of the specific activity of PatBSUMO on pH was determined by assaying O-acetyltransferase activity (in triplicate) with p-nitrophenyl acetate (pNP-Ac) and chitotriose as substrates (19) in the following buffers: 50 mm ammonium acetate (pH 4 and 5), 50 mm sodium phosphate (pH 6–8), and 50 mm Tris·HCl (pH 8.5 and 9). Activity was calculated based on the total consumption of chitotriose as measured by graphitized carbon HPLC (19).
Q-TOF MS was used to analyze the initial transacetylation reaction products generated by PatBSUMO. Enzyme (1 μm) in 50 mm sodium phosphate buffer, pH 6.5, was incubated with 1 mm chitopentaose and 4 mm pNP-Ac for 1 min, and then 10 μl of this reaction mixture was infused directly into the mass spectrometer.
The requirement of various metals for PatBSUMO activity was assessed by performing the assay as described above with the inclusion of 5 mm CaCl2, MgCl2, FeSO4, CuCl2, ZnSO4, or MnSO4. The effect of the metal chelator EDTA was also tested at a concentration of 5 mm. Other compounds assayed for their impact on PatBSUMO activity included NaCl (100 mm) and BSA (5 mg·ml−1).
Determination of PatBSUMO Activity on Soluble Muropeptides
To assess the ability of PatB to catalyze transfer of acetyl groups to soluble PG fragments, microplate assays were performed as described above with soluble muropeptides replacing chitotriose at a final concentration of 2 mm (based on muramic acid content). Completed reactions were pooled, and muropeptides were isolated from the reaction mixtures by solid phase extraction (described below). Eluted reaction products were analyzed by either MALDI-TOF, ESI, or Q-TOF mass spectrometry.
Solid Phase Extraction
Purification of assay reaction products for analysis by MS was achieved by solid phase extraction on graphitized carbon (SPEGC) as described previously (19) with some modifications. Reaction mixtures were added to SPEGC columns that had been previously washed with one column volume of acetonitrile (ACN) and then with three column volumes of water. Flow was achieved using a vacuum manifold (Phenomenex), and all eluted fractions were collected for analysis. Elution of non-acidic sugars was achieved with 50% ACN, whereas acidic sugars were recovered with 50% ACN in 0.1% trifluoroacetic acid (TFA). PG-derived muropeptides were eluted with a 3:1 mixture of 2-propanol/ACN in 0.1% TFA, followed by 50% tetrahydrofuran to remove any remaining bound material.
Steady-state Kinetics
Routine kinetic assays of the O-acetyltransferase activity catalyzed by PatBSUMO involved incubation of the enzyme (1.0 or 3.0 μm) in 50 mm sodium phosphate buffer, pH 7, at 37 °C with 4 mm pNP-Ac (in 5% ethanol, final volume) and 1 mm chitopentaose as substrates. The progress of reactions was monitored spectrophotometrically at 420 nm for the release of p-nitrophenol (19). The rates of pNP-Ac hydrolysis (combined spontaneous and enzyme-catalyzed) were obtained from negative control reactions that lacked added chitopentaose, and they were subtracted from complete reaction rates (19). The ability of PatB to utilize other chromogenic acetyl donors was investigated by substituting pNP-Ac with 0.2–5 mm α-naphthyl acetate (αN-Ac; in 5% ethanol, final concentration) and 4-methylumbelliferyl acetate (MU-Ac; in 5% dimethyl sulfoxide, final concentration). The release of α-naphthol and 4-methylumbelliferone was monitored spectrophotometrically at 320 and 355 nm, respectively. The release of co-enzyme A (CoA) from acetyl-, propionyl-, butyryl-, isobutyryl, and hexanoyl-CoA was monitored using 5,5′-dithiobis(2-nitrobenzoic acid) at 420 nm, and the production of O-acetyl sugars was determined either by graphitized carbon HPLC or MS following reaction cleanup by SPEGC. The ability of PatB to utilize different acceptors was investigated by substituting chitopentaose with other chitooligosaccharides (DP 2–6), a hexamer of chitosan (β-(1→4)-linked glucosamine), cellooligosaccharides (β-(1→4)-linked glucose; DP 4 and 5), xylooligosaccharides (β-(1→4)-linked xylose; DP 4 and 6), and maltotriose (trimer of α-(1→4)-linked glucose).
Michaelis-Menten parameters (kcat and Km) were determined by non-linear regression analysis of plots of initial velocity as a function of substrate concentration. In all experiments, n ≥ 3.
Mass Spectrometry
All MS analyses were conducted using instruments at the Mass Spectrometry Facility of the Advanced Analysis Centre, University of Guelph. MALDI-TOF MS was performed using 2,5-dihidroxybenzoic acid as a matrix for non-amino sugars and 5-chloro-2-mercaptobenzothiazole for amino sugars or PG derivatives. Spectra were collected in positive mode on a Bruker Reflex III MALDI-TOF mass spectrometer in reflectron mode using a 337-nm nitrogen laser (set to 109–121 μJ output). Statistical analyses were carried out using the GraphPad Prism 5 software package.
ESI-MS analyses were conducted with an Amazon SL ion trap mass spectrometer (Bruker Daltonics, Billerica, MA). Sample was applied by direct infusion at a flow rate of 5 μl·min−1. The mass spectrometer electrospray capillary voltage was maintained at 4.5 kV, and the drying temperature was set at 300 °C with a flow rate of 8 liters·min−1. Nebulizer pressure was 40 p.s.i., and nitrogen was used as both nebulizing and drying gas with helium used as collision gas at 60 p.s.i. The mass/charge ratio was scanned across the m/z range 15–2,000 in enhanced resolution positive ion mode. The resulting mass spectra were analyzed using the open source mMass software package (22–24).
Q-TOF MS was performed on an Agilent UHD 6530 Q-TOF mass spectrometer. The flow rate was maintained at 0.2 ml·min−1, and the MS electrospray capillary voltage was maintained at 4.0 kV with the drying gas temperature set to 350 °C with a flow rate of 13 liters·min−1. Nebulizer pressure was at 40 p.s.i. The mass/charge ratio was scanned across the range of 300–2000 m/z in 4 GHz in extended dynamic range positive ion mode. The instrument was externally calibrated with the ESI TuneMix (Agilent). For manual MS/MS of acetylated muropeptides, precursor ions were selected with an isolation width of 1.5 m/z and a collision-induced dissociation reaction amplitude of 1 V.
Other Analytical Techniques
Protein concentrations were determined using a bicinchoninic acid assay (Pierce). SDS-PAGE on 12% acrylamide gels was conducted by the method of Laemmli (25) with Coomassie Brilliant Blue staining and Western immunoblot analysis as described previously (19).
RESULTS
Production and Purification of PatB
Previously, we reported the production of a relatively soluble, truncated form of PatB (PatBΔ36) lacking its N-terminal 36-amino acid signal sequence and membrane anchor, which we could purify in only modest yields (<1 mg·liter−1 culture) (9). This enzyme was demonstrated to function as PG O-acetyl-transferase in vivo, but efforts to use this enzyme in reproducible in vitro assays were problematic due to protein instability and precipitation despite the inclusion of surfactants and other stabilizing agents in buffers. Given this, alternative protein constructs were sought.
One of the major degradation products of the wild-type form of PatB produced from pACPM19 was a protein fragment with an apparent molecular mass of 29.4 kDa as determined by MALDI-TOF MS (data not shown). The missing segment was determined to be from the N-terminal end of the protein based on Western immunoblot analysis showing the presence of the C-terminal His6 tag (data not shown). It was hypothesized that this PatB fragment might be both active and stable because it would still possess the predicted catalytic residues or conserved motifs (9). Thus, a new vector (pACPM30) was generated using pACPM22 as a template, which provided a further truncation of the patB gene by 123 codons. The protein product, PatB lacking its N-terminal 77 amino acid residues, PatBΔ77, was purified by a combination of affinity and anion exchange chromatographies on Ni2+-NTA-agarose and Source Q, respectively, with substantial yields (∼15 mg·liter−1 culture) and relatively little apparent degradation (Fig. 1A). Preliminary activity assays performed with PatBΔ77 indicated that it possessed both esterase and transferase activity. However, concerns were raised over the impact of the 77-amino acid truncation, and indeed, further analysis indicated that its transferase activity was reduced as compared with PatBΔ26 (data not shown). Consequently, another alternative expression construct was sought.
FIGURE 1.
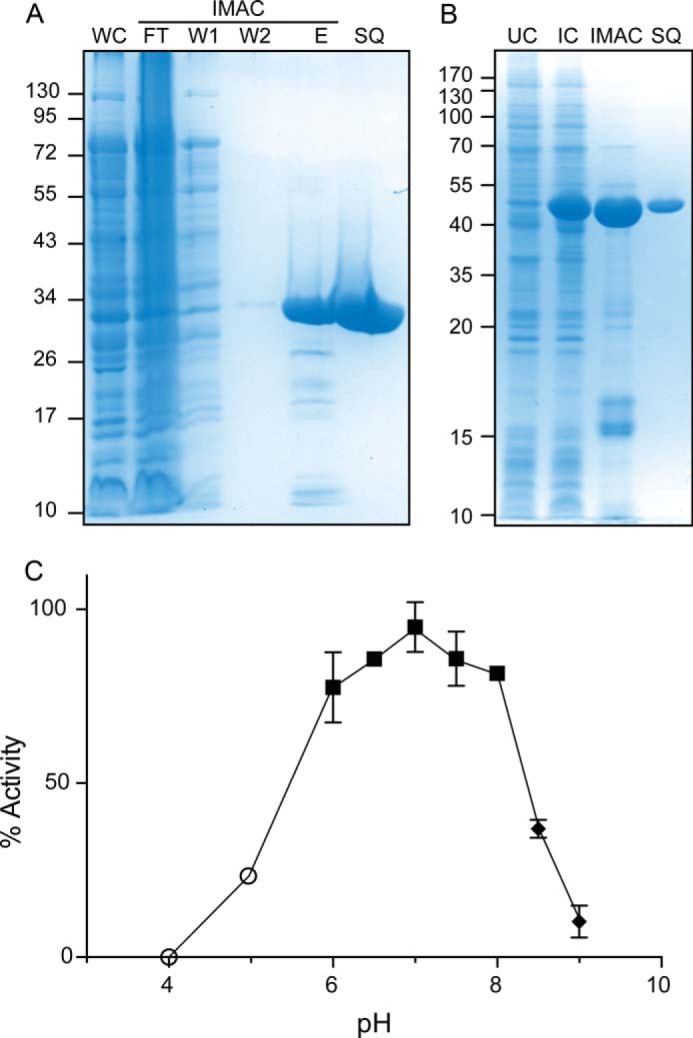
Purification and dependence of PatB activity on pH. A, Coomassie Brilliant Blue stained SDS-polyacrylamide gel of fractions taken at different points during the purification of PatBΔ77 by immobilized metal affinity chromatography (IMAC) and anion exchange chromatography on Source Q. The E and SQ fractions were purposely overloaded to demonstrate the respective presence and absence of low abundance contaminants. WC, whole cell fraction; FT, flow-through fraction; W1 and W2, wash 1 and 2, respectively; E, fraction eluted with imidazole; SQ, fraction from Source Q chromatography. B, Coomassie Brilliant Blue-stained SDS-polyacrylamide gel of fractions taken at different points during the purification of PatBSUMO. The expected molecular mass of PatBSUMO is 46,532.5 Da. UC, uninduced whole cell fraction; IC, induced whole cell fraction. The migration of molecular mass markers (in kDa) is presented on the left of each panel. C, PatBSUMO activity was assayed by graphitized carbon HPLC using acetyl-CoA and chitotriose as co-substrates in 50 mm ammonium acetate (○), 50 mm sodium phosphate (■), and 50 mm Tris-HCl (♦) at the pH values indicated. Error bars, S.D. (n ≥ 2).
Recognizing its ability to stabilize the production of recombinant proteins (27), PatB was engineered as a fusion protein with SUMO. As with the generation of pACPM30 described above, pACPM22 served as the template for this gene construction. Consequently, the PatBSUMO product lacked the 26-amino acid, periplasm-localizing signal sequence of PatB while possessing an N-terminal fusion to His6-SUMO. Again, the combination of affinity and anion exchange chromatographies was used to purify PatBSUMO to >95% homogeneity, as judged by SDS-PAGE and MS analysis (Fig. 1B). However, efforts to remove the SUMO were met with difficulty with respect to maintaining PatB stability. First, the removal of SUMO using SUMO protease resulted in the production of several PatB fragments that could not be resolved from one another. This was surprising, given the reported specificity of the SUMO protease (27), so the limited digestion observed probably occurred spontaneously following cleavage of SUMO from PatB (data not shown). Second, the removal of SUMO led to significant precipitation of protein, resulting in very low protein yields. As before, repeated efforts to stabilize PatB by inclusion of surfactants, different buffer salts, and glycerol were unsuccessful. Given these issues, SUMO had to be retained on the recombinant PatB for its biochemical and kinetic characterization, and significant efforts were still required to prevent precipitation of the protein during its purification. Even with these precautions, the half-life of the purified enzyme in 50 mm sodium phosphate buffer, pH 7, at 25 °C was ∼12 h, thus requiring that all assays be conducted immediately after its preparation. Given this, an accurate measurement of specific activity for the liberated PatB was impossible to obtain.
Determination of Assay Conditions
The dependence of PatBSUMO activity on pH was assessed by determining its specific activity as an O-acetyltransferase in different buffers ranging from pH 4 to 9 using chitotriose and pNP-Ac as co-substrates with the porous graphitized carbon HPLC-based assay. Under the conditions employed, the activity profile was relatively broad and bell-shaped with maximal activity at pH 7.0 (Fig. 1C). No activity was detected at pH 4.0. The bell shape of the plot suggests that at least two ionizable groups on the enzyme are essential for catalytic activity (both the acetyl donor and acceptor substrates used in this assay are uncharged).
The addition of the divalent metals tested (viz. Mg2+, Ca2+, Mn2+, Fe2+, Cu2+, and Zn2+) was found to be detrimental to PatBSUMO activity, with most leading to the precipitation of the protein. Although the addition of CaCl2 and MgCl2 did not appear to precipitate the protein, they were found to decrease specific activity by 43 and 80%, respectively, compared with control enzyme. The inclusion of 5 mm EDTA during purification steps had little impact on specific activity (increase by <5%), indicating that metals are unlikely to be involved in catalysis. The addition of 100 μg·ml−1 BSA reduced specific activity by 34%, whereas 50 mm NaCl resulted in a decrease of 21%. Given these data, all further routine assays for activity were conducted using a simple buffer of 50 mm sodium phosphate, pH 7.0.
Specificity of PatBSUMO for Different Acetyl Donors
Despite the proposed role of the integral membrane protein PatA as the translocator and donor of acetyl groups to PatB for the O-acetylation of PG in vivo (9, 10), PatB was recently discovered to utilize the simple activated acetyl substrates acetyl-CoA and pNP-Ac for in vitro transacetylations (19). The assay that was subsequently developed exploits the chromogenic properties of the liberated p-nitrophenylate co-product as the acetyl group is transferred from pNP-Ac to an acceptor co-substrate.
The possibility that other chromogenic substrates may serve as better co-substrates for PatB was investigated by monitoring hydrolytic reactions. Thus, as with pNP-Ac, the differences between the rates of spontaneous and PatBSUMO-catalyzed hydrolysis of αN-Ac and MU-Ac were determined with varying concentrations of the chromogens. As seen in Table 2, the enzyme was most efficient with pNP-Ac as substrate, as reflected by the determined kcat/Km values. Consequently, this chromogen was used for all further kinetic analyses.
TABLE 2.
Kinetic parameters of esterase and O-acetyltransferase activities of PatBSUMO
| Substrate | Km | kcat | kcat/Km |
|---|---|---|---|
| mm | s−1 | m−1·s−1 | |
| Hydrolysis activitya | |||
| Ac-CoA | 0.99 ± 0.08 | 0.011 ± 0.0003 | 11 ± 0.90 |
| Propionyl-CoA | 2.9 ± 0.50 | 0.0043 ± 0.00033 | 1.5 ± 0.31 |
| αN-Ac | 5.5 ± 2.7 | 0.34 ± 0.15 | 62 ± 40 |
| MU-Ac | 0.44 ± 0.04 | 0.11 ± 0.004 | 260 ± 26 |
| pNP-Acb | 0.96 ± 0.10 | 0.76 ± 0.032 | 797 ± 88 |
| Transferase activityc | |||
| GlcNAc2 | |||
| GlcNAc2-O-benzyl | |||
| GlcNAc3b | 2.63 ± 0.22 | 0.74 ± 0.02 | 280 ± 25 |
| GlcNAc4 | 1.84 ± 0.14 | 0.52 ± 0.01 | 283 ± 23 |
| GlcNAc5 | 0.95 ± 0.10 | 0.65 ± 0.02 | 681 ± 74 |
| GlcNAc6 | 0.73 ± 0.05 | 0.58 ± 0.02 | 791 ± 59 |
a Reactions conducted in 50 mm sodium phosphate buffer, pH 6.5, at 37 °C.
b Data from Ref. 19.
c Same reaction conditions with 4 mm pNP-Ac serving as acetyl donor and 0.2–8 mm chitooligosaccharides as acceptors (0.2–2.5 mm chitohexaose).
To evaluate the ability of PatBSUMO to utilize other acyl groups as substrate, the hydrolysis of propionyl-, butyryl-, isobutyryl, and hexanoyl-CoA was tested. Of these, the enzyme was only able to utilize propionyl-CoA. A kinetic analysis of this hydrolysis provided a Km value of 2.9 mm, which is only 3-fold higher than that for Ac-CoA, and a kcat value 2.6-fold lower (Table 2). Overall, however, these data indicate almost an order of magnitude difference in catalytic efficiency between the two CoA substrates. Nonetheless, PatBSUMO was able to transfer the proprionyl to an acceptor sugar, such as chitooligosaccharides (Fig. 2).
FIGURE 2.

MALDI-TOF MS analysis of PatB reaction products with propionyl-CoA as donor substrate. Enzyme (1.0 μm) in 50 mm sodium phosphate buffer (pH 6.5) was incubated with 4 mm propionyl-CoA and chitotetraose at 25 °C. After 30 min of incubation, samples were desalted using SPEGC and applied to a Bruker Reflex III mass spectrometer using 5-chloro-2- mercaptobenzothiazole as the matrix. Contamination of chitotetraose with chitooligosaccharides of other DPs (as supplied by the manufacturer) is apparent; all identified species are sodium adducts. a.i., absolute intensity.
Acceptor Substrate Specificity
Various commercially available oligosaccharides were tested under standard reaction conditions to identify the minimal acceptor substrate requirements for PatBSUMO in vitro. As expected, the enzyme was unable to transfer the acetyl to oligomers (DP = 4 and 6) of xylan, homopolymers of d-xylose, which thus lack a C-6 hydroxyl; the natural O-acetylation of the C-3 hydroxyl of xylan occurs in plants (26). However, polymers of glucose (DP = 4 and 6), which differ from xylose by the presence of a C-6 and its corresponding hydroxyl moiety, also did not serve as a substrate. Likewise, no acetylated product was observed in MALDI-TOF MS spectra of reaction mixtures when the amino sugar derivative of cellohexaose, chitosan (DP = 6), was tested as a potential acceptor substrate. On the other hand, and as previously described (19), PatBSUMO did catalyze the transfer of acetyl to the N-acetyled derivatives of chitosan oligomers, chitooligosaccharides with DP ≥ 3; no product was found for the disaccharide chitobiose as analyzed for by MS (Table 3). At extended incubation times (>10 min), reaction products were observed with multiple acetylations, although the number of modifications to an individual oligosaccharide was always less than its DP (19).
TABLE 3.
Oligosaccharide substrate specificity of PatBSUMO
Potential substrates in 50 mm sodium phosphate buffer, pH 6.5, containing 4 mm pNP-Ac were incubated with 1.0 μm PatBSUMO, and samples of reaction mixtures were subjected to MALDI-TOF MS analysis. ND, not detected.
| Potential substrate |
m/z [m + Na] of potential O-acetylated product |
Difference | |
|---|---|---|---|
| Theoretical | Detected | ||
| m/z | m/z | ||
| PG metabolites | |||
| GlcNAc | 286.09 | ND | |
| MurNAc | 316.10 | ND | |
| GlcNAc-MurNAc | 561.19 | ND | |
| GlcNAc-MurNAc-dipeptide | 759.28 | ND | |
| Chitooligosaccharides | |||
| GlcNAc2 | 489.17 | ND | |
| GlcNAc2-O-benzyl | 579.22 | 579.45 | 0.23 |
| GlcNAc3 | 692.25 | 692.45 | 0.20 |
| GlcNAc4 | 895.33 | 894.54 | −0.79 |
| GlcNAc5 | 1,098.41 | 1098.85 | 0.44 |
| GlcNAc6 | 1,301.49 | 1302.18 | 0.69 |
| Other oligosaccharides | |||
| β-1,4 Glc6 | 1,055.33 | ND | |
| β-1,4 Glc4 | 731.22 | ND | |
| α-1,4 Glc3 | 569.17 | ND | |
| Xylose6 | 875.26 | ND | |
| Xylose4 | 611.18 | ND | |
| GlcN6 | 1,049.42 | ND | |
To determine the nature of the initial reaction products of PatBSUMO acting on chitooligosaccharides, samples of reaction mixtures incubated for 1 min were infused directly into the Q-TOF mass spectrometer for analysis. As depicted in Fig. 3 for the reaction with chitopentaose, a single product peak was detected with an m/z consistent with singly acetylated chitopentaose; no products were observed at the expected m/z for multiple acetylated species. Nonetheless, separation of reaction products by adsorption chromatography on graphitized carbon indicated that PatBSUMO produced two structurally distinct initial products (Fig. 3). To probe this apparent discrepancy, fractionated products from reactions with chitotriose, chitotetraose, and chitohexaose as substrates were collected and subjected to ion trap MS/MS. These analyses indicated that indeed two unique but singly acetylated products were produced and resolved by the graphitized carbon HPLC column. With each substrate, the difference between the two reaction products was the positioning of the transferred acetyl. Thus, with chitotetraose and chitohexaose, acetylations occurred exclusively at either the third or fourth residues of the respective oligomers, whereas only the first or second residue of chitotriose served as acceptor sites.
FIGURE 3.

Identification of initial PatBSUMO reaction products. PatBSUMO (1 μm) in 50 mm sodium phosphate buffer, pH 7.0, was incubated with 4 mm Ac-CoA and 1 mm chitotetraose (A) and chitohexaose (B). Following 1 min at 25 °C, reactions were quenched by acidification, and products were subjected to adsorption HPLC on graphitized carbon. The column was equilibrated in water, and a linear gradient to 100% acetonitrile (1%/min for chitotetraose; 2%/min for chitohexaose) was applied to elute solutes. Fractions of interest were collected, dried under vacuum, and resuspended in 100 μl of 50:50 water/methanol for Q-TOF MS analysis in positive ion mode. C, Q-TOF-MS analysis of PatBSUMO O-acetyltransferase activity with 1 mm chitopentaose and 4 mm pNP-Ac at 25 °C. After 1 min of incubation, 10 μl of the reaction mixture was directly infused into the mass spectrometer. The expected positions of the (absent) doubly and triply acetylated species are also denoted. mAU, milliabsorbance units; a.i., absolute intensity.
Assay of PG O-Acetylation in Vitro
A variety of commercially available PG-related metabolites were tested as acceptors for PatBSUMO in vitro. These included the monosaccharide MurNAc, the disaccharide GlcNAc-β-1,4-MurNAc, and its dipeptide derivative GlcNAc-β-1,4-MurNAc-l-Ala-d-iso-Gln. No O-acetyl product was observed for any of these sugars, further demonstrating the enzyme's need for oligomers of carbohydrate with DP ≥ 3 (as seen with the use of chitooligosaccharides). Unfortunately, such PG-related metabolites are not commercially available.
With the lack of commercial products, we prepared a pool of soluble muropeptides (viz. GlcNAc-MurNAc peptides) by digesting insoluble PG with a mixture of mutanolysin (a muramidase) and PBPs 4 and 7 (peptidases) from E. coli. Initial O-acetyltransferase reactions with this heterogeneous mixture of solubilized muropeptides resulted in precipitation of PatBSUMO, probably due to buffer components (MgCl2, NaCl, etc.) that were required to stabilize the hydrolases used for the prior PG digestion. Consequently, we developed a novel isolation procedure to rapidly desalt and concentrate the solubilized muropeptides. Taking advantage of the high binding capacity of SPEGC, adsorption of the muropeptides to the graphitized carbon and elution in volatile solvent successfully removed buffer salts and resulted in the production of a pool of diverse and highly concentrated muropeptides as characterized by ESI-MS and quantified by high performance anion exchange chromatography (data not shown). This protocol provided concentrations of muropeptides as high as 26 mm (as determined by MurNAc content) that involved mixtures of di-, tri-, and tetrapeptide stems (GM2, GM3, and GM4, respectively, where G and M represent GlcNAc and MurNAc, respectively) but predominantly muro-tetrapeptides (GM4).
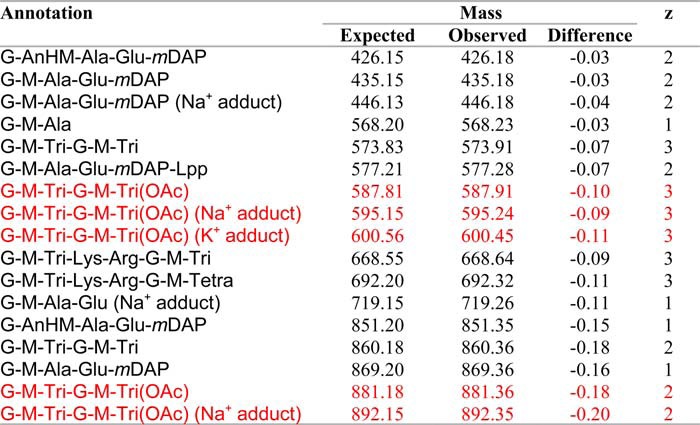
Using final concentrations of 2 mm, PatBSUMO was observed to transfer acetyl groups to these muropeptides (Fig. 4A and Table 4). Further MS analysis of these products indicated the predominance of acetylated muro-tetrapeptides, both di- and tetrasaccharides (GM4 and GM4-GM4), which was not surprising given the enrichment of the corresponding substrates in the PG digests. Importantly, similar results were obtained with PatB liberated from SUMO by prior digestion with SUMO protease, although the instability of this form of the enzyme required significantly higher concentrations to be used (viz. 5 μm PatB versus 1 μm PatBSUMO). To investigate further this apparent specificity for muro-tetrapeptides, the soluble muropeptide pool was digested with Helicobacter pylori Csd6, a carboxypeptidase with specificity for the terminal d-Ala of these metabolites (28). MS analysis of this digestion confirmed that the resulting pool of muropeptides was almost exclusively composed of muro-tripeptides (both di- and tetrasaccharides; GM3 and GM3-GM3). Unlike with GM4, PatBSUMO did not transfer the acetyl to GM3 (Fig. 4B and Table 5). However, the dimer GM3-GM3 did serve as an acceptor for the enzyme because a single O-acetylation was observed. It should be noted, however, that even in the presence of high concentrations of substrate, this activity was relatively weak.
FIGURE 4.

O-Acetyltransferase activity of PatBSUMO on solubilized PG-derived muropeptides as detected by ESI-ion trap MS. pNP-Ac (1 mm) in 50 mm sodium phosphate buffer, pH 7.0, and 2 mm PG, previously solubilized by treatment with hen egg white lysozyme and E. coli PBPs 4 and 7 in the absence (A) and presence (B) of H. pylori Csd6, were incubated in the absence (blue) and presence (red) of PatBSUMO (1 μm). After mixing for 10 min at 25 °C, samples were withdrawn, desalted by SPEGC, and subjected to ESI-ion trap MS analysis. r. int., relative intensity.
TABLE 4.
Muropeptide products of PatBSUMO activity on PG identified by ESI - ion trap MS
PG digested with lysozyme and E. coli PBPs 4 and 7 in 50 mm sodium phosphate buffer, pH 7.0, was incubated with 4 mm pNP-Ac in the absence and presence of 1 μm PatBSUMO and subjected to MS analysis. Muropeptides listed in black were present in both reaction product mixtures, whereas those in red were unique to reactions in the presence of PatBSUMO.
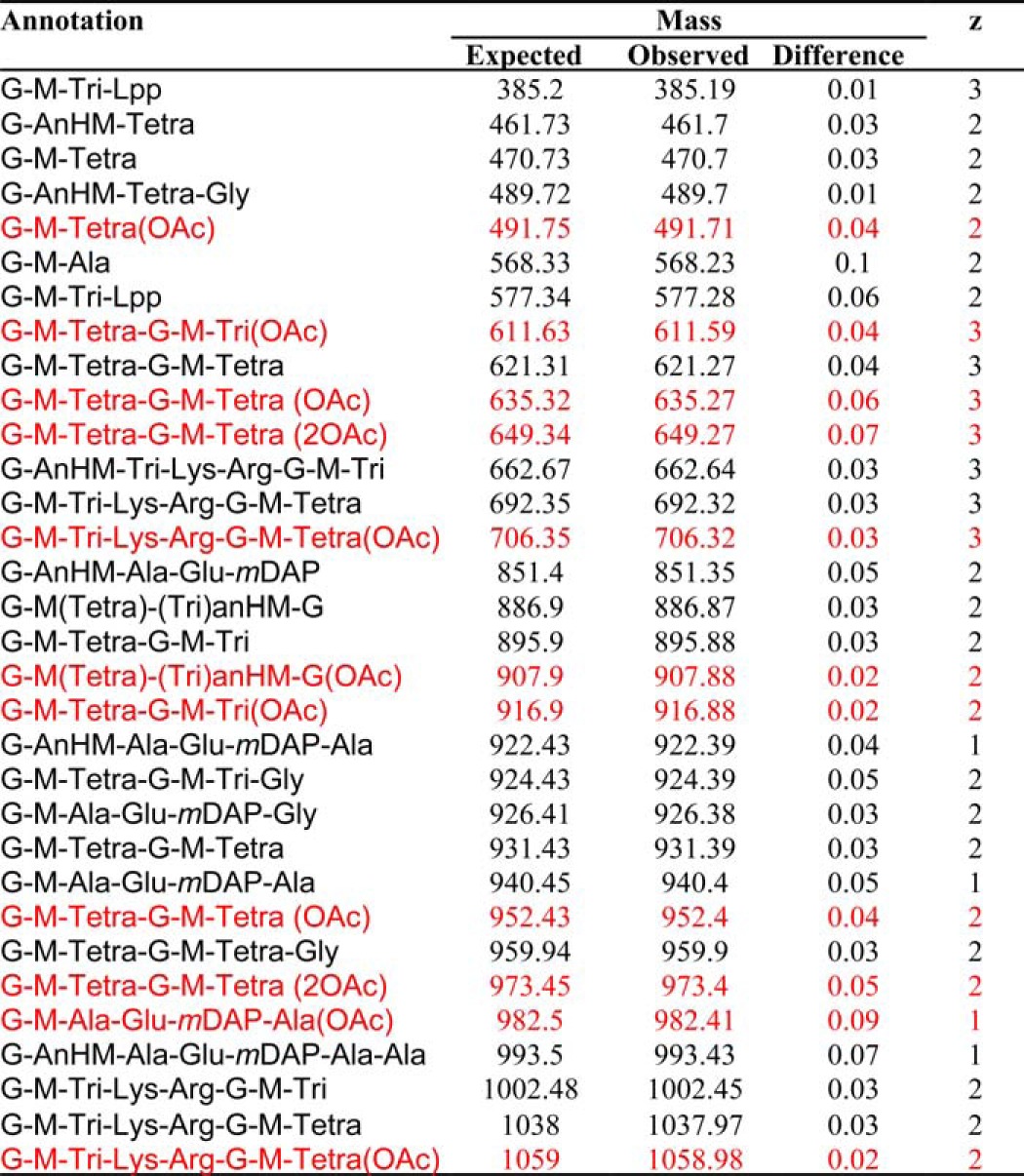
TABLE 5.
Muropeptide products of PatBSUMO activity on PG enriched with muro-tripeptides identified by ESI - ion trap MS
PG digested with lysozyme, E. coli PBPs 4 and 7, and H. pylori Csd6 in 50 mm sodium phosphate buffer, pH 7.0, was incubated with 4 mm pNP-Ac in the absence and presence of 1 μm PatBSUMO and subjected to MS analysis. Muropeptides listed in black were present in both reaction product mixtures, whereas those in red were unique to reactions in the presence of PatBSUMO.
Ion trap MS/MS was used again to confirm both the identity the reaction products of reactions with the tetrasaccharide substrates and the positioning of the transferred acetyl specifically to muramoyl residues (Fig. 5), as occurs naturally on the PG of N. gonorrhoeae. These analyses also revealed that the acetylations occurred on the internal MurNAc residues of the tetrasaccharides, thus confirming specificity for the C-6 hydroxyl group; this internal MurNAc does not have another available hydroxyl moiety for acetylation. Finally, there was no evidence of O-acetylated GlcNAc in the reaction products.
FIGURE 5.

Ion trap MS/MS analysis of O-Ac-GM4-GM4 (A) and O-Ac-GM3-GM3 (B) identified in Fig. 4. The MS/MS spectrum of m/z = 952.43 (of Fig. 4A) and m/z = 881.18 (of Fig. 4B) indicates that the PatBSUMO reaction products are O-acetylated on the C-6 hydroxyl moiety of MurNAc on the non-reducing MurNAc residue for both tripeptide- and tetrapeptide-containing species.
Steady State Kinetics of PatBSUMO
Our recently developed chromogenic assay (19) was employed to determine the Michaelis-Menten parameters for the O-acetylation of chitooligosaccharides by PatBSUMO. This assay monitors the release of p-nitrophenol from pNP-Ac as the acetyl group is transferred to either water or an acceptor chitooligosaccharide; transacetylase activity is calculated from the difference between the two rates. Representative plots of the hydrolysis of pNP-Ac in the absence and presence of acceptor ligand are presented in Fig. 6A. Initial rates of activity were determined at pH 7, holding the concentration of pNP-Ac constant at 4 mm, a concentration that is ∼4 times Km (and just below the solubility limit of the chromogen) (Fig. 6B). Values of kcat were similar for the series of chitooligosaccharides tested, whereas Km values decreased slightly from chitotriose through to chitohexaose, with the greatest difference occurring between chitotetraose and chitopentaose (Table 2). This difference is more apparent in the calculated efficiency constants kcat/Km.
FIGURE 6.

Example assays of O-acetyltransferase activity of PatB. A, PatBSUMO (1 μm) in 50 mm sodium phosphate buffer, pH 7.0, was incubated at 25 °C with 3 mm pNP-Ac hydrolysis in the presence (●) and absence (○) of 2 mm chitopentaose. The rates of p-NP production were monitored spectrophotometrically at 410 nm, and O-acetyltransferase activity was determined by the difference between the two. B, initial rates of reaction of PatBSUMO incubated with 4 mm pNP-Ac and varying concentrations of chitopentaose. Error bars, S.D. (n = 3).
DISCUSSION
We report herein the first kinetic analysis of PatB, a major virulence determinant found in numerous bacterial pathogens, both Gram-positive and Gram-negative (5, 17, 29, 30), and a potential new target for antibacterial therapy. Our elucidation of the substrate profile for PatBSUMO in vitro provides valuable insight for the development of an assay amenable to the high throughput screening for inhibitors that may serve as new leads for antibiotic development.
Despite being identified almost 10 years ago (10, 11, 13), a kinetic analysis of the enzymes involved in PG O-acetylation had not been possible due to the significant technical limitations associated with the complexity of attempting to analyze a peripheral membrane protein that receives its acetyl co-substrate from a transmembrane protein, which it then donates to a completely insoluble heteropolymer. As presented herein, we were able to overcome these limitations by using a chemical biological approach. In an earlier study, we demonstrated that PatB is capable of catalyzing transfer of acetyl from pNP-Ac (19), thus dispensing with the need for the natural partner of PatB, the integral membrane protein and putative translocator of acetyl, PatA (13, 19). In the current study, we were able to expand upon this and kinetically characterize the family of acetyl donors and acceptors that PatB is able to utilize. The pool of donors includes the non-O-linked acetyl-CoA in addition to two other O-linked donors (MU-Ac and αN-AC), suggesting significant active site plasticity. MU-Ac is a fluorogenic substrate/acetyl donor, and as such, it will serve as an exceptional tool for high throughput screening of this family of enzymes, given that many inhibitory compounds are often not amenable to colorimetric assays.
The finding that PatB is able to catalyze transfer from thioesters was surprising, given the absence of acetyl-CoA in the periplasm/extracellular space of bacteria. This apparent promiscuity of acetyl-donor specificity is consistent with our earlier observation that PatB was able to receive acetyl groups from WecH (9), an integral membrane protein involved in the O-acetylation of enterobacterial common antigen in E. coli. Such flexibility for the relatively small acetyl donors may not be too surprising, however, when considering that the proposed natural source of acetyl for PatB is acetyl-bound PatA. If indeed this postulate is correct, and that there is no intermediary carrier molecule involved in the transfer process between the two proteins, PatB must complex with PatA and thus have a binding site/cleft to accommodate both the binding elements of PatA and its linked acetyl. Regarding a PatA-PatB complex, its formation, involving the reaction centers of both proteins, would probably preclude free water from accessing the active site and thereby minimize, if not prevent, the hydrolysis of the translocated acetyl group that we observe in vitro (and exploited herein). This exclusion of water would thus ensure the efficiency of the O-acetylation process that occurs outside of the cytoplasm and prevent the wasteful loss of acetate.
That PatB may have a relatively flexible active site was further exemplified by the finding that it could transfer propionyl groups to acceptors in addition to acetyls, albeit at a much reduced rate. Recently, the modification of muropeptides for use as adjuvants has been explored (reviewed in Ref. 31). One of the limiting factors in this research is the relatively low lipophilicity of these molecules. The capacity of PatB to transfer propionyl groups to oligosaccharides could be further exploited through reaction optimization and perhaps protein engineering to make possible the transfer of longer-chain acyl groups.
Initial studies with PG digestion products as acceptor substrate led to our collection of inconsistent data, probably due to the heterogeneous nature of the polymer. Indeed, muramidase digests of insoluble PG from a single bacterial source lead to the production of over 70 distinct muropeptides, as first demonstrated by Glauner (32). In our search for a more consistent and soluble co-substrate for in vitro studies, we found that the minimum requirements were for oligomers of GlcNAc with a DP ≥ 3. Chitooligosaccharides are simple structural mimics of PG oligomers, and that they serve as pseudosubstrates for PatB is analogous to the ability of lysozyme to also utilize them as substrate, albeit weakly (33, 34). The observation that the DP of the chitooligosaccharides had to be ≥3 to serve as acceptor for PatB is consistent with early reports concerning the O-acetylation of PG in N. gonorrhoeae, Proteus mirabilis, and S. aureus. These earlier biochemical studies demonstrated that O-acetylation occurs shortly after transglycosylation and transpeptidation of Lipid II (undecaprenyl-bound GlcNAc-MurNAc-pentapeptide) (29, 35–39), thus involving PG chains and not the precursor disaccharide. The modest improvement in the Km and specificity constant for the chitooligosaccharides with increasing DP to 5/6 suggests that PatB, like many other carbohydrate-active enzymes, including lysozyme (40), probably has several sugar binding subsites in its active site. The multiacetylations observed with the chitooligosaccharides would reflect the relative structural simplicity of the substrate because presumably it is able to bind at these subsites in different orientations.
Despite the complexities of working with PG-derived ligands, a clear specificity of PatB was observed in vitro with these more complex acceptor substrates, where a preference was found for any muropeptides possessing tetrapeptides or longer chain muropeptides (DP ≥ 2) with tripeptide stems. This again is consistent with the earlier in vivo studies indicating that O-acetylation follows transpeptidation of growing PG chains (29, 35–39); the formation of cross-links in PG by transpeptidases results in the release of the terminal d-Ala from stem pentapeptides, leaving tetrapeptides on muramyl residues. Whereas high resolution data are not available for the structure of all muropeptides generated by N. gonorrhoeae, the apparent specificity of PatB for muro-tetrapeptides is also consistent with the known PG composition of closely related N. meningitidis, which encodes homologs of PatA and PatB, both with 96% identity to the N. gonorrhoeae proteins and with complete conservation of the consensus motifs (19). Analysis of N. meningitidis PG has indicated that O-acetylation is found predominantly on muro-tetrapeptides (41, 42), and a phenotypic study involving mutants deficient in O-acetylation suggested that the modification occurred mostly on muropeptides containing tetra- or pentapeptide stems (30). Given the relatedness of N. gonorrhoeae, it is likely that its PG possesses a similar composition, but this, together with a more detailed analysis of PatB substrate specificity in vivo, will form the basis of a future study.
A model for the role of O-acetylation in regulation of PG chain length in N. meningitidis has recently been proposed. According to Veyrier et al. (30), the O-acetyl-PG esterase Ape1 only deacetylates MurNAc residues containing tripeptide stems in vivo. They propose that newly deacetylated muro-tripeptides serve as substrate for a lytic transglycosylase, thus limiting PG chain length. Consistent with this model, our data show that PatB has specificity for muro-tetrapeptides in vitro, thereby preserving these regions of PG from lytic activity. If the model of Veyrier et al. (30) is correct, it would be expected that a carboxypeptidase (PBP?) with specificity for O-acetyl-MurNAc-tetrapeptide would cleave the tetrapeptide as required, thereby generating the tripeptide substrate for Ape1, which in turn releases the O-acetyl group to provide an accessible lytic transglycosylase substrate (Fig. 7). A zymographic method for determining substrate specificity of lytic enzymes has been developed (43), but unfortunately it has not yet been applied for the detection of O-acetyl-PG-specific enzymes from N. gonorrhoeae.
FIGURE 7.

Proposed pathway for the metabolism of PG in the periplasm. Acetyl (▴) is translocated from the cytoplasm to the periplasm by PatA for PatB-catalyzed transfer to MurNAc residues in PG possessing tetrapeptide stems (previously generated by PBP-catalyzed transpeptidation) (step 1). The carboxypeptidase (CP) activity of a PBP trims the tetrapeptide stem of O-acetyl-MurNAc residues to a tripeptide (step 2), providing the appropriate substrate for O-acetyl-PG esterase, Ape1. Ape1 catalyzes the de-O-acetylation of PG (step 3), thereby exposing MurNAc residues to cleavage by lytic transglycosylases, which generate/release 1,6-anhydro-MurNAc products (step 4).
In this study, we have demonstrated that, in vitro, PatB is preferentially active on polymerized glycans containing N-acetyl moieties or muroglycans containing tetrapeptide stems. This information will be valuable for the identification and development of PG O-acetyltransferase inhibitors that could represent potential leads to novel classes of antibiotics that are desperately needed by clinicians combating bacterial infections and diseases. Indeed, in its 2013 Threat Report, the Centers for Disease Control and Prevention listed drug-resistant N. gonorrhoeae, together with carbapenem-resistant Enterobacteriaceae and Clostridium difficile, as its three urgent (highest level) threats in the United States (44). Finally, this study will make possible, for the first time, in vitro characterization of other related acetyltransferases, yielding important insights into the role of glycan modification in extracellular polysaccharides.
Acknowledgments
We thank K. Young (University of Arkansas School of Medicine) and N. Salama (University of Washington) for generous gifts of E. coli PBPs 5 and 7 and H. pylori Csd6, respectively. We also thank both Dyanne Brewer and Armen Charchoglyan of the Mass Spectrometry Facility (Advanced Analysis Centre, University of Guelph) for expert technical assistance and advice and David Sychantha for helpful discussions.
This work was supported by Canadian Institutes of Health Research Team Grant TGC114045 (to A. J. C.) and an Ontario Graduate Scholarship from the Province of Ontario (to P. J. M.).
- PG
- peptidoglycan
- MurNAc
- N-acetylmuramic acid
- GlcNAc
- N-acetylglucosamine
- Ni2+-NTA
- Ni2+-nitrilotriacetic acid
- SPEGC
- solid phase extraction on graphitized carbon
- ACN
- acetonitrile
- pNP-Ac
- p-nitrophenol acetate
- MU-Ac
- 4-methylumbelliferyl acetate
- αN-Ac
- α-naphthyl acetate
- DP
- degree of polymerization
- GM2
- GlcNAc-MurNAc-dipeptide
- GM3
- GlcNAc-MurNAc-tripeptide
- GM4
- GlcNAc-MurNAc-tetrapeptide
- SUMO
- small ubiquitin-like modifier
- ESI
- electrospray ionization
- PBP
- penicillin-binding protein.
REFERENCES
- 1. Moynihan P. J., Clarke A. J. (2011) O-Acetylated peptidoglycan: controlling the activity of bacterial autolysins and lytic enzymes of innate immune systems. Int. J. Biochem. Cell Biol. 43, 1655–1659 [DOI] [PubMed] [Google Scholar]
- 2. Scheurwater E., Reid C. W., Clarke A. J. (2008) Lytic transglycosylases: bacterial space-making autolysins. Int. J. Biochem. Cell Biol. 40, 586–591 [DOI] [PubMed] [Google Scholar]
- 3. Höltje J.-V., Mirelman D., Sharon N., Schwarz U. (1975) Novel type of murein transglycosylase in Escherichia coli. J. Bacteriol. 124, 1067–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bernard E., Rolain T., Courtin P., Guillot A., Langella P., Hols P., Chapot-Chartier M.-P. (2011) Characterization of O-acetylation of N-acetylglucosamine: a novel structural variation of bacterial peptidoglycan. J. Biol. Chem. 286, 23950–23958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Laaberki M.-H., Pfeffer J., Clarke A. J., Dworkin J. (2011) O-Acetylation of peptidoglycan is required for proper cell separation and S-layer anchoring in Bacillus anthracis. J. Biol. Chem. 286, 5278–5288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Abrams A. (1958) O-Acetyl groups in the cell wall of Streptococcus faecalis. J. Biol. Chem. 230, 949–959 [PubMed] [Google Scholar]
- 7. Brumfitt W., Wardlaw A. C., Park J. T. (1958) Development of lysozyme-resistance in Micrococcus lysodiekticus and its association with an increased O-acetyl content of the cell wall. Nature 181, 1783–1784 [DOI] [PubMed] [Google Scholar]
- 8. Clarke A. J., Strating H., Blackburn N. T. (2000) in Glycomicrobiology (Doyle R. J., ed) pp. 187–223, Plenum Publishing Co. Ltd., New York [Google Scholar]
- 9. Moynihan P. J., Clarke A. J. (2010) O-Acetylation of peptidoglycan in Gram-negative bacteria: identification and characterization of peptidoglycan O-acetyltransferase in Neisseria gonorrhoeae. J. Biol. Chem. 285, 13264–13273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dillard J. P., Hackett K. T. (2005) Mutations affecting peptidoglycan acetylation in Neisseria gonorrhoeae and Neisseria meningitidis. Infect. Immun. 73, 5697–5705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bera A., Herbert S., Jakob A., Vollmer W., Götz F. (2005) Why are pathogenic staphylococci so lysozyme resistant? The peptidoglycan O-acetyltransferase OatA is the major determinant for lysozyme resistance of Staphylococcus aureus. Mol. Microbiol. 55, 778–787 [DOI] [PubMed] [Google Scholar]
- 12. Crisóstomo M. I., Vollmer W., Kharat A. S., Inhülsen S., Gehre F., Buckenmaier S., Tomasz A. (2006) Attenuation of penicillin resistance in a peptidoglycan O-acetyl transferase mutant of Streptococcus pneumoniae. Mol. Microbiol. 61, 1497–1509 [DOI] [PubMed] [Google Scholar]
- 13. Weadge J. T., Pfeffer J. M., Clarke A. J. (2005) Identification of a new family of enzymes with potential O-acetylpeptidoglycan esterase activity in both Gram-positive and Gram-negative bacteria. BMC Microbiol. 5, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Weadge J. T., Clarke A. J. (2006) Identification and characterization of O-acetylpeptidoglycan esterase: a novel enzyme discovered in Neisseria gonorrhoeae. Biochemistry 45, 839–851 [DOI] [PubMed] [Google Scholar]
- 15. Weadge J. T., Clarke A. J. (2007) Neisseria gonorrheae O-acetylpeptidoglycan esterase, a serine esterase with a Ser-His-Asp catalytic triad. Biochemistry 46, 4932–4941 [DOI] [PubMed] [Google Scholar]
- 16. Pfeffer J. M., Clarke A. J. (2012) Identification of the first known inhibitors of O-acetylpeptidoglycan esterase: a potential new antibacterial target. Chembiochem 13, 722–731 [DOI] [PubMed] [Google Scholar]
- 17. Bera A., Biswas R., Herbert S., Götz F. (2006) The presence of peptidoglycan O-acetyltransferase in various staphylococcal species correlates with lysozyme resistance and pathogenicity. Infect. Immun. 74, 4598–4604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aubry C., Goulard C., Nahori M.-A., Cayet N., Decalf J., Sachse M., Boneca I. G., Cossart P., Dussurget O. (2011) OatA, a peptidoglycan O-acetyltransferase involved in Listeria monocytogenes immune escape, is critical for virulence. J. Infect. Dis. 204, 731–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moynihan P. J., Clarke A. J. (2013) Assay for peptidoglycan O-acetyltransferase: a potential new antibacterial target. Anal. Biochem. 439, 73–79 [DOI] [PubMed] [Google Scholar]
- 20. Kellogg D. S., Jr., Peacock W. L., Jr., Deacon W. E., Brown L., Pirkle C. I. (1963) Neisseria gonorrhoeae I. Virulence genetically linked to clonal variation. J. Bacteriol. 85, 1274–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pagotto F. J., Salimnia H., Totten P. A., Dillon J. R. (2000) Stable shuttle vectors for Neisseria gonorrhoeae, Haemophilus spp., and other bacteria based on a single origin of replication. Gene 244, 13–19 [DOI] [PubMed] [Google Scholar]
- 22. Strohalm M., Hassman M., Kosata B., Kodícek M. (2008) mMass data miner: an open source alternative for mass spectrometric data analysis. Rapid Commun. Mass Spectrom. 22, 905–908 [DOI] [PubMed] [Google Scholar]
- 23. Strohalm M., Kavan D., Novák P., Volný M., Havlícek V. (2010) mMass 3: a cross-platform software environment for precise analysis of mass spectrometric data. Anal. Chem. 82, 4648–4651 [DOI] [PubMed] [Google Scholar]
- 24. Niedermeyer T. H., Strohalm M. (2012) mMass as a software tool for the annotation of cyclic peptide tandem mass spectra. PLoS One 7, e44913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 26. Kiefer L. L., York W. S., Darvill A. G., Albersheim P. (1989) Xyloglucan isolated from suspension-cultured sycamore cell walls is O-acetylated. Phytochemistry 28, 2105–2107 [Google Scholar]
- 27. Peroutka R. J., 3rd, Orcutt S. J., Strickler J. E., Butt T. R. (2011) SUMO fusion technology for enhanced protein expression and purification in prokaryotes and eukaryotes. Methods Mol. Biol. 705, 15–30 [DOI] [PubMed] [Google Scholar]
- 28. Sycuro L. K., Rule C. S., Petersen T. W., Wyckoff T. J., Sessler T., Nagarkar D. B., Khalid F., Pincus Z., Biboy J., Vollmer W., Salama N. R. (2013) Flow cytometry-based enrichment for cell shape mutants identifies multiple genes that influence Helicobacter pylori morphology. Mol. Microbiol. 90, 869–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lear A. L., Perkins H. R. (1986) O-Acetylation of peptidoglycan in Neisseria gonorrhoeae: investigation of lipid-linked intermediates and glycan chains newly incorporated into the cell wall. J. Gen. Microbiol. 132, 2413–2420 [DOI] [PubMed] [Google Scholar]
- 30. Veyrier F. J., Williams A. H., Mesnage S., Schmitt C., Taha M.-K., Boneca I. G. (2013) De-O-acetylation of peptidoglycan regulates glycan chain extension and affects in vivo survival of Neisseria meningitidis. Mol. Microbiol. 87, 1100–1112 [DOI] [PubMed] [Google Scholar]
- 31. Fujimoto Y., Pradipta A. R., Inohara N., Fukase K. (2012) Peptidoglycan as Nod1 ligand: fragment structures in the environment, chemical synthesis, and their innate immunostimulation. Nat. Prod. Rep. 29, 568–579 [DOI] [PubMed] [Google Scholar]
- 32. Glauner B. (1988) Separation and quantification of muropeptides with high performance liquid chromatography. Anal. Biochem. 172, 451–464 [DOI] [PubMed] [Google Scholar]
- 33. Berger L. R., Weiser R. S. (1957) The β-glucosaminidase activity of egg-white lysozyme. Biochim. Biophys. Acta 26, 517–521 [DOI] [PubMed] [Google Scholar]
- 34. Banerjee S. K., Holler E., Hess G. P., Rupley J. A. (1975) Reaction of N-acetylglucosamine oligosaccharides with lysozyme: temperature, pH, and solvent deuterium isotope effects: equilbrium, steady state, and pre-steady state measurements. J. Biol. Chem. 250, 4355–4367 [PubMed] [Google Scholar]
- 35. Gmeiner J., Kroll H. P. (1981) Murein biosynthesis and O-acetylation of N-acetylmuramic acid during the cell-division cycle of Proteus mirabilis. Eur. J. Biochem. 117, 171–177 [DOI] [PubMed] [Google Scholar]
- 36. Gmeiner J., Sarnow E. (1987) Murein biosynthesis in synchronized cells of Proteus mirabilis: quantitative analysis of O-acetylated murein subunits and of chain terminators incorporated into the sacculus during the cell cycle. Eur. J. Biochem. 163, 389–395 [DOI] [PubMed] [Google Scholar]
- 37. Lear A. L., Perkins H. R. (1983) Degrees of O-acetylation and cross-linking of the peptidoglycan of Neisseria gonorrhoeae during growth. J. Gen. Microbiol. 129, 885–888 [DOI] [PubMed] [Google Scholar]
- 38. Lear A. L., Perkins H. R. (1987) Progress of O-acetylation and cross-linking of peptidoglycan in Neisseria gonorrhoeae grown in the presence of penicillin. J. Gen. Microbiol. 133, 1743–1750 [DOI] [PubMed] [Google Scholar]
- 39. Snowden M. A., Perkins H. R. (1991) Cross-linking and O-acetylation of peptidoglycan in Staphylococcus aureus (strains H and MR-1) grown in the presence of sub-growth-inhibitory concentrations of β-lactam antibiotic. J. Gen. Microbiol. 137, 1661–1666 [DOI] [PubMed] [Google Scholar]
- 40. Blake C. C., Koenig D. F., Mair G. A., North A. C., Phillips D. C., Sarma V. R. (1965) Structure of hen egg-white lysozyme: a three-dimensional Fourier synthesis at 2 Å resolution. Nature 206, 757–761 [DOI] [PubMed] [Google Scholar]
- 41. Antignac A., Rousselle J.-C., Namane A., Labigne A., Taha M.-K., Boneca I. G. (2003) Detailed structural analysis of the peptidoglycan of the human pathogen Neisseria meningitidis. J. Biol. Chem. 278, 31521–31528 [DOI] [PubMed] [Google Scholar]
- 42. Zarantonelli M. L., Skoczynska A., Antignac A., El Ghachi M., Deghmane A.-E., Szatanik M., Mulet C., Werts C., Peduto L., d'Andon M. F., Thouron F., Nato F., Lebourhis L., Philpott D. J., Girardin S. E., Vives F. L., Sansonetti P., Eberl G., Pedron T., Taha M. K., Boneca I. G. (2013) Penicillin resistance compromises Nod1-dependent proinflammatory activity and virulence fitness of Neisseria meningitidis. Cell Host. Microbe. 13, 735–745 [DOI] [PubMed] [Google Scholar]
- 43. Strating H., Clarke A. J. (2001) Differentiation of bacterial autolysins by zymogram analysis. Anal. Biochem. 291, 149–154 [DOI] [PubMed] [Google Scholar]
- 44. Centers for Disease Control and Prevention (2013) Antibiotic Resistance Threats in the United States, 2013, United States Department of Health and Human Services, Atlanta, GA [Google Scholar]