

Background: Sphingolipid metabolism is functionally linked to the proteolytic processing of APP.
Results: Inhibition of S1P-lyase decreases APP degradation in lysosomes, and mobilization of Ca2+ can partially rescue the accumulation of APP.
Conclusion: S1P-lyase is critically involved in the regulation of lysosomal activity and degradation of APP.
Significance: Alterations in S1P metabolism could play important roles in the pathogenesis of Alzheimer disease.
Keywords: Alzheimer Disease, Amyloid, Amyloid Precursor Protein (APP), Lysosome, Sphingolipid, Sphingosine-1-phosphate (S1P)
Abstract
Progressive accumulation of the amyloid β protein in extracellular plaques is a neuropathological hallmark of Alzheimer disease. Amyloid β is generated during sequential cleavage of the amyloid precursor protein (APP) by β- and γ-secretases. In addition to the proteolytic processing by secretases, APP is also metabolized by lysosomal proteases. Here, we show that accumulation of intracellular sphingosine-1-phosphate (S1P) impairs the metabolism of APP. Cells lacking functional S1P-lyase, which degrades intracellular S1P, strongly accumulate full-length APP and its potentially amyloidogenic C-terminal fragments (CTFs) as compared with cells expressing the functional enzyme. By cell biological and biochemical methods, we demonstrate that intracellular inhibition of S1P-lyase impairs the degradation of APP and CTFs in lysosomal compartments and also decreases the activity of γ-secretase. Interestingly, the strong accumulation of APP and CTFs in S1P-lyase-deficient cells was reversed by selective mobilization of Ca2+ from the endoplasmic reticulum or lysosomes. Intracellular accumulation of S1P also impairs maturation of cathepsin D and degradation of Lamp-2, indicating a general impairment of lysosomal activity. Together, these data demonstrate that S1P-lyase plays a critical role in the regulation of lysosomal activity and the metabolism of APP.
Introduction
Alzheimer disease is the most common form of dementia and is characterized by the progressive accumulation of extracellular plaques containing the amyloid-β peptide (Aβ)3 (1). Aβ derives from proteolytic processing of the amyloid precursor protein (APP) by β- and γ-secretases (2). Alternatively, APP can also be cleaved by α-secretase within the Aβ domain preventing the generation of Aβ generation (2). APP and the secretases are integral membrane proteins, and the processing occurs throughout secretory or endocytic vesicular transport routes (1, 2). APP is transported from the endoplasmic reticulum (ER) to the plasma membrane, where it is predominantly cleaved by α-secretase resulting in the generation of soluble APPα and corresponding C-terminal fragments (CTF-α) (2, 3). APP not cleaved by α-secretase can be internalized from the cell surface into endosomes where β-secretory cleavage can occur (2, 4). β-Secretase has been identified as the membrane-bound aspartic protease BACE1 (β-site APP cleaving enzyme-1) (5–8). The cleavage of APP by BACE1 generates soluble APPβ and APP-βCTF that could then be processed by γ-secretase, resulting in the release of Aβ peptides (2, 3). Importantly, significant fractions of APP and its CTFs are also targeted to lysosomes where they are degraded by lysosomal hydrolases (9–11).
Recently, sphingosine-1-phosphate (S1P) has been shown to increase the generation of Aβ by directly activating BACE1 (12). S1P is a bioactive signaling molecule regulating cell proliferation and survival as well as differentiation and motility (13, 14). S1P derives during degradation of sphingolipids from the cleavage of ceramide into fatty acid and sphingosine (15). Sphingosine is then phosphorylated by sphingosine kinases 1 and 2 (Sphk1 and Sphk2) to produce S1P (16). S1P can then be irreversibly cleaved by the S1P-lyase to phosphoethanolamine and hexadecenal (17). Alternatively, S1P can be dephosphorylated by sphingosine phosphatases back to sphingosine and thereby recycled for ceramide formation (16). S1P-lyase, sphingosine phosphatase, and Sphk2 are localized at the ER and regulate intracellular levels of S1P (16, 18, 19). Because S1P and its metabolic precursor sphingosine exert opposing functions in the regulation of cell survival (20), the activities of these enzymes are critical for determination of the cellular fate (21).
The importance of S1P-lyase is demonstrated by the severe phenotype of S1P-lyase knock-out mice that die within the first weeks after birth, hardly surviving the weaning period (19, 22). Also, cerebellar neurons derived from S1PL-KO mice, which have 20-fold increased concentrations of intracellular S1P, are more susceptible to certain stressors and undergo early cell death (23–25). Deficiency of S1P-lyase is also associated with elevated levels of basal intracellular Ca2+ concentrations and increased stimulated Ca2+ release from intracellular stores (25, 26).
In this work, we show that the genetic and pharmacological inhibition of the S1P-lyase strongly impairs the metabolism of APP by affecting the activities of β- and γ-secretase, as well as the degradation in lysosomes. Importantly, the accumulation of APP and CTFs in S1P-lyase-deficient cells could be partially reversed by starvation or the stimulated release of Ca2+ from ER or lysosomal stores. Together, these data demonstrate the importance of S1P-lyase in the cellular metabolism of APP. Thus, this enzyme represents a potential target to modulate the generation of Aβ.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
4-[4-(4-chloro-phenyl)-thiazol-2-ylamino]-phenol (SKI II) was purchased from Sigma-Aldrich. S1P was from Biomol (Hamburg, Germany). Thapsigargin was obtained from Invitrogen. Glycylphenylalanine 2-naphthylamide (GPN) was purchased from Santa Cruz Biotechnology (Dallas, TX).
The following antibodies were used: anti-APP c-terminal C1/6.1 (Covance, Princeton, NH), mouse specific anti-Aβ m3.2 (Covance), human specific anti-Aβ 6E10 (Covance), anti-Actin AC-15 (Sigma-Aldrich), anti-Lamp2 Abl-93 (DSHB, University of Iowa), anti-Calnexin H-70 (Santa Cruz Biotechnology), anti-LC3 (MBL, Woburn), anti-GFP (Roche), anti-Cathepsin D (kind gift of Prof. Dr. Stefan Höning, Cologne), anti-rat Alexa488 and anti-mouse Alexa546 (Life Technologies), secondary Abs coupled to HRP anti-mouse-HRP, anti-rabbit-HRP (Sigma-Aldrich), and anti-rat-HRP (Rockland, Gilbertsville, PA).
Cell Culture
Embryonic fibroblasts from WT and S1P-lyase-KO (S1PL-KO) mice were previously described by Ihlefeld et al. (27). HEK293 cells stably overexpressing human APP695 were described previously (28). The cells were cultured in DMEM supplemented with 10% (MEF and HEK293) or in RPMI supplemented with 15% (SH-SY5Y) fetal calf serum (PAN Biotech) and 1% penicillin/streptomycin (Invitrogen). Stably transfected HEK-APP695 cells were selected with 200 μg/ml G418. Cells were grown until 70% confluence prior to treatment. Starvation was induced by culturing cells in Earle's balanced salt solution (Invitrogen).
Cell Viability Tests
Cells were seeded into 96-well plates 1 day prior to the treatment and grown until 70% confluency (as described above). After 24 h cells were treated with compounds and reagents for respective times in 100 μl of culturing medium. Later the cells were first incubated with 550 ng/μl MTT for 4 h in the conditioned medium and subsequently solubilized overnight by adding 100 μl of 10% SDS in 0.001 m HCl to the medium. The metabolization of MTT was then measured at 570 nm and statistically analyzed.
Viral Transduction of Cells
Human APP695 cDNA with the Swedish mutation (APPswe) was cloned into a lentiviral rrl-CMV-vector. The construct also drives the separate expression of GFP by an internal ribosomal entry site. Cells were seeded in 6-well plates 1 day before the transduction to a 70% confluence in DMEM medium supplemented with 10% FCS, 1% penicillin/streptomycin. Next day, the cells were transduced with lentiviral particles at 1 × 106 IP/100,000 cells for 15 h. Later cells were washed four times with DMEM and cultured for an additional 48 h.
Reverse siRNA Transfection
25 μl of Sgpl1 targeting or control siRNA (10 μm) was pipetted into a individual wells of a 24-well plate, followed by addition of 100 μl of diluted HiPerfect transfection reagent (95:5% H2O:HiPerfect), and incubated for 15 min. Then murine N9 cells (150,000 cells/well) were seeded into the wells. After 6 h of transfection, medium was replaced by fresh DMEM. Cells were lysed after 30 h, and proteins were detected by Western immunoblotting.
Protein Extraction and Western Immunoblotting
For extraction of proteins, cell were washed three times in PBS and lysed in STEN lysis buffer (50 mm Tris-HCl, pH 7.6, 250 mm NaCl, 20 mm EDTA, 1.2% Nonidet P-40, and 1% Triton X-100) containing Complete® protease inhibitor (Hoffmann-La Roche, Basel, Switzerland). For isolation of cellular membranes, the cells were briefly washed with PBS and collected by centrifugation. The cells were then incubated for 10 min in hypotonic buffer (10 mm Tris, 1 mm EDTA, 1 mm EGTA). After repeated resuspension through a 0.6-mm cannula, the mixture was centrifuged at 1300 rcf for 5 min to remove cellular debris and nuclei. The remaining supernatant was centrifuged for 60 min at 16,100 rcf, and the resulting membrane pellet was solved in STEN lysis buffer containing Complete® protease inhibitor. Proteins were separated by SDS-PAGE and detected by Western immunoblotting using ECL imaging (Bio-Rad).
Subcellular Fractionation
Isolated membranes were resuspended in hypotonic buffer containing protease inhibitor mixture and incubated overnight at 4 °C with constant stirring. Vesicles were separated on a stepwise iodixanol (OptiPrep, Sigma) gradient (50–2,5%), diluted with a sucrose buffer (0.25 M sucrose, 6 mm EDTA, 60 mm HEPES-NaOH, pH 7.4).
Measurement of Aβ Variants
Cells were grown on 24-well culture plates until 70% confluency in DMEM as described above. For collection of Aβ, 500 μl of fresh medium was added overnight. Conditioned media were cleared by centrifugation and then analyzed by electrochemiluminescence technology (MesoScale Discovery) for Aβ40 and Aβ42 according top the manufacturer's protocol.
Measurement of Secretase Activity
Detection of secretase activities in living cells was performed as described previously with slight modifications (29, 30). Shortly, after incubation, cells were washed two times with prewarmed life cell imaging solution (HEPES buffer, pH 7,4). Buffer was removed, and 50 μl of life cell imaging solution containing 30 μm β-and 12 μm γ-secretase fluorogenic substrate (Calbiochem, Darmstadt, Germany) was added. Fluorescence was measured continuously at an excitation wavelength of 355 ± 10 nm and an emission wavelength of 440 ± 10 nm for γ-secretase or 345 ± 5 nm/500 ± 2.5 nm for β-secretase at 37 °C under light exclusion using a Safire Infinity Fluorometer (Tecan, Crailsheim, Germany).
In Vitro γ-Secretase Assay
Assay was performed similar to published protocol (31). Purified cellular membranes were reconstituted in citrate buffer (150 mm sodium citrate in H2O, pH 6.4) and incubated for the indicated time periods at 37 °C. Incubation at 4 °C for 3 h served as control. Proteins were detected by Western immunoblotting.
Extraction and Quantification of S1P
S1P measurements were performed according to an established protocol using liquid chromatography coupled to triple-quadruple mass spectrometry (LC/MS/MS) (32). Cell suspensions in 1 ml of PBS and 1 ml of supernatants were transferred into glass centrifuge tubes. After addition of C17-base internal standards of the analytes (300 pmol/sample; Avanti Polar Lipids) samples were mixed with 200 μl of 6 n hydrochloric acid and 1 ml of methanol and vigorously vortexted for 5 min in the presence of 2 ml of chloroform. Aqueous and chloroform phases were separated by centrifugation for 3 min at 1900 rcf, and the lower chloroform phase was transferred into a new glass centrifuge tube. After a second round of lipid extraction with additional 2 ml of chloroform, the two chloroform phases were combined and vacuum-dried at 50 °C for 50 min using a vacuum concentrator. The extracted lipids were dissolved in 100 μl of methanol/chloroform (4:1, v/v) and stored at −20 °C. Detection was performed with the QTrap triple-quadrupole mass spectrometer (AB Sciex, Framingham, MA) interfaced with the Merck-Hitachi Elite LaChrom series 3.1.3 chromatograph and autosampler (VWR International). Positive electrospray ionization LC/MS/MS analysis was used for detection of all analytes. The ion source conditions and gas settings for positive electrospray ionization LC/MS/MS analysis were as follows: ion spray voltage, 5500; ion source heater temperature, 450 °C; collision gas setting, medium; ion source gases 1 and 2, settings 30 and 60, respectively; curtain gas setting, 45. Multiple reaction monitoring transitions were as follows: S1P m/z, 380/264; C17-S1P m/z, 366/250; and C17-sphingosine m/z, 286/268. Liquid chromatographic resolution of all analytes was achieved using a 2 × 60-mm MultoHigh C18 reversed phase column with 3-μm particle size (CS-Chromatography Service). The column was equilibrated with 10% methanol and 90% of 1% formic acid in H2O for 10 min, followed by sample injection and 26-min elution with 100% methanol with a flow rate of 300 μl/min. Standard curves were generated by adding increasing concentrations of the analytes to 300 pmol of the internal standard. Linearity of the standard curves and correlation coefficients were obtained by linear regression analyses. Data analyses were performed using Analyst 1.4 (AB Sciex).
Immunocytochemistry
Cells were cultured on glass coverslips until 70% confluency and fixed in 4% paraform aldehyde. After washing with PBS, cells were first permeabilized in 0.25% Triton X-100 for 10 min and blocked with 10% BSA containing 0.125% Triton X-100 in PBS for 1 h. Primary and secondary antibodies were incubated for 1 h in 5% BSA with 0.125% Triton X-100 in PBS with three repetitive washing steps for 10 min in between (0.125% Triton X-100 in PBS). The nuclei were stained with DAPI for 10 min in PBS. Cells were embedded on a microscope slide with ImmuMount (Thermo Scientific) and analyzed by fluorescence microscopy (AxioVert 200; Zeiss; equipped with a plan-Apochromat 63×/0.75 objective and an Axiocam MRm camera). Images were acquired and processed using AxioVision 4.8 software (Zeiss).
Densitometric Quantification and Statistical Analysis
Western blot signals were quantified by densitometric analysis using Quantity One® software (Bio-Rad). Two-sided Student's t test was used for statistical analysis using Exel 2010 (Microsoft). p values were classified and indicated as follows: *, p < 0.05; **, p < 0,01; and ***, p < 0,001.
RESULTS
Modulation of Intracellular S1P Levels Affects APP Metabolism
To assess the role of S1P in the metabolism of APP, we first used the sphingosine kinase inhibitor SKI II to reduce intracellular levels of S1P in native HEK293 or HEK293 cells overexpressing human APP695 (Fig. 1A). The inhibition of SphK induced a decrease of APP-CTFs in a dose-dependent manner, whereas APP-FL showed few, if any, changes (Fig. 1B). Cell viability was analyzed by MTT reduction assay and was not impaired by treatment with SKI II (data not shown). Next, we analyzed the expression of APP and CTFs in embryonic fibroblasts from wild-type mice and from mice with a genetic deletion of the S1P-lyase (Fig. 1C). Mass spectrometry showed a selective increase of intracellular S1P in S1P-lyase KO cells as compared with WT cells (Fig. 1D), whereas concentrations of extracellular S1P were not increased in conditioned media of S1P-lyase KO cells (Fig. 1E), suggesting that deletion of the S1P degrading enzyme predominantly affects the intracellular pool of S1P. Interestingly, levels of full-length and particularly that of APP-CTFs were strongly increased in S1P-lyase KO cells (Fig. 1F). The use of specific antibodies against APP-CTFs showed that all three variants of APP-CTFs, αCTF, βCTF, and βCTF*, are increased in S1P-lyase-deficient cells. APP-βCTF* is an additional cleavage product of APP generated by alternative BACE1 processing at position Glu-11 within the Aβ domain (33, 34). Consistent with a previously described conversion of βCTF and βCTF* into αCTF by α-secretase (35), the highest accumulation was observed for αCTF.
FIGURE 1.

Modulation of S1P levels affects the metabolism of APP. A, schematic showing the metabolism of S1P and enzymes involved. B, native HEK293 expressing endogenous APP751/770 isoforms (left panels) or HEK293 cells overexpressing the human neuron-specific APP695 isoform (right panels) were treated with the indicated concentrations of SKI II for 24 h. APP and its CTFs were detected by Western immunoblotting. APP695 im and APP695 m denote immature and mature forms of overexpressed APP. * and ** indicate migration of endogenous immature and mature APP isoforms. C, expression of S1P-lyase was analyzed in embryonic fibroblasts of WT or S1P-lyase knock-out (S1PL-KO) mice by Western immunoblotting. D, intracellular concentration of S1P measured by LC/MS-MS (*, p < 0.05; n = 3). E, extracellular concentration of S1P measured by LC/MS-MS (n.s., p > 0.05; n = 3). F, expression of endogenous APP and different CTFs was analyzed in WT and S1P-lyase KO by Western immunoblotting. Full-length APP (APP-FL) and different CTFs generated by β-or γ-secretase cleavage were detected with specific antibodies against the C terminus or the Aβ domain of APP (see “Experimental Procedures”). G, Western immunoblotting of APP-FL and S1P-lyase in murine N9 cells upon RNAi-mediated knock down of S1P-lyase. H and I, quantitative analysis of S1P-lyase (H; **, p < 0.01; n = 3) and APP-FL (I; **, p < 0.01; n = 3) by ECL imaging.
In addition to the use of MEFs from WT and S1P-lyase KO mice, we also targeted S1P-lyase by siRNA in mouse N9 cells. Although levels of APP CTFs were below the detection limit, levels of full-length APP significantly increased upon knockdown of S1P-lyase (Fig. 1, G and H), thereby confirming the effects observed in S1P-lyase KO cells.
We also determined the levels of sphingosine that could derive from dephosphorylation of S1P in WT and S1P-lyase KO cells. Consistent with previous results (23), sphingosine levels were significantly increased in S1P-lyase KO cells (Fig. 2A). Thus, we tested the effect of sphingosine on APP and its CTFs. Incubation of both WT and S1P-lyase KO cells with sphingosine led to an increase in full-length APP and CTFs (Fig. 2B). In contrast, cell incubation with extracellular S1P exerted little, if any, effect on APP or APP CTFs (Fig. 2C). Because sphingosine efficiently penetrates the plasma membrane and could be phosphorylated by intracellular SphK to S1P, these data suggest that the accumulation of APP in S1P-lyase KO cells might be caused by intracellular rather than extracellular S1P or sphingosine.
FIGURE 2.

Involvement of intracellular S1P in the accumulation of APP. A, determination of intracellular sphingosine concentration in WT and S1PL-KO cells by LC/MS-MS (**, p < 0.01; n = 3). B and C, WT and S1P-lyase KO cells were treated with 10 μm sphingosine or 10 μm S1P for the indicated time periods, and APP-FL and its CTF were detected by Western immunoblotting. D, WT and S1PL-KO MEFs were treated with 5 μm SKI II for 24 h followed by detection of APP-FL and CTFs in isolated membranes and of secreted APPα in conditioned media.
We next tested the effect of the SphK inhibitor SKI II in both WT and S1P-lyase KO cells. Mass spectrometry revealed a significant increase of endogenous sphingosine in both WT (control, 3.63 ± 1.34 pmol/sample; SKI II, 17.83 ± 4.55 pmol/sample; p < 0.01) and S1P-lyase KO cells (control, 16.18 ± 1.99 pmol/sample; SKI II, 42.92 ± 11.20 pmol/sample; p < 0.01), respectively, indicating inhibition of sphingosine phosphorylation. Interestingly, SKI II strongly reduced levels of APP-FL and CTFs in S1P-lyase knock-out as well as in WT cells (Fig. 2D). Notably, the secretion of soluble APPα was also reduced upon treatment with SKI II in both WT and S1P-lyase KO cells, indicating that the decrease in cellular APP and APP-CTF levels was not caused by increased secretion of this protein (Fig. 2D).
S1P-Lyase Deficiency Modulates the Generation of Aβ and Secretase Activities
To determine whether S1P-lyase deficiency also affects the generation of Aβ, we stably transduced WT and S1P-lyase KO MEFs with lentivirus containing the human APP695 cDNA with the Swedish mutation (APPswe). The construct also drives the separate expression of GFP by an internal ribosomal entry site. WT and S1P-lyase KO cells showed similar expression of GFP, indicating similar transduction and expression of the construct (Fig. 3A). The specific detection of transgenic human APPswe with the human specific antibody 6E10 revealed strongly increased levels of APPswe in S1P-lyase KO as compared with S1P-lyase WT cells (Fig. 3A). These data thereby confirm the observations for the endogenous APP (Fig. 1, D and E) and further indicate an impaired metabolism of APP in S1P-lyase-deficient cells.
FIGURE 3.

Altered Aβ generation in S1P-lyase-deficient cells. A, WT and S1P-lyase KO MEFs were stably transduced with a human APP695swedish-internal ribosomal entry site-GFP construct. Expression of APP and GFP was analyzed by Western immunoblotting. B and C, Aβ levels in conditioned media were determined by electrochemiluminescence and normalized to GFP expression (see “Experimental Procedures”). D and E, Aβ secretion was normalized to cellular APP levels. (***, p < 0.001; n = 3).
We then measured levels of different Aβ species secreted by the transduced cells. Levels of Aβ40 were ∼20-fold higher than those of Aβ42. Interestingly, the levels of Aβ40 were only slightly but significantly increased in S1P-lyase KO as compared with WT cells (Fig. 3B). Aβ42 levels were not significantly different (Fig. 3C). Notably, when Aβ levels were normalized to the total cellular APP, a significant decrease in secreted Aβ40 and Aβ42 was evident in S1P-lyase-deficient cells (Fig. 3, D and E), suggesting that the accumulated APP-CTFs were not efficiently processed by γ-secretase. Direct measurements of γ-secretase with a fluorogenic substrate indeed revealed slightly but significantly reduced activity of this enzyme in S1P-lyase KO cells as compared to cells with functional S1P expression (Fig. 4A). To further prove decreased γ-secretase activity in S1P-lyase KO cells, we performed an in vitro γ-secretase assay as described previously (31). Incubation of purified membranes for 3 h at 37 °C showed efficient production of APP intracellular domain (AICD) in both WT and S1P-lyase KO samples. However, at 1 h of incubation, CTFs were still detected in samples from S1P-lyase KO samples, whereas very few, if any, CTFs were detectable in WT samples (Fig. 4B). Quantification of CTFs and AICD showed statistically significant differences (Fig. 4C). These data confirm the measurements with the fluorometric assay on reduced γ-secretase activity in S1P-lyase KO. However, the decrease in γ-secretase activity in S1P-lyase-deficient cells was low compared with the strong accumulation of APP CTFs.
FIGURE 4.
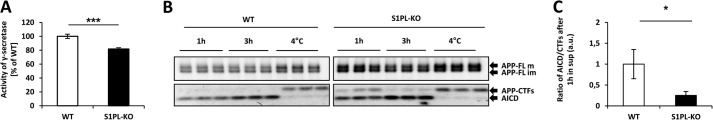
Decreased γ-secretase activity in S1P-lyase-deficient cells. A, γ-secretase activity in living cells was determined by a fluorometric assay (see “Experimental Procedures”). γ-Secretase is decreased by ∼20% in S1PL-KO as compared with WT cells (***, p < 0.001; n = 9). B and C, in vitro γ-secretase assay with purified membranes of WT and S1P-lyase KO cells (see “Experimental Procedures”). APP, CTFs, and AICD were detected by Western immunoblotting after the indicated incubation times (B). The ratios of AICD and CTFs after 1 h of incubation at 37 °C were determined by densitometry (*, p < 0.05; n = 3). Incubation of 3 h at 4 °C efficiently blocked the cleavage of CTFs to AICD (C).
Accumulation of APP-CTFs in Lysosomal Compartments of S1P-lyase KO Cells
In addition to the processing by γ-secretase, APP-CTFs can also be degraded by lysosomal proteases, including cathepsins (9, 10, 36, 37). Thus, we specifically tested whether S1P-lyase deficiency affects the lysosomal degradation of APP-CTFs. First, the association of APP CTFs with lysosomes was investigated by double staining of cells with antibodies against the lysosome-associated membrane protein-2 (Lamp-2) and the C-terminal domain of APP, respectively. As compared with WT cells, the intensity and size of Lamp-2-positive structures appeared increased in S1P-lyase-deficient cells, indicative for impaired lysosomal activity. Importantly, S1P-lyase KO cells also showed increased reactivity for the APP C terminus in vesicular structures that partly co-localized with Lamp-2. In contrast, WT cells revealed only little co-localization of APP and Lamp-2 (Fig. 5A, red frames), likely because of efficient degradation of APP and its CTFs in lysosomes.
FIGURE 5.

APP-CTFs accumulate in lysosomal compartments of S1PL-KO. A, immunocytochemical analysis of Lamp-2 and APP in WT and S1P-lyase KO MEFs. Cells were co-stained with antibodies against Lamp-2 and APP C terminus (APP c-ter). Red frames indicate the enlarged areas (right panels). Arrowheads indicate accumulations of APP in lysosomal compartments. Scale bar, 20 μm. B, subcellular fractionation of WT and S1PL-KO MEFs by density gradient centrifugation (see “Experimental Procedures”). The distribution of APP-CTFs (lower panels) and cathepsin D (Cat. D) in the different fractions was analyzed by Western immunoblotting. Note the strong accumulation of APP-CTFs in cathepsin D-positive fractions from S1PL-KO cells.
To further prove accumulation of APP-CTFs in lysosomal compartments, we next performed subcellular fractionations. Only very low amounts of APP-CTFs were detected in cathepsin D-positive fractions in WT cells (Fig. 5B). In contrast, APP-CTFs were strongly increased in cathepsin D-positive fractions of S1P-lyase-deficient cells (Fig. 5B), indicating their selective accumulation in lysosomes.
To test whether S1P-lyase deficiency affects other lysosomal proteins, we analyzed the expression of cathepsin D and Lamp-2 in more detail. Western immunoblotting revealed decreased levels of the mature active form (25 kDa) of cathepsin D, whereas those of the immature pro-/intermediate-forms (55–45 kDa) were slightly elevated in S1P-lyase KO cells. Accordingly, the ratio of mature to immature forms of cathepsin D in S1P-lyase KO cells was significantly reduced by ∼30% as compared with WT cells (Fig. 6, A and B). Reduced ratios of mature/immature forms of cathepsin D were also demonstrated upon RNAi-mediated knockdown of S1P-lyase (Fig. 6, C and D).
FIGURE 6.

Impaired lysosomal activity in S1P-lyase KO cells. A–D, Western blot analysis of cathepsin D in WT and S1PL-KO (A and B) and in siRNA transfected murine N9 cells (C and D). The ratio of active to immature (pro-/intermediate-) forms of cathepsin D is decreased in S1PL-KO (***, p < 0,001; n = 6) and upon knockdown of S1P-lyase (**, p < 0.01; n = 3). E–H, detection of lysosomally degraded Lamp-2 (E and F) and Gm2 activator protein (Gm2a) (G and H) in WT and S1PL-KO cell lysates by Western immunoblotting. Levels of Lamp-2 (F) and Gm2a (H) are increased in S1PL-KO cells, indicative for an impairment of lysosomal activity. Quantitative analysis was done by ECL imaging (E; *, p < 0.05; n = 3; and G; *, p < 0.05; n = 3). Cat. D or Cath D, cathepsin D.
To further demonstrate a lysosomal impairment in S1P-lyase-deficient cells, we also detected Lamp-2 and the GM2 activator protein, two proteins also degraded in the lysosome (38). Both proteins were slightly but significantly increased by ∼ 20–30% in S1P-lyase KO cells (Fig. 6, E–H). Together, these data strongly indicate that S1P-lyase deficiency and intracellular S1P accumulation impair lysosomal turnover of proteins.
We showed previously that induction of autophagy by cell starvation promotes the degradation of APP-CTFs (9). Thus, we tested the effects of starvation on the clearance of APP-CTFs in S1P-lyase and WT cells. Starvation strongly decreased the levels of both APP-FL and APP-CTFs in WT, as well as S1P-lyase KO cells (Fig. 7). However, APP-CTF levels remained higher in S1P-lyase KO cells as compared with that of WT cells at each time point. To check the induction of autophagy, we monitored conversion of LC3-I to LC3-II. After 2 h of starvation, we detected almost exclusively LC3-II in both cell types, indicating efficient induction of autophagy in WT, as well as in S1P-lyase KO cells. After 6 h of starvation, levels of LC3-II were strongly decreased in WT cells, indicating efficient degradation during autophagic flux (Fig. 7). Interestingly, LC3-II showed higher stability in S1P-lyase KO cells after 6 h of starvation. Together, the data indicate that S1P-lyase deficiency did not inhibit the induction of autophagy but rather impairs the final consumption of substrates in lysosomal compartments.
FIGURE 7.
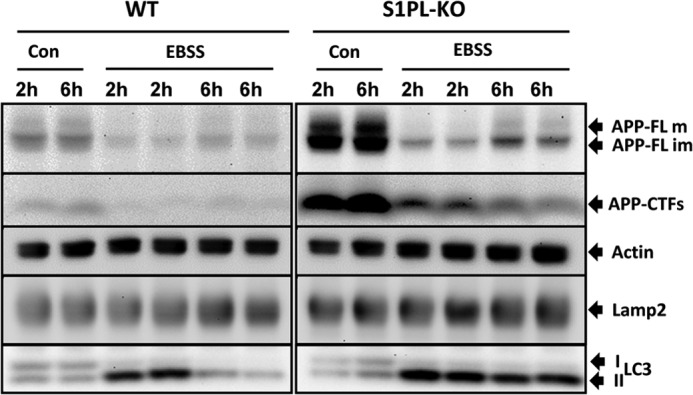
Impaired autophagic flux in S1P-lyase-deficient cells. Starvation of WT and S1PL-KO cells was induced by incubation in Earle's balanced salt solution (EBSS) for the indicated periods of time. The indicated proteins were detected by Western immunoblotting. APP and APP-CTFs strongly decrease upon starvation in WT and S1PL-KO cells. Both cell types show a conversion of LC3-I to LC3-II after 2 h of incubation in Earle's balanced salt solution, indicative for induction of autophagy. Notably, whereas LC3-II is efficiently consumed after 6 h in WT cells, S1PL-KO cells show decreased consumption of this protein. Con, control.
Lysosomal Ca2+ Release Affects Metabolism of APP-FL and APP-CTFs
S1P-lyase-deficient cells have elevated intracellular Ca2+ levels but also increased Ca2+ release from the endoplasmic reticulum, indicating impaired regulation of intracellular Ca2+ concentrations (26). To assess whether the increased levels of APP-FL and APP-CTFs in S1PL-KO cells could be related to altered Ca2+ concentrations, we first treated cells with thapsigargin to block the reuptake of cytosolic Ca2+ into the ER. Time course analysis revealed that thapsigargin treatment efficiently decreased the levels of APP-CTFs in both WT and S1P-lyase-deficient cells already after 1 h of thapsigargin treatment (Fig. 8A). At 2–4 h of treatment, APP-CTFs remained at a very low level in WT cells and continuously decreased in S1P-lyase-deficient cells. However, at each time point, APP-CTFs were higher in S1P-lyase KO cells than in WT cells. Thus, relative changes in cytosolic Ca2+ partially reversed the accumulation of APP CTFs.
FIGURE 8.

Mobilization of Ca2+ promotes the degradation of APP CTFs. A and B, cells were incubated in the presence or absence of thapsigargin (A) or GPN (B) for the indicated periods of time and levels of APP and APP-CTFs analyzed by Western immunoblotting. The treatment with thapsigargin and GPN strongly promoted the degradation of APP-CTFs, although the effects in S1PL-KO cells were less pronounced.
Notably, lysosomal dysfunction in a Niemann-Pick type C (NPC) model for lipid storage was attributed to impaired Ca2+ mobilization from lysosomal stores (39). To specifically assess the role of lysosomal Ca2+, we treated cells with GPN, which causes a release of Ca2+ from lysosomal stores and partially reverses lysosomal cholesterol trafficking in NPC cell models (39, 40). GPN treatment increased levels of full-length APP after 2–16 h in WT cells but had little, if any, effect on full-length APP in S1P KO cells (Fig. 8B). Interestingly, APP-CTFs showed biphasic changes upon GPN treatment. After 1 h, APP-CTF levels strongly decreased and then continuously recovered during the following chase period until 16 h in WT cells (Fig. 8B). In S1P-lyase-deficient cells, GPN treatment also decreased levels of APP-CTFs in the first 1–2 h. As observed with WT cells, levels of APP-CTFs steadily increased after the initial decline upon longer incubation times (Fig. 8B). The combined data indicate that mobilization of Ca2+ from ER or lysosomal stores strongly promotes the degradation of APP-CTFs. However, the magnitude and time course of effects strongly differ between WT and S1P-lyase-deficient cells, indicating that the altered metabolism of APP-CTFs involves aberrant regulation of intracellular Ca2+ concentrations.
DISCUSSION
Our data demonstrate involvement of the S1P-lyase in lysosomal APP metabolism. Genetic inhibition of the enzyme results in increased levels of intracellular S1P and impaired degradation of APP and its CTFs in lysosomal compartments. Although S1P-lyase deficiency led to a slight increase in the total secretion of Aβ, we found a strongly decreased product-precursor relationship when Aβ levels were normalized to that of APP CTFs. In line with this observation, the absolute activity of γ-secretase was lower in S1P-lyase KO cells as compared with WT cells.
S1P has been shown to increase the secretion of Aβ from neuronal cells, and this effect was attributed to the direct stimulation of the β-secretase BACE1 (12, 41). Indeed, we also observed increased β-secretase activity when S1P was added to isolated membranes of SH-SY5Y cells.4 Because the stimulatory effect of S1P has also been observed for an isolated BACE1 variant without the transmembrane domain, S1P likely exerts allosteric effects upon interaction with the catalytic ectodomain of the enzyme (12, 41).
The molecular mechanisms underlying the effects of S1P on γ-secretase remain unclear. However, cleavage of APP-CTFs to the intracellular AICD was slightly reduced in isolated membranes from S1PL-KO cells. Thus, it would be interesting to further assess the role of S1P or sphingosine in the regulation of γ-secretase activity. It has been shown that γ-secretase activity could be modulated by membrane lipids (42–46). However, whether these effects are caused by direct interaction of the respective lipids with γ-secretase components and its substrates or via more global changes in membrane fluidity is unknown.
In this study, we identified a more general effect of S1P-lyase deficiency on lysosomal activity, resulting in the strong accumulation of APP-CTFs in lysosomal compartments of S1P-lyase-deficient cells. The impairment of lysosomal activity was also evident from the lower maturation of cathepsin D and the accumulation of Lamp-2. Both proteins are well accepted markers to evaluate lysosomal function (38, 47, 48). In addition, we also observed accumulation of the GM2 activator protein that is also localized in lysosomes and facilitates lysosomal degradation of sphingolipids (49). Several observations in this study indicate that the impaired lysosomal function is caused by alterations in intracellular S1P levels. First, mass spectrometry revealed a selective increase of intracellular S1P in S1P-lyase KO cells, whereas extracellular levels were not significantly different as compared with WT cells. Second, addition of extracellular S1P did not affect APP levels, whereas incubation with sphingosine induced a strong accumulation of APP and CTFs. Sphingosine could have increased membrane permeability than S1P and can be subsequently phosphorylated by intracellular sphingosine kinases to increase the intracellular S1P concentration. Third, the inhibition of sphingosine kinase by SKI II decreased the levels of APP and its CTFs in both WT and S1P-lyase KO cells. Because SKI II further increased cellular sphingosine levels, it is unlikely that the accumulation of APP is directly caused by intracellular sphingosine. Thus, rather intracellular S1P concentrations appear to be critical for lysosomal function. However, the data do not rule out a contribution of other sphingolipid metabolites, including the cleavage products of the S1P-lyase reaction, phosphoethanolamine and hexadecenal. It would be interesting to further investigate the role of intracellular S1P and its derivatives in more detail.
Our experiments on the effects of starvation showed that both S1P-lyase-deficient cells and WT cells respond with similar induction of autophagy as indicated by efficient conversion of LC3-I to LC3-II and initial degradation of APP and its CTFs. However, whereas LC3-II and APP CTFs were almost completely cleared during starvation in WT cells, the same proteins were much more stable in the S1P-lyase KO cells. These data also strongly support inefficient protein degradation in lysosomes, despite induction of autophagy in S1P-lyase KO cells.
Delivery of molecules to lysosomes depends on transport and fusion of endocytic and secretory vesicles with the lysosomes and involves mobilization of Ca2+ from intracellular stores (50–54). Interestingly, NPC cells have defective Ca2+ mobilization from lysosomes (39) associated with impaired trafficking and metabolism of cholesterol in endolysosomal compartments. This deficit could be partially restored by elevating the cytosolic Ca2+ concentration (55). Of interest, NPC cells, as well as other models of lysosomal storage disorders, also show accumulation of APP and CTFs (9, 36, 56–59) similar to the S1P-lyase KO. We thus assessed the effect of Ca2+ mobilization in the S1P-lyase KO model. Indeed, the elevation of cytosolic Ca2+ concentration by thapsigargin efficiently decreased APP-CTF levels in S1P-lyase-deficient cells. More interestingly, the selective release of Ca2+ from lysosomal stores by GPN acutely promoted the degradation of APP-CTFs within 1 h of treatment. These data thus indicate impaired mobilization of Ca2+ and lysosomal function in S1P-lyase-deficient cells. In line with this findings, it has been demonstrated previously by Ca2+ imaging that S1P-lyase KO cells have aberrantly high levels of Ca2+ in the cytosol but also in intracellular stores (26).
The present study revealed an important role for the S1P-lyase in lysosomal function. It will be interesting to further dissect the molecular mechanisms underlying these effects and also explore the potential of S1P-lyase modulation and lysosomal Ca2+ mobilization to promote APP degradation and thereby decreased Aβ generation.
Acknowledgments
We thank Drs. S. Höning and Konrad Sandhoff for providing antibodies against cathepsin D and GM2a, respectively, and Dr. N. Hagen-Euteneuer for helpful discussion.
This work was supported by the Deutsche Forschungsgemeinschaft by Priority Program 1267 Grants WA1477/8-1 (to J. W.), EC-118/6-1 (to G. v. E. D.), HA2985/6-2 (to T. H. and M. O. G.), and GR1943/2-2 (to M. H. G.); by Collaborative Research Center 645 Grant TP A7 (to J. W.); and by Clinical Research Group KFO177 Grant WA1477/4-2 (to J. W.).
I. Karaca, I. Y. Tamboli, K. Glebov, J. Richter, L. H. Fell, M. O. Grimm, V. J. Haupenthal, T. Hartmann, M. H. Gräler, G. van Echten-Deckert, and J. Walter, unpublished observations.
- Aβ
- amyloid β
- APP
- amyloid precursor protein
- S1P
- sphingosine-1-phosphate
- CTF
- C-terminal fragment
- ER
- endoplasmic reticulum
- SKI II
- 4-[4-(4-chloro-phenyl)-thiazol-2-ylamino]-phenol
- GPN
- glycylphenylalanine 2-naphthylamide
- AICD
- APP intracellular domain
- Lamp-2
- lysosome-associated membrane protein-2
- NPC
- Niemann-Pick type C
- MEF
- mouse embryonic fibroblast
- rcf
- relative centrifugal force.
REFERENCES
- 1. Selkoe D. J. (2001) Alzheimer's disease: genes, proteins, and therapy. Physiol. Rev. 81, 741–766 [DOI] [PubMed] [Google Scholar]
- 2. Walter J., Kaether C., Steiner H., Haass C. (2001) The cell biology of Alzheimer's disease: uncovering the secrets of secretases. Curr. Opin. Neurobiol. 11, 585–590 [DOI] [PubMed] [Google Scholar]
- 3. Thinakaran G., Koo E. H. (2008) Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 283, 29615–29619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kandalepas P. C., Vassar R. (2012) Identification and biology of β-secretase. J. Neurochem. 120, 55–61 [DOI] [PubMed] [Google Scholar]
- 5. Hussain I., Powell D., Howlett D. R., Tew D. G., Meek T. D., Chapman C., Gloger I. S., Murphy K. E., Southan C. D., Ryan D. M., Smith T. S., Simmons D. L., Walsh F. S., Dingwall C., Christie G. (1999) Identification of a novel aspartic protease (Asp 2) as β-secretase. Mol. Cell Neurosci. 14, 419–427 [DOI] [PubMed] [Google Scholar]
- 6. Sinha S., Anderson J. P., Barbour R., Basi G. S., Caccavello R., Davis D., Doan M., Dovey H. F., Frigon N., Hong J., Jacobson-Croak K., Jewett N., Keim P., Knops J., Lieberburg I., Power M., Tan H., Tatsuno G., Tung J., Schenk D., Seubert P., Suomensaari S. M., Wang S., Walker D., Zhao J., McConlogue L., John V. (1999) Purification and cloning of amyloid precursor protein β-secretase from human brain. Nature 402, 537–540 [DOI] [PubMed] [Google Scholar]
- 7. Vassar R., Bennett B. D., Babu-Khan S., Kahn S., Mendiaz E. A., Denis P., Teplow D. B., Ross S., Amarante P., Loeloff R., Luo Y., Fisher S., Fuller J., Edenson S., Lile J., Jarosinski M. A., Biere A. L., Curran E., Burgess T., Louis J. C., Collins F., Treanor J., Rogers G., Citron M. (1999) β-Secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741 [DOI] [PubMed] [Google Scholar]
- 8. Yan R., Bienkowski M. J., Shuck M. E., Miao H., Tory M. C., Pauley A. M., Brashier J. R., Stratman N. C., Mathews W. R., Buhl A. E., Carter D. B., Tomasselli A. G., Parodi L. A., Heinrikson R. L., Gurney M. E. (1999) Membrane-anchored aspartyl protease with Alzheimer's disease β-secretase activity. Nature 402, 533–537 [DOI] [PubMed] [Google Scholar]
- 9. Tamboli I. Y., Hampel H., Tien N. T., Tolksdorf K., Breiden B., Mathews P. M., Saftig P., Sandhoff K., Walter J. (2011) Sphingolipid storage affects autophagic metabolism of the amyloid precursor protein and promotes Aβ generation. J. Neurosci. 31, 1837–1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haass C., Koo E. H., Mellon A., Hung A. Y., Selkoe D. J. (1992) Targeting of cell-surface β-amyloid precursor protein to lysosomes: alternative processing into amyloid-bearing fragments. Nature 357, 500–503 [DOI] [PubMed] [Google Scholar]
- 11. van Echten-Deckert G., Walter J. (2012) Sphingolipids: Critical players in Alzheimer's disease. Prog. Lipid Res. 51, 378–393 [DOI] [PubMed] [Google Scholar]
- 12. Takasugi N., Sasaki T., Suzuki K., Osawa S., Isshiki H., Hori Y., Shimada N., Higo T., Yokoshima S., Fukuyama T., Lee V. M., Trojanowski J. Q., Tomita T., Iwatsubo T. (2011) BACE1 activity is modulated by cell-associated sphingosine-1-phosphate. J. Neurosci. 31, 6850–6857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang H., Desai N. N., Olivera A., Seki T., Brooker G., Spiegel S. (1991) Sphingosine-1-phosphate, a novel lipid, involved in cellular proliferation. J. Cell Biol. 114, 155–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Spiegel S., Milstien S. (2003) Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 4, 397–407 [DOI] [PubMed] [Google Scholar]
- 15. van Echten-Deckert G., Herget T. (2006) Sphingolipid metabolism in neural cells. Biochim. Biophys. Acta 1758, 1978–1994 [DOI] [PubMed] [Google Scholar]
- 16. Le Stunff H., Giussani P., Maceyka M., Lépine S., Milstien S., Spiegel S. (2007) Recycling of sphingosine is regulated by the concerted actions of sphingosine-1-phosphate phosphohydrolase 1 and sphingosine kinase 2. J. Biol. Chem. 282, 34372–34380 [DOI] [PubMed] [Google Scholar]
- 17. Van Veldhoven P. P. (2000) Sphingosine-1-phosphate lyase. Methods Enzymol. 311, 244–254 [DOI] [PubMed] [Google Scholar]
- 18. Liu H., Sugiura M., Nava V. E., Edsall L. C., Kono K., Poulton S., Milstien S., Kohama T., Spiegel S. (2000) Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J. Biol. Chem. 275, 19513–19520 [DOI] [PubMed] [Google Scholar]
- 19. Schmahl J., Raymond C. S., Soriano P. (2007) PDGF signaling specificity is mediated through multiple immediate early genes. Nat. Genet. 39, 52–60 [DOI] [PubMed] [Google Scholar]
- 20. Hannun Y. A., Obeid L. M. (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 9, 139–150 [DOI] [PubMed] [Google Scholar]
- 21. Payne S. G., Milstien S., Spiegel S. (2002) Sphingosine-1-phosphate: dual messenger functions. FEBS Lett. 531, 54–57 [DOI] [PubMed] [Google Scholar]
- 22. Bektas M., Allende M. L., Lee B. G., Chen W., Amar M. J., Remaley A. T., Saba J. D., Proia R. L. (2010) Sphingosine 1-phosphate lyase deficiency disrupts lipid homeostasis in liver. J. Biol. Chem. 285, 10880–10889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hagen-Euteneuer N., Lütjohann D., Park H., Merrill A. H., Jr., van Echten-Deckert G. (2012) Sphingosine 1-phosphate (S1P) lyase deficiency increases sphingolipid formation via recycling at the expense of de novo biosynthesis in neurons. J. Biol. Chem. 287, 9128–9136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hagen N., Van Veldhoven P. P., Proia R. L., Park H., Merrill A. H., Jr., van Echten-Deckert G. (2009) Subcellular origin of sphingosine 1-phosphate is essential for its toxic effect in lyase-deficient neurons. J. Biol. Chem. 284, 11346–11353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hagen N., Hans M., Hartmann D., Swandulla D., van Echten-Deckert G. (2011) Sphingosine-1-phosphate links glycosphingolipid metabolism to neurodegeneration via a calpain-mediated mechanism. Cell Death Differ. 18, 1356–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Claas R. F., ter Braak M., Hegen B., Hardel V., Angioni C., Schmidt H., Jakobs K. H., Van Veldhoven P. P., zu Heringdorf D. M. (2010) Enhanced Ca2+ storage in sphingosine-1-phosphate lyase-deficient fibroblasts. Cell Signal. 22, 476–483 [DOI] [PubMed] [Google Scholar]
- 27. Ihlefeld K., Claas R. F., Koch A., Pfeilschifter J. M., Meyer Zu Heringdorf D. (2012) Evidence for a link between histone deacetylation and Ca2+ homoeostasis in sphingosine-1-phosphate lyase-deficient fibroblasts. Biochem. J. 447, 457–464 [DOI] [PubMed] [Google Scholar]
- 28. Wahle T., Thal D. R., Sastre M., Rentmeister A., Bogdanovic N., Famulok M., Heneka M. T., Walter J. (2006) GGA1 is expressed in the human brain and affects the generation of amyloid β-peptide. J. Neurosci. 26, 12838–12846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grimm M. O., Haupenthal V. J., Rothhaar T. L., Zimmer V. C., Grösgen S., Hundsdörfer B., Lehmann J., Grimm H. S., Hartmann T. (2013) Effect of different phospholipids on α-secretase activity in the non-amyloidogenic pathway of Alzheimer's disease. Int. J. Mol. Sci. 14, 5879–5898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rothhaar T. L., Grösgen S., Haupenthal V. J., Burg V. K., Hundsdörfer B., Mett J., Riemenschneider M., Grimm H. S., Hartmann T., Grimm M. O. (2012) Plasmalogens inhibit APP processing by directly affecting γ-secretase activity in Alzheimer's disease. ScientificWorldJournal 2012, 141240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sastre M., Steiner H., Fuchs K., Capell A., Multhaup G., Condron M. M., Teplow D. B., Haass C. (2001) Presenilin-dependent γ-secretase processing of β-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep. 2, 835–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bode C., Gräler M. H. (2012) Quantification of sphingosine-1-phosphate and related sphingolipids by liquid chromatography coupled to tandem mass spectrometry. Methods Mol. Biol. 874, 33–44 [DOI] [PubMed] [Google Scholar]
- 33. Liu K., Doms R. W., Lee V. M. (2002) Glu11 site cleavage and N-terminally truncated Aβ production upon BACE overexpression. Biochemistry 41, 3128–3136 [DOI] [PubMed] [Google Scholar]
- 34. Haass C., Schlossmacher M. G., Hung A. Y., Vigo-Pelfrey C., Mellon A., Ostaszewski B. L., Lieberburg I., Koo E. H., Schenk D., Teplow D. B. (1992) Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature 359, 322–325 [DOI] [PubMed] [Google Scholar]
- 35. Fluhrer R., Capell A., Westmeyer G., Willem M., Hartung B., Condron M. M., Teplow D. B., Haass C., Walter J. (2002) A non-amyloidogenic function of BACE-2 in the secretory pathway. J. Neurochem. 81, 1011–1020 [DOI] [PubMed] [Google Scholar]
- 36. Tamboli I. Y., Tien N. T., Walter J. (2011) Sphingolipid storage impairs autophagic clearance of Alzheimer-associated proteins. Autophagy 7, 645–646 [DOI] [PubMed] [Google Scholar]
- 37. Agholme L., Hallbeck M., Benedikz E., Marcusson J., Kågedal K. (2012) Amyloid-β secretion, generation, and lysosomal sequestration in response to proteasome inhibition: involvement of autophagy. J. Alzheimers Dis. 31, 343–358 [DOI] [PubMed] [Google Scholar]
- 38. Yanagawa M., Tsukuba T., Nishioku T., Okamoto Y., Okamoto K., Takii R., Terada Y., Nakayama K. I., Kadowaki T., Yamamoto K. (2007) Cathepsin E deficiency induces a novel form of lysosomal storage disorder showing the accumulation of lysosomal membrane sialoglycoproteins and the elevation of lysosomal pH in macrophages. J. Biol. Chem. 282, 1851–1862 [DOI] [PubMed] [Google Scholar]
- 39. Lloyd-Evans E., Morgan A. J., He X., Smith D. A., Elliot-Smith E., Sillence D. J., Churchill G. C., Schuchman E. H., Galione A., Platt F. M. (2008) Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 14, 1247–1255 [DOI] [PubMed] [Google Scholar]
- 40. Shen D., Wang X., Li X., Zhang X., Yao Z., Dibble S., Dong X. P., Yu T., Lieberman A. P., Showalter H. D., Xu H. (2012) Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat. Commun. 3, 731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kalvodova L., Kahya N., Schwille P., Ehehalt R., Verkade P., Drechsel D., Simons K. (2005) Lipids as modulators of proteolytic activity of BACE: involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J. Biol. Chem. 280, 36815–36823 [DOI] [PubMed] [Google Scholar]
- 42. Osenkowski P., Ye W., Wang R., Wolfe M. S., Selkoe D. J. (2008) Direct and potent regulation of γ-secretase by its lipid microenvironment. J. Biol. Chem. 283, 22529–22540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Holmes O., Paturi S., Ye W., Wolfe M. S., Selkoe D. J. (2012) Effects of membrane lipids on the activity and processivity of purified γ-secretase. Biochemistry 51, 3565–3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Walter J., van Echten-Deckert G. (2013) Cross-talk of membrane lipids and Alzheimer-related proteins. Mol. Neurodegener. 8, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Di Paolo G., Kim T. W. (2011) Linking lipids to Alzheimer's disease: cholesterol and beyond. Nat. Rev. Neurosci. 12, 284–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Winkler E., Kamp F., Scheuring J., Ebke A., Fukumori A., Steiner H. (2012) Generation of Alzheimer disease-associated amyloid β42/43 peptide by γ-secretase can be inhibited directly by modulation of membrane thickness. J. Biol. Chem. 287, 21326–21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Benes P., Vetvicka V., Fusek M. (2008) Cathepsin D: many functions of one aspartic protease. Crit. Rev. Oncol. Hematol. 68, 12–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bankowska A., Gacko M., Chyczewska E., Worowska A. (1997) Biological and diagnostic role of cathepsin D. Rocz Akad Med. Bialymst. 42, 79–85 [PubMed] [Google Scholar]
- 49. Sandhoff K., Kolter T. (1998) Processing of sphingolipid activator proteins and the topology of lysosomal digestion. Acta Biochim. Pol. 45, 373–384 [PubMed] [Google Scholar]
- 50. Luzio J. P., Bright N. A., Pryor P. R. (2007) The role of calcium and other ions in sorting and delivery in the late endocytic pathway. Biochem. Soc. Trans. 35, 1088–1091 [DOI] [PubMed] [Google Scholar]
- 51. Luzio J. P., Poupon V., Lindsay M. R., Mullock B. M., Piper R. C., Pryor P. R. (2003) Membrane dynamics and the biogenesis of lysosomes. Mol. Membr. Biol. 20, 141–154 [DOI] [PubMed] [Google Scholar]
- 52. Lloyd-Evans E., Platt F. M. (2011) Lysosomal Ca2+ homeostasis: role in pathogenesis of lysosomal storage diseases. Cell Calcium 50, 200–205 [DOI] [PubMed] [Google Scholar]
- 53. Buxbaum J. D., Ruefli A. A., Parker C. A., Cypess A. M., Greengard P. (1994) Calcium regulates processing of the Alzheimer amyloid protein precursor in a protein kinase C-independent manner. Proc. Natl. Acad. Sci. U.S.A. 91, 4489–4493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bastow E. R., Last K., Golub S., Stow J. L., Stanley A. C., Fosang A. J. (2012) Evidence for lysosomal exocytosis and release of aggrecan-degrading hydrolases from hypertrophic chondrocytes, in vitro and in vivo. Biol. Open 1, 318–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kiselyov K. K., Ahuja M., Rybalchenko V., Patel S., Muallem S. (2012) The intracellular Ca2+ channels of membrane traffic. Channels 6, 344–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Malnar M., Kosicek M., Lisica A., Posavec M., Krolo A., Njavro J., Omerbasic D., Tahirovic S., Hecimovic S. (2012) Cholesterol-depletion corrects APP and BACE1 misstrafficking in NPC1-deficient cells. Biochim. Biophys. Acta 1822, 1270–1283 [DOI] [PubMed] [Google Scholar]
- 57. Boland B., Smith D. A., Mooney D., Jung S. S., Walsh D. M., Platt F. M. (2010) Macroautophagy is not directly involved in the metabolism of amyloid precursor protein. J. Biol. Chem. 285, 37415–37426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Keilani S., Lun Y., Stevens A. C., Williams H. N., Sjoberg E. R., Khanna R., Valenzano K. J., Checler F., Buxbaum J. D., Yanagisawa K., Lockhart D. J., Wustman B. A., Gandy S. (2012) Lysosomal dysfunction in a mouse model of Sandhoff disease leads to accumulation of ganglioside-bound amyloid-β peptide. J. Neurosci. 32, 5223–5236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jin L. W., Shie F. S., Maezawa I., Vincent I., Bird T. (2004) Intracellular accumulation of amyloidogenic fragments of amyloid-β precursor protein in neurons with Niemann-Pick type C defects is associated with endosomal abnormalities. Am. J. Pathol. 164, 975–985 [DOI] [PMC free article] [PubMed] [Google Scholar]