

Background: KCNE3 is tentatively associated with inner ear diseases, but its functions are unknown.
Results: Null deletion of Kcne3 produces profound changes in the excitability of auditory neurons.
Conclusion: KCNE3 regulates the magnitude of K+ conductances responsible for maintaining the electrical phenotype of auditory neurons.
Significance: This study is the first to examine the in vivo functions of KCNE3 in the auditory system.
Keywords: Gene Knock-out, Hearing, Neurobiology, Neurochemistry, Potassium Channels, Auditory Neuron, Deafness, KCNE Subunits
Abstract
The KCNE3 β-subunit interacts with and regulates the voltage-dependent gating, kinetics, and pharmacology of a variety of Kv channels in neurons. Because a single neuron may express multiple KCNE3 partners, it is impossible to predict the overall functional relevance of the single transmembrane domain peptide on the pore-forming K+ channel subunits with which it associates. In the inner ear, the role of KCNE3 is undefined, despite its association with Meniere disease and tinnitus. To gain insights on the functional significance of KCNE3 in auditory neurons, we examined the properties of spiral ganglion neurons (SGNs) in Kcne3 null mutant neurons relative to their age-matched controls. We demonstrate that null deletion of Kcne3 abolishes characteristic wide variations in the resting membrane potentials of SGNs and yields age-dependent alterations in action potential and firing properties of neurons along the contour of the cochlear axis, in comparison with age-matched wild-type neurons. The properties of basal SGNs were markedly altered in Kcne3−/− mice compared with the wild-type controls; these include reduced action potential latency, amplitude, and increased firing frequency. Analyses of the underlying conductance demonstrate that null mutation of Kcne3 results in enhanced outward K+ currents, which is sufficient to explain the ensuing membrane potential changes. Additionally, we have demonstrated that KCNE3 may regulate the activity of Kv4.2 channels in SGNs. Finally, there were developmentally mediated compensatory changes that occurred such that, by 8 weeks after birth, the electrical properties of the null mutant neurons were virtually indistinguishable from the wild-type neurons, suggesting that ion channel remodeling in auditory neurons progresses beyond hearing onset.
Introduction
Potassium (K+) represents one of the most tightly regulated ions in the body within a limited physiological range. The functional importance of K+, which ranges from regulation of ionic compartments and conductance to cell division and cell death (1–3), cannot be overstated. The diversity of K+-dependent functions is made possible by several K+ channels that facilitate the repolarization phase of excitable cells via K+ efflux in response to cellular depolarization (4–6). Therefore, as reflected by their physiological importance, K+ channels represent the most varied ion channel (7, 8). These channels are subject to further regulation at the level of their subunits, including several α-subunit gene families (7), and also by a multitude of regulatory β-subunits. β-Subunits associate with K+ channel α-subunits to modulate their subcellular trafficking and several aspects of their channel behavior (9).
One K+ channel β-subunit family consists of the MinK-related peptides encoded by the Kcne gene family. This ancillary subunit family includes five known human genes, namely Kcne1–5. KCNE subunits display a single transmembrane-spanning domain coupled with an intracellular C terminus and an extracellular N terminus (10, 11); therefore, they cannot form functional channels themselves (12, 13). Indeed, interactions between subunits have been established to a large extent. One must bear in mind that the complexity of such heteromeric interaction between α- and β-subunits is compounded further at two levels as follows: 1) KCNE subunits co-assemble with and modulate α-subunits, resulting in a diverse set of K+ channel phenotypes; and 2) one K+ channel family member can be regulated by different KCNE types. Such heterologous systems suggest unorthodox, yet flexible, stoichiometric dynamics in subunit assembly and therefore present difficulties in dissecting their precise and subunit-specific functions.
Nevertheless, the co-assembly of KCNE1 with Kv7.1 is the best studied due to the physiological importance of this channel. Findings from Barhanin et al. (15) and Sanguinetti and Jurkiewicz (14) established that the voltage-gated K+ (Kv)4 channel Kv7.1 α-subunit must co-assemble with a KCNE1 subunit to form a slowly activating cardiac repolarization current. This was later found to be crucial in the termination of cardiac action potentials. In addition, K+ channels with Kv7.1 and KCNE1 interaction also play a role in the maintenance of endolymph K+ homeostasis in the cochlea (16). The importance of these findings can be illustrated in KCNE1 mutations in human cardiac arrhythmia and deafness (17–19).
The situation is less clear for KCNE3, which is known to associate with the pore-forming α-subunits Kv7.1, Kv7.4, and Kv11.1 and a number of other Kv channels (e.g. Kv2.1, Kv3.2, Kv3.4, and Kv4.3). Where KCNE1 slows and increases depolarization-induced activation of Kv7.1 currents, KCNE3 abolishes the voltage dependence (15, 20). Moreover, Kv7.1/KCNE3 heteromers yield instantaneous, near-ohmic whole-cell currents. Such currents can be stimulated by cyclic AMP, at least in intestinal epithelial cells, and are shown to be involved in intestinal secretory processes and cystic fibrosis (21). Missense mutations have been identified in the Kcne3 gene in two families with skeletal muscle disorders, and they were found to form complexes with Kv3.4 to regulate the resting membrane potential (22). Although controversial, single nucleotide polymorphism analyses have shown an association between mutations/polymorphisms in the Kcne3 gene and the increased susceptibility for developing Meniere disease (23). Interestingly, deep sequencing in patients with chronic tinnitus found two polymorphisms in the Kcne3 gene (24). Because of the lack of statistical power in that study (24), the role of Kcne3 mutations in association with tinnitus could not be definitively concluded.
Here, we established the role of KCNE3 in murine auditory spiral ganglion neurons (SGNs). By using Kcne3 null mice, we were able to dissect the regulation of SGN firing properties by KCNE3 through electrophysiological means. Null deletion of Kcne3 eliminates variations in the resting membrane potentials (RMPs) of SGNs relative to wild-type pre-hearing mice. We further discovered that in the absence of functional KCNE3, there are age-dependent changes in action potential (AP) and firing properties of neurons, and this is diversified along the cochlear axis. In addition, basally localized SGNs from mutant pre-hearing animals have reduced AP latency, amplitudes, and firing rates. Conversely, these biophysical differences in SGNs were eliminated between null mutant and wild-type adult mice at 8 weeks of age, indicating compensatory changes and remodeling of K+ currents in auditory neurons.
EXPERIMENTAL PROCEDURES
Ethical Approval
Experiments described in this report were approved by the Institutional Animal Care and Use Committee of the University of California at Davis.
Isolation of SGNs
SGNs were isolated from the mouse inner ear using a combination of enzymatic and mechanical procedures (25). Male and female postnatal (P) (4–56 days old) C57BL/6N and Kcne3 null (C57BL/6N background) mice were sacrificed, and the temporal bones were removed in a solution containing minimum essential medium with Hanks' buffered saline solution (Invitrogen), 0.2 g/liter kynurenic acid, 10 mm MgCl2, 2% fetal bovine serum (FBS; v/v), and glucose (6 g/liter). The Kcne3 mouse strain used for this research project was created from ES cell clones (10053F-G10), obtained from the KOMP Repository and generated by Regeneron Pharmaceuticals, Inc. Methods used to create the VelociGene-targeted alleles have been described (26).
The central spiral ganglion tissue was dissected out and split into three segments, apical, middle, and basal, across the modiolar axis as described previously (25). The tissues were digested separately in an enzyme mixture containing collagenase type I (1 mg/ml) and DNase (1 mg/ml) at 37 °C for 20 min. After a series of gentle triturations and centrifugation in 0.45 m sucrose, the cell pellets were reconstituted in 500 μl of culture media (Neurobasal-A, supplemented with 2% B27 (v/v), l-glutamine (0.5 mm), penicillin (100 units/ml); Invitrogen) and filtered through a 40-μm cell strainer for cell culture and electrophysiological experiments. We cultured SGNs for ∼24–48 h to allow detachment of Schwann cells from neuronal membrane surfaces.
Electrophysiology
APs from SGNs were amplified (×100), filtered (bandpass 2–10 kHz), and digitized at 5–500 kHz. Extracellular solution for AP recording experiments contained (in mm) the following: 130 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2, 10 d-glucose, and 10 HEPES, pH 7.4, with NaOH. The recording electrodes contained (in mm) the following: 112 KCl, 2 MgCl2, 0.1 CaCl2, 10 HEPES, 1 EGTA, 5 K2ATP, and 0.5 Na2GTP, pH 7.3, with KOH. The Ca2+ concentrations in solutions were measured with a Ca2+-sensitive electrode as described (27).
For current measurements in SGNs, recordings were performed using the whole-cell configuration of the patch clamp technique. Signals were amplified using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA) and filtered (bandpass 2–10 kHz). Data were digitized using an analog-to-digital converter, Digidata 1322 (Molecular Devices). Patch electrodes were pulled from borosilicate glass capillaries with a Flaming/Brown microelectrode puller (P97, Sutter Instrument) and fire-polished to a final resistance of 2–3 megohms, when filled with internal solution. The internal solution for SGN recordings was as follows (in mm): 112 KCl, 2 MgCl2, 0.1 CaCl2, 5 K2ATP, 0.5 GTP-sodium, 1 EGTA, and 10 HEPES, pH 7.35, with KOH. The external solution for SGNs contained (in mm) the following: 130 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2, 10 d-glucose, and 10 HEPES, pH 7.4, with NaOH. Na+ and K+ ions were substituted for each other to maintain the osmolarity. The bath solution was perfused (2–3 ml/min) constantly. The liquid junction potentials were measured and corrected as described (28, 29). All recordings were carried out at room temperature (20–21 °C).
Immunostaining
For histological cryosection experiments, the cochleae were immersed into 4% paraformaldehyde in phosphate-buffered saline (PBS) overnight at 4 °C. After fixation, the cochleae were decalcified (10% EDTA, pH 7.4; 24–48 h; 4 °C). Cochleae were processed sequentially with 10 and 30% sucrose at 4 °C overnight and then embedded in OCT for cryosectioning. Sections were washed in PBS, permeabilized in 0.1% Triton X-100 for 25 min, and then incubated for 30 min in a blocking solution containing 1% bovine serum albumin and 1% normal goat serum. The 5–10-μm sections were incubated in primary antibody against KCNE3, residues 463–480 (Alomone Labs) at 1:100–1:200 dilutions overnight at 4 °C. To detect Kv4.2, we used an antibody against the extracellular S1-S2 loop 209–225 (Abcam, Cambridge, MA). Similarly, Kv4.3 was detected with an antibody against the C-terminal amino acids 415–636 (Abcam). To identify neurons, samples were counter-stained with an antibody against the neuronal marker Tuj1, as described previously (25). The rinsed sections were then incubated for 2 h at room temperature with an appropriate secondary antibody, followed by 5 min of DAPI nuclei staining with PBS washing. Slides were mounted with ProLong Gold mounting medium (Invitrogen). Images were captured with a Zeiss LSM 510 confocal microscope.
Genotyping
DNA was isolated from the tail and then PCR-amplified using primer sets. The size of the DNA fragments generated determined which mice in a litter were wild types, heterozygotes, and nulls. We used these primer for genotyping as follows: TDF, CGGGACTGAGACCTGGTACATG; R3, CCCACAGTGACGGCAAATAGG; NeoF, GCAGCCTCTGTTCCACATACACTTCA; and CD-R, CTGTGTCTCGAAAAATGTCTCATCC.
Data Analysis
Data were analyzed using pCLAMP 10.2 (Molecular Devices), Origin8.1 (MicroCal Software). All measurements are presented as mean ± S.D., and Student's t test was used to determine statistical significance.
RESULTS
Verification of Expression of KCNE3 in SGNs
Although previous reports have shown the lack of KCNE3 protein expression in tissues from the digestive tract of similar Kcne3−/− animal models (29), we cross-checked the expression pattern of the protein in isolated SGNs using immunocytochemical analysis. Early studies have suggested K+ channel β-subunit expression in the inner ear (also see Fig. 7) (30), which demonstrates that the membranes of SGNs are well decorated with a specific antibody directed against KCNE3. By contrast, SGNs isolated from Kcne3−/− mice reacted negatively toward a specific KCNE3 antibody (Fig. 1), reassuring the validity of using the animal model to test our intended hypothesis.
FIGURE 7.
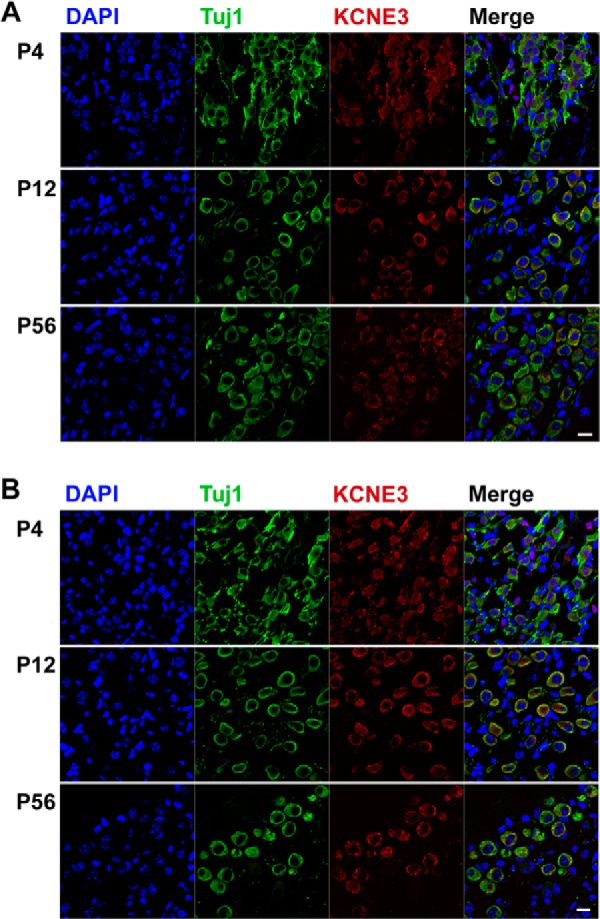
Identification of chronological expression of KCNE3 in SGNs. A, cryosections of P4 and P12 and 2 m (8 weeks (wk) or P56) apical SGNs were stained with an antibody directed against KCNE3 (in red), and neurons were identified with positive reactivity toward Tuj1, the neuronal marker. The nuclei were stained with DAPI. The expression of KCNE3 was seen from P4 to P56. B, similar observation was made for basal SGNs. These findings were observed from more than 200 sections from three preparations of each age. Scale bar, 20 μm.
FIGURE 1.
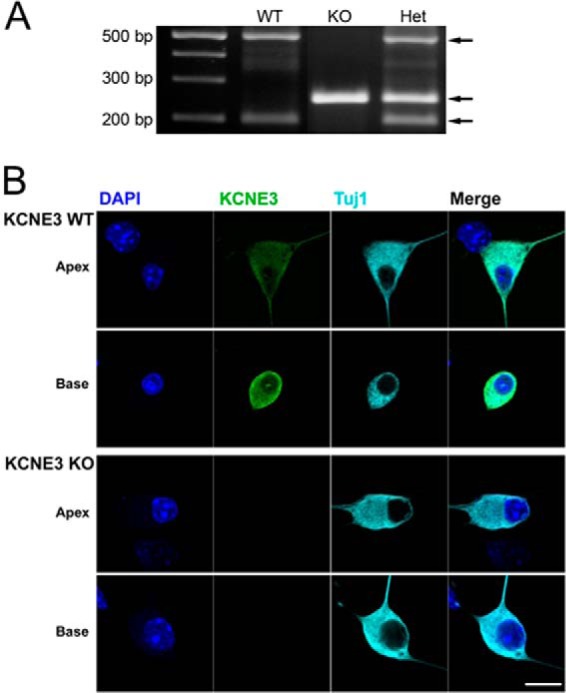
Identification of mutant mice by PCR analysis and KCNE3 protein in SGNs of Kcne3 null mice. Tail DNA was amplified with primers spanning the NeoR insertion site, and the wild-type and mutant chromosomes yield products of 204, 502, and 260 bp, respectively. Localization of KCNE3 in cultured SGNs (stained in green) isolated from P12 Kcne3+/+ and Kcne3−/− mice showed no reactivity toward a specific antibody directed against the protein. No positive reactivity was observed in 10 different samples from 10 different Kcne3−/− SGNs. In contrast, as shown, wild-type SGNs had positive reactivity toward the same antibody. SGNs were stained with antibodies against the neuronal marker, Tuj1 (in cyan), and the nuclei were stained with DAPI (in blue). The right panel is the merged image (scale bar, 10 μm).
Analyses of Membrane Properties of Pre-hearing Kcne3−/− SGNs
KCNE3 expression in developing and mature inner ear sensory cells has been demonstrated (30), and its regulatory effects on voltage-gated K+ (Kv) channels have been implicated in vestibular and auditory diseases. Yet the cellular and functional roles of the β-subunit of Kv channels in hair cells and SGNs are unknown. Because the KCNE3 subunit can associate with a variety of Kv α-subunits (7), a pragmatic strategy to examine the functions of the β-subunit in SGNs is to analyze the membrane properties of cells following null deletion of the gene. We first determined the properties of wild-type Kcne3+/+ SGNs. Fig. 2 illustrates action potentials (APs) recorded from fast and slowly adapting neurons from apical and basal aspects of the cochlear turn, respectively, from pre-hearing P4 Kcne3+/+ and Kcne3−/− neurons. We visibly confirmed the statistical analyses (Fig. 2, A–H, also see Table 1), and in contrast to the wild-type neurons, which have typical variability in the range of recorded RMPs (approximately −48 to −59 mV), null deletion of Kcne3 resulted in tightly ranging RMP values of basal SGNs (−56 to −58 mV; Fig. 2D). Also obvious were the findings that the first evoked AP amplitudes were statistically smaller in the Kcne3−/− apical neurons (Fig. 2G). The inter-spike intervals of spontaneously active P4 SGNs were markedly different between the wild-type and null mutant neurons (Fig. 2C). These findings were similar in both quickly and slowly adapting SGNs. Additional differences in the essential properties of APs between the wild-type and null mutant neurons at P4 are shown (Fig. 2, A--H, and Table 1).
FIGURE 2.

Marked differences in the membrane properties of Kcne3+/+versus Kcne3−/− in pre-hearing SGNs. A, evoked APs were recorded by injecting 0.1 nA current in P4 SGNs, which were isolated and kept in culture for 48 h. APs from fast adapting neurons isolated from the wild-type Kcne3+/+ mice at the apical aspects of the cochlear contour are shown in black. Similar recording obtained from the mutant Kcne3−/− mice is illustrated in blue (middle panel). The right panel shows merged traces of the Kcne3+/+ and Kcne3−/− data. At a glance, the amplitude of the first APs in this class of mutant neurons was substantially smaller than that of the wild-type neurons. The dashed lines represent 0 mV. B, examples of slowly adapting SGNs, which were isolated from the base of the cochlea and cultured for 48 h. C, for SGNs that were spontaneously active (see inset for Kcne3+/+ neurons (in black) and Kcne3−/−neurons (in blue)), wild-type neurons showed regular inter-spike intervals. In contrast, the mutant neurons firing pattern was irregular. D, shown is a box plot of the variations in RMPs of wild-type (n = 12; in black) and mutant (n = 8; in blue) basal neurons. E–H, examination of the ranges of RMPs of the two distinct classes of SGNs, namely fast and slowly adapting neurons, showed diverse patterns. Whereas the RMPs, AP threshold, and duration of the Kcne3+/+ SGNs were conserved in apical neurons, basal neurons had values that were statistically different. Only the amplitudes of apical neuronal APs were different between Kcne3+/+ and Kcne3−/− mice (see Table 1 for number of samples and summary data).
TABLE 1.
Properties of action potentials in control and Kcne3 null mice
| P4 |
P12 |
P56/2 m |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Apex |
Base |
Apex |
Base |
Apex |
Base |
|||||||
| Kcne3+/+ | Kcne3−/− | Kcne3+/+ | Kcne3−/− | Kcne3+/+ | Kcne3−/− | Kcne3+/+ | Kcne3−/− | Kcne3+/+ | Kcne3−/− | Kcne3+/+ | Kcne3−/− | |
| n = 12 | n = 12 | n = 10 | n = 11 | n = 15 | n = 12 | n = 19 | n = 9 | n = 13 | n = 10 | n = 8 | n = 6 | |
| RMP | −58.94 ± 3.02 | −57.25 ± 1.27 | −54.77 ± 3.01 | −57.68 ± 2.74a | −60.54 ± 3.85 | −59.28 ± 3.96 | −56.38 ± 3.07 | −58.46 ± 2.52 | −63.29 ± 5.58 | −63.37 ± 4.23 | −61.4 ± 6.55 | −59.42 ± 3.12 |
| Threshold | −37.21 ± 3.29 | −36.98 ± 1.74 | −34.61 ± 1.33 | −36.43 ± 2.03a | −40.29 ± 3.98 | −38.67 ± 4.55 | −35.06 ± 3.53 | −38.58 ± 3.1a | −42.94 ± 4.73 | −42.53 ± 3.15 | −41.49 ± 5.07 | −39.86 ± 4.18 |
| Latency | 2.12 ± 0.52 | 1.95 ± 0.39 | 2.17 ± 0.43 | 1.93 ± 0.31 | 2.11 ± 0.56 | 2.39 ± 0.73 | 1.96 ± 0.7 | 1.64 ± 0.67 | 2.19 ± 0.52 | 2.07 ± 0.56 | 1.69 ± 0.57 | 1.89 ± 0.6 |
| Duration | 2.16 ± 0.21 | 2.37 ± 0.33 | 2.09 ± 0.12 | 2.32 ± 0.31a | 1.84 ± 0.31 | 1.95 ± 0.41 | 1.97 ± 0.25 | 1.58 ± 0.22b | 1.66 ± 0.26 | 1.49 ± 0.31 | 1.86 ± 0.3 | 1.64 ± 0.31 |
| Amplitude | 86.37 ± 9.18 | 77.46 ± 6.54a | 89.61 ± 10.14 | 89.05 ± 7.58 | 88.34 ± 8.03 | 77.92 ± 12.02a | 89.37 ± 10.63 | 87.1 ± 10.32 | 83.6 ± 11.3 | 84.89 ± 10.31 | 89.29 ± 9.68 | 92.8 ± 15.58 |
| AHP | −25.76 ± 6.02 | −23.65 ± 3.79 | −29.89 ± 3.13 | −30.74 ± 7.34 | 25.48 ± 4.29 | 23.2 ± 3.34 | 24.49 ± 4.94 | 34.39 ± 6.96b | 25.67 ± 6.58 | 28.42 ± 6.17 | 25.32 ± 3.62 | 32.87 ± 6.89a |
a p < 0.05.
b p < 0.01.
Membrane Properties of Kcne3−/− SGNs at the Onset of Hearing and Post-hearing
To determine whether KCNE3 may have developmental attributes, we examined features of SGNs at P12, which is the onset of hearing in mice (31, 32). At P12, major differences between Kcne3+/+ and Kcne3−/− in AP properties were found in slowly adapting neurons isolated from the base of the cochlea. These include the following: 1) enhanced after hypolarization (AHP); 2) reduced AP duration, and 3) reduced threshold voltage (Fig. 3). Other features of APs in the wild-type and null mutant neurons were statistically indistinguishable.
FIGURE 3.

Membrane properties of Kcne3+/+versus Kcne3−/− SGNs at P12, the genesis of hearing. A, assessment of the evoked P12 SGN APs from rapidly adapting neurons isolated from the apical aspects of the cochlea from the Kcne3+/+ (left panel) and Kcne3−/− mice (middle panel) and superimposed traces (right panel). The recording conditions were identical in the wild-type and null mutant neurons. APs were evoked by injecting 0.1 nA current for ∼250 ms. B, representative recordings from slowly adapting SGNs from the base of the cochlea showing APs from Kcne3+/+(left panel) and Kcne3−/− (middle panel) neurons. We superimposed traces (right panel) for comparison. C–F, summary data from analyses of AP properties. C, threshold voltage; D, extent of AHP; E, AP amplitude, and F, AP duration. Significant differences are noted, where *, p < 0.05; **, p < 0.01. Also see Table 1 where the number of samples is listed.
To further ascertain potential K+ current re-modeling, we examined the AP properties in 2-month-old (2 m; ∼P56) SGNs. Surprisingly, in contrast to pre-hearing neurons and neurons at the onset of hearing, mature neuronal APs had characteristic features that were essentially similar in Kcne3+/+ and Kcne3−/− mice (Fig. 4). The exceptions were in Kcne3−/− basal neurons where the AHP remained statistically enhanced compared with wild-type neurons (Fig. 4C). Additionally, for basal and slowly adapting neurons, the inter-spike interval remained substantially reduced in the null mutant neurons (Fig. 4, D and E). For clarity, we have summarized chronological and cochlea-spatial similarities and differences in AP properties in SGNs between Kcne3+/+ and Kcne3−/− mice (Table 1).
FIGURE 4.

Close approximation of membrane properties of Kcne3+/+versus Kcne3−/− SGNs at post-hearing stages at 2 months old (8 weeks or P56). A and B, in accord with Figs. 2 and 3, we examined the AP properties of fast adapting (apical) and slowly adapting (basal) SGNs from Kcne3+/+ (in black) and Kcne3−/− (in blue) mice. The right panel shows merged data for comparison. C–E, with the exception of the magnitude of AHP (C) in basal SGNs in Kcne3−/− mice, and differences in the inter-spike intervals (D and E), AP properties of SGNs were comparable. Significant differences are noted, where *, p < 0.05.
Underlying Changes in Membrane Currents in Kcne3−/− SGNs from Pre-hearing to Post-hearing Era
KCNE3 has been shown to associate with and regulate the activity of several Kv α-subunits in heterologous expression systems (28, 33). Although these previous reports have established, to a large extent, the unrestrained nature of the β-subunit KCNE3 interaction with multiple Kv channels, it remains to be demonstrated which of these associations produces a physiological impact in SGNs. To begin to address the impact of KCNE3 on Kvs in SGNs, we performed whole-cell voltage clamp experiments under conditions when activation of outward K+ currents was ensured. Fig. 5 exemplifies outward K+ currents from apical and basal portions of the cochlea in P4 SGNs isolated from Kcne3+/+ and Kcne3−/− mice (Fig. 5, A–D). The recordings were obtained from a holding potential of −70 mV, and outward and inward K+ currents were activated using step potentials ranging from −120 to 30 mV with voltage increments of 10 mV. Two undisputable differences between the two genotypic SGNs were made, such that the peak whole-cell K+ current magnitude in the Kcne3 null mutant was ∼2-fold greater than the wild-type and that Kcne3−/− SGNs showed prominent transient current traces that were less apparent in the wild-type neurons. These observations are illustrated graphically by the difference current traces, shown as insets. A summary of the current density-voltage relationship for apical and basal SGNs is shown in Fig. 5, B and D. To account for the phenotypic differences between AP properties in Kcne3+/+ and Kcne3−/− neurons, we proceeded to examine the chronology of current density differences in P12 and P56 neurons (Fig. 6). At the onset of hearing (∼P12), whole-cell K+ currents in apical and basal SGNs from Kcne3+/+ and Kcne3−/− had undergone sufficient up-regulations (Fig. 6). However, the K+ current magnitude remained greater in the null mutant than wild-type neurons (Fig. 6, B and C), but the differences were not as profound at P12 compared with P4 neurons (Figs. 5 and 6). Indeed by P56, there were no overall differences between whole-cell K+ currents recorded from Kcne3+/+ and Kcne3−/− SGNs (Fig. 6).
FIGURE 5.

Enhanced transient current in pre-hearing SGNs from Kcne3−/− compared with Kcne3+/+ mice. A, voltage clamp recordings of P4 wild-type Kcne3+/+ apical SGNs (left panel) and null mutant Kcne3−/− neurons. SGNs were held at −70 mV and stepped to negative and positive voltages ranging from −120 to 30 mV using 10-mV increments. The wild-type current traces consisted of a small transient and large sustained outward component. In null mutant neurons, the whole-cell outward current was enhanced, and the transient component was bolstered. Shown below is the difference current representing the profile of outward current traces produced after null deletion of Kcne3. B, summary of the current density-voltage relationship from nine apical SGNs showing the peak and steady-state current magnitude. C and D, current traces of P4 basal SGNs from Kcne3+/+ (left panel) and Kcne3−/− mice. The difference transient current is shown below, and the current density-voltage relationships assembled from 14 basal SGNs are summarized in D. pF, picofarad.
FIGURE 6.

Changes in outward K+ current in SGNs during onset of hearing (P12) and post-hearing (P56 or 2 months old) in Kcne3−/− compared with Kcne3+/+ mice. A and B, using standard activation protocol from a holding potential of −70 mV and stepped to voltages ranging from −120 to 40 mV, we recorded outward currents from P12 SGNs from apical (A) and basal (B) aspects of the cochlea. The tail currents were recorded at −80 mV. We compared data from Kcne3+/+ and Kcne3−/− mice, and the difference current traces are shown below. Similar to P4 SGNs, P12 neurons from Kcne3−/− mice expressed greater whole-cell K+ current magnitude than Kcne3+/+ mice. The difference in current magnitude was observed in both apical and basal neurons (see E for the summary data of apical and basal SGNs from Kcne3+/+ and Kcne3−/− mice; n = 10 for each group). C and D, similar recordings and analyses were performed for P56 SGNs from Kcne3+/+ and Kcne3−/− mice from apical (C) and basal (D) aspects of the cochlea. By P56, we could not identify differences in the current magnitude between Kcne3+/+ and Kcne3−/− mice in both apical and basal contours of the cochlea. F, summary data of the current density-voltage relationship (n = 9). pF, picofarad.
Examination on the Chronology of KCNE3 Expression
A simple, but testable, hypothesis for the present findings is that KCNE3 is highly expressed in developing SGNs but later undergoes substantial down-regulation. To obtain traces of this assertion, we examined time-dependent expression of the Kv β-subunit protein in SGNs. Qualitatively, we were unable to identify obvious distinct changes in membrane expression of KCNE3 in SGNs from P4 to P56 (Fig. 7). Alternatively, the findings may reflect compensatory mechanisms associated with null deletions of specific genes (34) and/or altered time-dependent expression of the prominent Kv α-subunit in SGNs, which is under KCNE3 modulation. However, these studies have provided a glimpse of the properties of the most likely Kv α-subunit, which is under the regulation of KCNE3 in SGNs. To extend our study further, we rationalized that at least one of the Kv channels, under the regulation of KCNE3, would generate a transient current. Among the transient current channels in SGNs is Kv4.2 (35, 36). We expressed mouse Kv4.2 in CHO cells, either singly or co-jointly with KCNE3. As expected, transfection of Kv4.2 alone yielded robust transient K+ current (Fig. 8A). Co-transfection of Kv4.2 and KCNE3, at different ratios, resulted in whole-cell K+ current with reduced transient but increased sustained components (Fig. 8, A and B). We cross-checked the Kv4.2 and KCNE3 expression and localization in SGNs (Fig. 9). Indeed, SGNs reacted positively to antibodies against Kv4.2 and KCNE3, and the two proteins were co-localized. In contrast, we did not detect positive staining with antibodies against Kv4.3, consistent with the results of previous studies (35, 36).
FIGURE 8.

Effects of KCNE3 on the current properties of Kv4.2. CHO cells were transfected with mKv4.2 DNA alone and co-jointly with mKCNE3 DNA at ratios (Kv4.2/KCNE3) of 2:1 and 1:1. A, representative current traces show whole-cell recordings from CHO cells for mKv4.2 alone (left panel), Kv4.2/KCNE3 (2:1) (middle panel), and Kv4.2/KCNE3 (1:1) (right panel) using a holding potential of −70 mV and step voltages ranging from −90 to 40 mV. B, graphic representation of the ratio of the sustained to the transient current density, illustrating the extent of inhibition of the transient current by co-expression with KCNE3.
FIGURE 9.

Effects of KCNE3 on the current properties of Kv4.2. Cultured SGNs express multiple Kv4.2 channels, KCNE3 but not Kv4.3. A, cultured (48 h in culture) apical and basal SGNs were fixed and labeled with antibodies against Kv4.2 and Kv4.3 (green) and KCNE3 (red). A, shown are the staining of Kv4.2, and the nuclei were stained with DAPI (blue) as well as the merged images. Apical and basal SGNs stained positively for Kv4.2 and KCNE3 (red). B, similar data were obtained for apical and basal neurons using antibody against Kv4.3. We did not detect positive reactivity toward Kv4.3. The intensity profiles are shown in the plots below. C, analyses of the axons of SGNs showed similar results. Scale bar, 10 μm.
DISCUSSION
Previous studies have established that the K+ channel plays a crucial role in the maintenance of ion homeostasis. Recently, K+ channel β-subunits KCNE have been found as a novel family of regulators of K+ channels. KCNE3 was previously reported to be expressed in the cardiovascular, digestive, respiratory, skeletal, and nervous systems (37). Of particular interest, KCNE3 expression was exclusively confined to the cochlear epithelial lining, starting at embryonic day 14.5 (37). However, its function was not reported. Here, we demonstrated for the first time that KCNE3 is expressed in the cochlear SGNs of pre-hearing and hearing mice (P4 to P56). We further found that KCNE3 is crucial in the regulation of SGN membrane properties, as well as development of its electrophysiological signature.
We have focused our studies on KCNE3 due to previous studies identifying their association with chronic tinnitus and Meniere disease. Both of these conditions have underlying hearing disorders, thus underscoring KCNE3's importance in auditory physiology. Both population studies, however, were met with some significant setbacks; for instance, the lack of statistical power in one study could not rule out the effects of KCNE3 on the risk of developing chronic tinnitus. Nevertheless, we have used a Kcne3 null mouse to allow dissection of its physiological function, specifically in the auditory neurons, SGNs. Such an animal model provides significant insight due to one main reason, the promiscuity of this β-subunit's interaction with a wide variety of K+ channels. As mentioned previously, whether these partnerships are physiologically significant remains a matter of debate. Here, examining a potential neuronal role for mammalian KCNE3, we stressed physiological relevance rather than heterologous screening and examination of the functional consequences of each possible pairing of KCNE3 and α-subunits in in vitro systems. Whereas Sand et al. (24) and Doi et al. (23) described a possible association with hearing disorders, in our case, the lack of KCNE3 in mice demonstrated no obvious hearing phenotype, as evaluated through auditory brain stem recordings (data not shown). Therefore, this study argues against a role for KCNE3 in hearing disorders.
The absence of obvious hearing and balance disorders in our mutant mice does not exclude underlying developmental changes. Indeed, our studies have found, in pre-hearing mice, that APs recorded from Kcne3 null mutant SGNs have significantly narrow variability in RMPs relative to wild type. Because KCNE3 β-subunits cannot form channels of their own (12, 13), such changes in RMP suggest it is strongly involved in regulating membrane conductance. RMPs are determined predominantly by high permeability to K+. In our case, with the observed reduction in the range of SGN RMPs from Kcne3 null mutant mice, it indicates that the ancillary subunit may regulate K+ currents. Indeed, in the absence of KCNE3, we further saw a drastic change in K+ currents where the peak of K+ current magnitude was greater in the Kcne3 null mutant SGNs relative to the wild-type SGNs. Kcne3 null mutant SGNs also showed prominent transient current traces. KCNE β-subunits co-assemble with a wide array of K+ channels and alter their behavior. For instance, Kv7.1 can be “converted” to a voltage-independent, constitutively active “leak” channel by associating with KCNE2 and KCNE3 (21, 38–40). Such dichotomic behavior of Kv channels is crucial for providing K+ leak conductance in the colon and stomach (21, 41–43). However, such a phenomenon is exclusive to Kv7.1 due to the requirement of the S4 domain (44); other Kv7 subfamily α-subunits that reportedly interact with KCNEs do not become leak channels (i.e. Kv7.2/3 with KCNE2 and Kv7.4 with KCNE3 (21, 41)). Although a significant reduction was seen in the activation of Kv3.1 and -3.2 by KCNE1 and KCNE3, there were minimal changes in voltage dependence (33). Moreover, voltage dependence of Kv3.4 activation is negatively shifted by KCNE3 but without adoption of a constitutive current (22). Therefore, the propensity of Kv7.1 for conversion to leak mode by KCNE3 is Kv7.1-specific. The Kv7 family of Kv channels was reported to be associated with hearing disorders. Mutations in Kv7.1 are associated with Jervell and Lange-Nielsen syndrome with congenital deafness due to faulty K+ recycling via KCNE1/Kv7.1-associated current in marginal cells of the stria vascularis (45). Moreover, Kv7.4 mutation, which is mainly expressed in hair cells and SGNs, leads to autosomal dominant progressive hearing loss, otherwise called DFNA2 (39, 46). The potential association of Kv4.2 channels with KCNE3 in SGNs and regulation revealed yet another complexity of understanding the mechanisms of Kv channel regulation in native neurons.
We further examined SGNs of Kcne3 null mice at the onset of hearing and beyond. Here, we reported more pronounced differences in AP firing properties at P12, when the onset of hearing occurs. These changes in AP properties without KCNE3 were localized in specific regions and neuron populations; we observed enhanced AHP, reduced AP duration and threshold voltage in slowly adapting SGNs from the base of the cochlea, and a smaller amplitude of first spike in fast adapting SGNs from the apex. Such selective differences are not surprising, considering numerous reports demonstrating differential expression of K+ channels along the length of the cochlea (36). Such heterogeneity reflects the complexity in firing characteristics that are associated with an orderly tonotopic map in the cochlea. SGNs isolated from the base fire promptly with an AP of short duration, whereas SGNs from the apex fire a delayed AP of longer duration (36). Moreover, the same study revealed that some Kv channels, such as KCa, Kv3.1, and Kv1.1 subunits, were distributed predominantly in the basal SGNs and Kv4.2 predominantly in the apical SGNs (36). Later studies revealed other channels, such as Kv7.4 (25, 47), also demonstrate an expression gradient along the cochlear axis. Therefore, specialized electrophysiological signatures along the cochlear axis are governed by differential ion channel distributions in the cochleae. Compounding this complex heterogeneity, we found that β-subunit KCNE3 (or the lack thereof) may also contribute to the distinct electrophysiological features in SGNs. One can then hypothesize that KCNE3 can co-assemble with α-subunits of a wide repertoire of K+ channels to mediate refinement of neuronal output. Further and closer examination of localization between pore-forming and ancillary subunits will provide insights to the functional outcome of their interactions.
Although newborn mice are responsive to sound within the first 2 weeks after birth, they do not yet achieve full auditory maturity until adulthood at 2 months old (48). Maturation of the tonotopic map in the cochlea is not uniform; it progresses at different rates along the cochlear axis, starting with the basal and then the apical region (49). We discovered that SGNs derived from Kcne3 null mice have differences in AP characteristics, and it is possible that some of the changes we observed in mice during onset of hearing reflect a potential role for KCNE3 in the developmental progression in attaining electrophysiological maturation.
It is also instructive to compare the firing properties of SGNs in older animals (2 months old) when they attain full maturation. To our surprise, most of the AP characteristics were normalized between Kcne3 null mice and wild-type mice. Additionally, the differences in K+ currents became less pronounced between the null mutant and wild-type SGNs. Given that β-subunits form promiscuous interactions, genetic ablation of Kcne3 may result in subsequent effects on other interacting subunits. Up-regulation in compensatory response to the loss of a single component, altered stoichiometry of the remaining channel complexes, and/or elimination of associated subunits due to the aberrant nature of the resultant complexes are all possible outcomes responsible for the near-normalization of firing properties in SGNs from older mice.
Our results collectively indicated that intrinsic electrical properties of SGNs are dependent on KCNE3 in mice during pre-hearing and onset of hearing stages. These differences are not uniform; lack of KCNE3 in fast adapting basal SGNs revealed an enhanced AHP, reduced AP duration, and threshold voltage, whereas apical fast adapting SGNs only changed in the amplitude of the first spike of the AP. Although the differences in electrophysiological properties between 2-month-old Kcne3 mutant and wild-type mice were negligible, it is suggestive of a possible adaptive compensatory response whereby ion channel remodeling in auditory neurons progresses beyond maturation of hearing.
Acknowledgment
We thank members of our laboratory for their constructive comments.
This work was supported, in whole or in part, by National Institutes of Health Grant DC-010386 (to E. N. Y.) from NIDCD. This work was also supported by National Natural Science Foundation of China Grant 1200746 (to P. L.).
- Kv
- voltage-gated K+
- SGN
- spiral ganglion neuron
- RMP
- resting membrane potential
- AP
- action potential
- AHP
- after hyperpolarization.
REFERENCES
- 1. Bortner C. D., Hughes F. M., Jr., Cidlowski J. A. (1997) A primary role for K+ and Na+ efflux in the activation of apoptosis. J. Biol. Chem. 272, 32436–32442 [DOI] [PubMed] [Google Scholar]
- 2. Wei L., Xiao A. Y., Jin C., Yang A., Lu Z. Y., Yu S. P. (2004) Effects of chloride and potassium channel blockers on apoptotic cell shrinkage and apoptosis in cortical neurons. Pflugers Arch. 448, 325–334 [DOI] [PubMed] [Google Scholar]
- 3. DeCoursey T. E., Chandy K. G., Gupta S., Cahalan M. D. (1984) Voltage-gated K+ channels in human T lymphocytes: a role in mitogenesis? Nature 307, 465–468 [DOI] [PubMed] [Google Scholar]
- 4. Nin F., Hibino H., Doi K., Suzuki T., Hisa Y., Kurachi Y. (2008) The endocochlear potential depends on two K+ diffusion potentials and an electrical barrier in the stria vascularis of the inner ear. Proc. Natl. Acad. Sci. U.S.A. 105, 1751–1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nin F., Hibino H., Murakami S., Suzuki T., Hisa Y., Kurachi Y. (2012) Computational model of a circulation current that controls electrochemical properties in the mammalian cochlea. Proc. Natl. Acad. Sci. U.S.A. 109, 9191–9196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Giebisch G. (2001) Renal potassium channels: function, regulation, and structure. Kidney Int. 60, 436–445 [DOI] [PubMed] [Google Scholar]
- 7. Gutman G. A., Chandy K. G., Grissmer S., Lazdunski M., McKinnon D., Pardo L. A., Robertson G. A., Rudy B., Sanguinetti M. C., Stühmer W., Wang X. (2005) International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol. Rev. 57, 473–508 [DOI] [PubMed] [Google Scholar]
- 8. Wulff H., Castle N. A., Pardo L. A. (2009) Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov. 8, 982–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li Y., Um S. Y., McDonald T. V. (2006) Voltage-gated potassium channels: regulation by accessory subunits. Neuroscientist 12, 199–210 [DOI] [PubMed] [Google Scholar]
- 10. Sanguinetti M. C. (2000) Long QT syndrome: ionic basis and arrhythmia mechanism in long QT syndrome type 1. J. Cardiovasc. Electrophysiol. 11, 710–712 [DOI] [PubMed] [Google Scholar]
- 11. McCrossan Z. A., Abbott G. W. (2004) The MinK-related peptides. Neuropharmacology 47, 787–821 [DOI] [PubMed] [Google Scholar]
- 12. Blumenthal E. M., Kaczmarek L. K. (1994) The minK potassium channel exists in functional and nonfunctional forms when expressed in the plasma membrane of Xenopus oocytes. J. Neurosci. 14, 3097–3105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang K. W., Goldstein S. A. (1995) Subunit composition of minK potassium channels. Neuron 14, 1303–1309 [DOI] [PubMed] [Google Scholar]
- 14. Sanguinetti M. C., Jurkiewicz N. K. (1990) Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J. Gen. Physiol. 96, 195–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barhanin J., Lesage F., Guillemare E., Fink M., Lazdunski M., Romey G. (1996) K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 384, 78–80 [DOI] [PubMed] [Google Scholar]
- 16. Vetter D. E., Mann J. R., Wangemann P., Liu J., McLaughlin K. J., Lesage F., Marcus D. C., Lazdunski M., Heinemann S. F., Barhanin J. (1996) Inner ear defects induced by null mutation of the isk gene. Neuron 17, 1251–1264 [DOI] [PubMed] [Google Scholar]
- 17. Tyson J., Tranebjaerg L., Bellman S., Wren C., Taylor J. F., Bathen J., Aslaksen B., Sørland S. J., Lund O., Malcolm S., Pembrey M., Bhattacharya S., Bitner-Glindzicz M. (1997) IsK and KvLQT1: mutation in either of the two subunits of the slow component of the delayed rectifier potassium channel can cause Jervell and Lange-Nielsen syndrome. Hum. Mol. Genet. 6, 2179–2185 [DOI] [PubMed] [Google Scholar]
- 18. Splawski I., Tristani-Firouzi M., Lehmann M. H., Sanguinetti M. C., Keating M. T. (1997) Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat. Genet. 17, 338–340 [DOI] [PubMed] [Google Scholar]
- 19. Schulze-Bahr E., Wang Q., Wedekind H., Haverkamp W., Chen Q., Sun Y., Rubie C., Hördt M., Towbin J. A., Borggrefe M., Assmann G., Qu X., Somberg J. C., Breithardt G., Oberti C., Funke H. (1997) KCNE1 mutations cause Jervell and Lange-Nielsen syndrome. Nat. Genet. 17, 267–268 [DOI] [PubMed] [Google Scholar]
- 20. Sanguinetti M. C., Curran M. E., Zou A., Shen J., Spector P. S., Atkinson D. L., Keating M. T. (1996) Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 384, 80–83 [DOI] [PubMed] [Google Scholar]
- 21. Schroeder B. C., Waldegger S., Fehr S., Bleich M., Warth R., Greger R., Jentsch T. J. (2000) A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature 403, 196–199 [DOI] [PubMed] [Google Scholar]
- 22. Abbott G. W., Goldstein S. A. (2001) Potassium channel subunits encoded by the KCNE gene family: physiology and pathophysiology of the MinK-related peptides (MiRPs). Mol. Interv. 1, 95–107 [PubMed] [Google Scholar]
- 23. Doi K., Sato T., Kuramasu T., Hibino H., Kitahara T., Horii A., Matsushiro N., Fuse Y., Kubo T. (2005) Meniere's disease is associated with single nucleotide polymorphisms in the human potassium channel genes, KCNE1 and KCNE3. ORL J. Otorhinolaryngol. Relat. Spec. 67, 289–293 [DOI] [PubMed] [Google Scholar]
- 24. Sand P. G., Langguth B., Kleinjung T. (2011) Deep resequencing of the voltage-gated potassium channel subunit KCNE3 gene in chronic tinnitus. Behav. Brain Funct. 7, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lv P., Wei D., Yamoah E. N. (2010) Kv7-type channel currents in spiral ganglion neurons: involvement in sensorineural hearing loss. J. Biol. Chem. 285, 34699–34707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Valenzuela D. M., Murphy A. J., Frendewey D., Gale N. W., Economides A. N., Auerbach W., Poueymirou W. T., Adams N. C., Rojas J., Yasenchak J., Chernomorsky R., Boucher M., Elsasser A. L., Esau L., Zheng J., Griffiths J. A., Wang X., Su H., Xue Y., Dominguez M. G., Noguera I., Torres R., Macdonald L. E., Stewart A. F., DeChiara T. M., Yancopoulos G. D. (2003) High-throughput engineering of the mouse genome coupled with high-resolution expression analysis. Nat. Biotechnol. 21, 652–659 [DOI] [PubMed] [Google Scholar]
- 27. Yamoah E. N., Lumpkin E. A., Dumont R. A., Smith P. J., Hudspeth A. J., Gillespie P. G. (1998) Plasma membrane Ca2+-ATPase extrudes Ca2+ from hair cell stereocilia. J. Neurosci. 18, 610–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim H. J., Lv P., Sihn C. R., Yamoah E. N. (2011) Cellular and molecular mechanisms of autosomal dominant form of progressive hearing loss, DFNA2. J. Biol. Chem. 286, 1517–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lv P., Sihn C. R., Wang W., Shen H., Kim H. J., Rocha-Sanchez S. M., Yamoah E. N. (2012) Posthearing Ca2+ currents and their roles in shaping the different modes of firing of spiral ganglion neurons. J. Neurosci. 32, 16314–16330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Strutz-Seebohm N., Seebohm G., Fedorenko O., Baltaev R., Engel J., Knirsch M., Lang F. (2006) Functional coassembly of KCNQ4 with KCNE-β-subunits in Xenopus oocytes. Cell. Physiol. Biochem. 18, 57–66 [DOI] [PubMed] [Google Scholar]
- 31. Sonntag M., Englitz B., Typlt M., Rübsamen R. (2011) The calyx of held develops adult-like dynamics and reliability by hearing onset in the mouse in vivo. J. Neurosci. 31, 6699–6709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang Z., Dou Y., Goodchild S. J., Es-Salah-Lamoureux Z., Fedida D. (2013) Components of gating charge movement and S4 voltage-sensor exposure during activation of hERG channels. J. Gen. Physiol. 141, 431–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lewis A., McCrossan Z. A., Abbott G. W. (2004) MinK, MiRP1, and MiRP2 diversify Kv3.1 and Kv3.2 potassium channel gating. J. Biol. Chem. 279, 7884–7892 [DOI] [PubMed] [Google Scholar]
- 34. Wu T., Lv P., Kim H. J., Yamoah E. N., Nuttall A. L. (2010) Effect of salicylate on KCNQ4 of the guinea pig outer hair cell. J. Neurophysiol. 103, 1969–1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Adamson C. L., Reid M. A., Davis R. L. (2002) Opposite actions of brain-derived neurotrophic factor and neurotrophin-3 on firing features and ion channel composition of murine spiral ganglion neurons. J. Neurosci. 22, 1385–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Adamson C. L., Reid M. A., Mo Z. L., Bowne-English J., Davis R. L. (2002) Firing features and potassium channel content of murine spiral ganglion neurons vary with cochlear location. J. Comp. Neurol. 447, 331–350 [DOI] [PubMed] [Google Scholar]
- 37. de Castro M. P., Aránega A., Franco D. (2006) Protein distribution of Kcnq1, Kcnh2, and Kcne3 potassium channel subunits during mouse embryonic development. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 288, 304–315 [DOI] [PubMed] [Google Scholar]
- 38. Wang H. S., Pan Z., Shi W., Brown B. S., Wymore R. S., Cohen I. S., Dixon J. E., McKinnon D. (1998) KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282, 1890–1893 [DOI] [PubMed] [Google Scholar]
- 39. Kubisch C., Schroeder B. C., Friedrich T., Lütjohann B., El-Amraoui A., Marlin S., Petit C., Jentsch T. J. (1999) KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell 96, 437–446 [DOI] [PubMed] [Google Scholar]
- 40. Jentsch T. J. (2000) Neuronal KCNQ potassium channels: physiology and role in disease. Nat. Rev. Neurosci. 1, 21–30 [DOI] [PubMed] [Google Scholar]
- 41. Tinel N., Diochot S., Borsotto M., Lazdunski M., Barhanin J. (2000) KCNE2 confers background current characteristics to the cardiac KCNQ1 potassium channel. EMBO J. 19, 6326–6330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Heitzmann D., Grahammer F., von Hahn T., Schmitt-Gräff A., Romeo E., Nitschke R., Gerlach U., Lang H. J., Verrey F., Barhanin J., Warth R. (2004) Heteromeric KCNE2/KCNQ1 potassium channels in the luminal membrane of gastric parietal cells. J. Physiol. 561, 547–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Roepke T. K., Anantharam A., Kirchhoff P., Busque S. M., Young J. B., Geibel J. P., Lerner D. J., Abbott G. W. (2006) The KCNE2 potassium channel ancillary subunit is essential for gastric acid secretion. J. Biol. Chem. 281, 23740–23747 [DOI] [PubMed] [Google Scholar]
- 44. Panaghie G., Abbott G. W. (2007) The role of S4 charges in voltage-dependent and voltage-independent KCNQ1 potassium channel complexes. J. Gen. Physiol. 129, 121–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Neyroud N., Tesson F., Denjoy I., Leibovici M., Donger C., Barhanin J., Fauré S., Gary F., Coumel P., Petit C., Schwartz K., Guicheney P. (1997) A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat. Genet. 15, 186–189 [DOI] [PubMed] [Google Scholar]
- 46. Oliver D., Knipper M., Derst C., Fakler B. (2003) Resting potential and submembrane calcium concentration of inner hair cells in the isolated mouse cochlea are set by KCNQ-type potassium channels. J. Neurosci. 23, 2141–2149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Beisel K. W., Rocha-Sanchez S. M., Morris K. A., Nie L., Feng F., Kachar B., Yamoah E. N., Fritzsch B. (2005) Differential expression of KCNQ4 in inner hair cells and sensory neurons is the basis of progressive high-frequency hearing loss. J. Neurosci. 25, 9285–9293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Werner L. A., Gray L. (1998) in Development of the Auditory System (Rubel E. W., Popper A. N., Fay R. R., eds) pp. 12–79, Springer-Verlag, New York [Google Scholar]
- 49. Romand R. (1983) Development in the frequency selectivity of auditory nerve fibers in the kitten. Neurosci. Lett. 35, 271–276 [DOI] [PubMed] [Google Scholar]