

Background: AICAr is a potent anti-proliferative compound, but its cellular effects are poorly characterized.
Results: Yeast and human AICAr transporters were identified and found to be critical for AICAr sensitivity.
Conclusion: AICAr uptake in yeast and mammalian cells occurs through nucleoside transport systems.
Significance: AICAr uptake occurs differently in yeast and mammalian cells.
Keywords: AMP-activated Kinase (AMPK), Nicotinamide, Nucleoside/Nucleotide Transport, Thiamine, Transporter
Abstract
5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAr) is the precursor of the active monophosphate form (AICAR), a small molecule with potent anti-proliferative and low energy mimetic properties. The molecular bases for AICAR toxicity at the cellular level are poorly understood. Here, we report the isolation and characterization of several yeast AICAr-hypersensitive mutants. Identification of the cognate genes allowed us to establish that thiamine transporters Thi7 and Thi72 can efficiently take up AICAr under conditions where they are overexpressed. We establish that, under standard growth conditions, Nrt1, the nicotinamide riboside carrier, is the major AICAr transporter in yeast. A study of AICAR accumulation in human cells revealed substantial disparities among cell lines and confirmed that AICAr enters cells via purine nucleoside transporters. Together, our results point to significant differences between yeast and human cells for both AICAr uptake and AICAR accumulation.
Introduction
Amino-imidazole carboxamide ribonucleotide monophosphate (AICAR)3 is a precursor of AMP in the purine de novo biosynthesis pathway and as such is naturally present in cells at low concentrations (micro molar range) (1). At higher concentrations, AICAR is a potent low energy mimetic that stimulates AMP-activated protein kinase (AMPK) by mimicking activation by AMP (2, 3). In vivo, this AMPK activating effect was shown to increase the endurance of sedentary mice (4). Because of its spectacular metabolic effects, AICAR is suspected to be used as a doping agent and since 2011 appears on the list of prohibited substances of the World Anti-Doping Agency. At high concentrations, AICAR induces cell cycle arrest and/or apoptosis (5). Importantly, it was found that AICAR-induced apoptosis was higher in trisomic mouse embryonic fibroblasts than in their euploid counterpart (6). Because most solid tumor cells are aneuploid, i.e. have an incorrect number of chromosomes, AICAR is a promising anti-tumor molecule.
AICAR is provided to mammalian cells in its riboside precursor form, AICAr, which is internalized and metabolized to AICAR monophosphate (7). AICAr was proposed to enter the cell through adenosine transporter(s) because its effects are reversed by dipyridamole, an adenosine transporter inhibitor (8, 9). The cognate AICAr transporters have not yet been identified. Once it has been internalized, AICAr is converted to its 5′-monophosphate form by adenosine kinase (1, 10). Importantly, the effects of AICAr on mammalian cells are generally fully reversed by inhibition of adenosine kinase activity with drugs such as 5-iodotubercidin, establishing that, in most cases, AICAr has to be metabolized to AICAR monophosphate to be active (as expected for an AMP-mimetic). Similarly, in yeast, the AICAr-induced transcriptional effects are abolished by a mutation in the adenosine kinase gene (ADO1), and AICAr toxicity is eliminated when it cannot be metabolized to the monophosphate form (1).
Although many effects of AICAR are at least partially dependent on AMPK, in many cases the effects of AICAR are AMPK-independent (11–15), suggesting the existence of additional AICAR targets. Interestingly, in yeast the AMPK homolog named Snf1 appears unaffected by AICAR (16). This likely reflects the fact that Snf1, contrary to its mammalian counterparts, is activated by ADP and not AMP (17). Yeast is therefore a very promising model to identify new AICAR targets and decipher the molecular mechanisms connecting AICAR monophosphate accumulation to its toxic effects. Here, we used a genetic approach to explore the molecular bases of AICAr sensitivity in this simple eukaryotic model. Our results revealed a link between AICAr sensitivity and thiamine metabolism and allowed us to identify Nrt1 as the major AICAr transporter in yeast.
EXPERIMENTAL PROCEDURES
Yeast Media and Strains
SD is a synthetic minimal medium containing 5% ammonium sulfate, 0.67% yeast nitrogen base (Difco), 2% glucose. SDcasaW is SD medium supplemented with 0.2% casamino acids (Difco) and tryptophan (0.2 mm). When indicated, adenine (0.3 mm) and/or uracil (0.3 mm) were added in SDcasaW medium, resulting in a medium named SDcasaWA (+ adenine), and SDcasaWAU (+ adenine + uracil). SC medium was prepared as described by Sherman et al. (18). SC complete medium is SC medium supplemented with adenine (0.3 mm), uracil (0.3 mm), histidine (0.06 mm), leucine (0.4 mm), lysine (0.06 mm), and tryptophan (0.2 mm). Yeast strains (see Table 1) belong to, or are derived from, a set of disrupted strains isogenic to BY4741 or BY4742 purchased from Euroscarf. Multimutant strains were obtained by crossing, sporulation, and micromanipulation of meiosis progeny.
TABLE 1.
Yeast strains
| Strain name | Genotype |
|---|---|
| BY4741 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 |
| BY4742 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 |
| Y175 | MATa his3Δ1 leu2Δ0 ura3Δ0 |
| Y2660 | MATa ade8::kanMX4 his1::kanMX4 his3Δ1 leu2Δ0 ura3Δ0 |
| Y2950 | MATα ade16::kanMX4 ade17::kanMX4 ade8::kanMX4 his1::kanMX4 his3Δ1 leu2Δ0 ura3Δ0 |
| Y3188 | MATα ade16::kanMX4 ade17::kanMX4 ade8::kanMX4 his1::kanMX4 ado1::LEU2 his3Δ1 leu2Δ0 ura3Δ0 |
| Y6986 | MATα ade16::kanMX4 ade17::kanMX4 ade8::kanMX4 his1::kanMX4 his3Δ1::HIS3-LEU2 leu2Δ0 ura3Δ0 |
| Y7242 | MATa ade16::kanMX4 ade17::kanMX4 ade8::kanMX4 his1::kanMX4 his3Δ1::HIS3-URA3 leu2Δ0 ura3Δ0 |
| Y7314 | MATα pdc2 (L634S) ade16::kanMX4 ade17::kanMX4 ade8::kanMX4 his1::kanMX4 his3Δ1::HIS3-LEU2 leu2Δ0 ura3Δ0 |
| Y7321 | MATα thi3 (S402F) ade16::kanMX4 ade17::kanMX4 ade8::kanMX4 his1::kanMX4 his3Δ1::HIS3-LEU2 leu2Δ0 ura3Δ0 |
| Y7506 | MATa thi80 (L90P) ade16::kanMX4 ade17::kanMX4 ade8::kanMX4 his1::kanMX4 his3Δ1::HIS3-URA3 leu2Δ0 ura3Δ0 |
| Y8750 | MATa thi7::KanMX4 ade16::kanMX4 ade17::kanMX4 ade8::kanMX4 his1::kanMX4 his3Δ1 leu2Δ0 ura3Δ0 |
| Y8755 | MATa thi72::KanMX4 ade16::kanMX4 ade17::kanMX4 ade8::kanMX4 his1::kanMX4 his3Δ1 leu2Δ0 ura3Δ0 |
| Y8843 | MATa pdc2 (L634S) ade16::kanMX4 ade17::kanMX4 ade8::kanMX4 his1::kanMX4 his3Δ1::HIS3-URA3 leu2Δ0 ura3Δ0 |
| Y8845 | MATα thi80 (L90P) ade16::kanMX4 ade17::kanMX4 ade8::kanMX4 his1::kanMX4 his3Δ1::HIS3-URA3 leu2Δ0 ura3Δ0 |
| Y8848 | MATa thi3 (S402F) ade16::kanMX4 ade17::kanMX4 ade8::kanMX4 his1::kanMX4 his3Δ1::HIS3-URA3 leu2Δ0 ura3Δ0 |
| Y9116 | MATα nrt1::KanMX4 ade16::kanMX4 ade17::kanMX4 ade8::kanMX4 his1::kanMX4 his3Δ1 leu2Δ0 ura3Δ0 lys2Δ0 |
| Y9188 | MATa nrt1::KanMX4 thi7::KanMX4 thi72::KanMX4 ade16::kanMX4 ade17::kanMX4 ade8::kanMX4 his1::kanMX4 his3Δ1 leu2Δ0 ura3Δ0 |
| Y9437 | MATa thi80 (L90P) nrt1::KanMX4 ade16::kanMX4 ade17::kanMX4 ade8::kanMX4 his1::kanMX4 his3Δ1::HIS3-URA3 leu2Δ0 ura3Δ0 |
Cell Culture and Proliferation Assay
U87 (HTB-14) and A549 (CCL-185) cells were from ATCC. SF188 and SF126 cells were a generous gift from M. Czabanka (Charité Universitätsmedizin, Berlin, Germany). NHA/TS cells were kindly provided by K. Sasai and S. Tanaka (Hokkaido University, Sapporo, Japan). Huh7 cells were given by E. Chevet (University of Bordeaux, France). Wild-type or knock-out mouse embryonic fibroblasts (MEFs) for both AMPKα1 and AMPKα2 subunits (AMPKα1/α2 KO) were generously provided by B. Viollet (Cochin Institute, INSERM, Paris, France). The cells were grown at 37 °C, 5% CO2 in complete medium: DMEM containing 4.5 g/liter glucose and supplemented with 10% FBS, l-glutamine, and antibiotics. The WST-1 cell proliferation assay was performed in 96-well plates according to the manufacturer's instructions (Roche). Briefly, the cells were plated at 5,000 cells/cm2 in 100 μl of complete culture medium. After 24 h of incubation, AICAr and/or inhibitors were added in fresh medium at the indicated concentrations. WST-1 assay was performed after 3 days.
Plasmids
The THI80 centromeric plasmid (p4878) was obtained by PCR amplification of the THI80 gene using S288C genomic DNA as template and oligonucleotides 3730 (5′-CGCGGATCCCAACAAATTAAAGCGGAGATC-3′) and 3731 (5′-CCGCTCGAGCCTTACTTTAGAATGATGACTTTAC-3′). The PCR product was digested with BamHI and XhoI and cloned in the pRS316 vector (19) digested with the same restriction enzymes. Plasmids allowing expression of the THI7 or THI72 genes under the control of a tetracycline repressible promoter were obtained by PCR amplification of the THI7 or THI72 coding regions using S288C genomic DNA as template and oligonucleotides 3176 (5′-CGCGGATCCAATATGAGTTTCGGTAGTAAAG-3′) and 3177 (5′-CCAATGCATCTAAGCAGCTTTTTCACTGGC-3′) for THI7 or oligonucleotides 3178 (5′-CGCGGATCCATTATGAGTTTCGGTACGAGAATC-3′) and 3179 (5′-CCAATGCATTTAGGCAATTTGTTTTTCACTGG-3′) for THI72. PCR products were digested with BamHI and NsiI and were cloned in the pCM189 vector (tetProm, CEN, URA3) (20) digested with BamHI and PstI, resulting in the tet-THI7 (p4678) and tet-THI72 (p4680) plasmids. The NRT1 overexpression plasmid p4926 (2μ, URA3) was constructed by PCR amplification of NRT1 using S288C genomic DNA as template and oligonucleotides 3825 (5′-CGCGGATCCCAAGACGGTTGGCAATAGGAG-3′) and 3826 (5′-CCGGAATTCCCCCACCGGATTCTCTTGC-3′). The PCR product was digested with BamHI and EcoRI and cloned in the YEpLac195 vector (21) digested with the same restriction enzymes. The NRT1 centromeric plasmid p4980 (CEN, URA3) was obtained by transferring the BamHI/EcoRI fragment containing NRT1 gene into YCpLac33 centromeric plasmid (21) opened with the same restriction enzymes. Finally, the THI7prom-LacZ fusion used for βGAL experiments (p4884) was obtained by PCR amplification of the THI7 promoter using S288C genomic DNA as template and oligonucleotides 3625 (5′-CGCGGATCCTATGACCGTGTCAAGGCATCC-3′) and 3626 (5′-ACGTCTGCAGCATATTGATATAATGCAATTGGCTC-3′). The PCR product was digested with BamHI and PstI and was cloned in the Yep367 vector (2μ, LEU2) (22) digested with the same restriction enzymes.
Isolation of AICAr Hypersensitive Mutants
To obtain AICAr-hypersensitive mutants, two ade16 ade17 ade8 his1 strains (Y6986 and Y7242) were plated on SD casa WAU medium, mutagenized with UV light for 20 s, and then grown for 72 h at 30 °C. The resulting clones (64,000) were then transferred by replica plating on the same medium containing various AICAr concentrations (1 or 2 mm at 30 °C and 0.5 or 1 mm at 37 °C). Thereby, 27 AICAr hypersensitive mutants were isolated from the two yeast strains (17 from Y6986 and 10 from Y7242), and 3 of these mutants were further studied. Each mutant was crossed with an ade16 ade17 ade8 his1 strain, and the meiotic progeny of the cross was phenotypically characterized. Genomic DNA extraction from pools of AICAr-resistant and -sensitive spores (see text for details) was carried out on cells grown to a final density of 5 × 107 cells/ml, as recommended by the genomic tips 100G kit supplier (Qiagen). Yeast genome resequencing and bioinformatics analyses were done by GATC Biotech (Konstanz, Germany). Once identified, mutations of interest in the mutant pools were verified on individual spores by Sanger sequencing (GATC Biotech).
Metabolite Extraction and Separation by Liquid Chromatography
Extraction of yeast metabolites was performed by the rapid filtration and ethanol boiling method as described (23). For mammalian cells, each extraction was done from one 78.5-cm2 Petri dish containing subconfluent cells (< 105 cells/cm2) grown in the complete medium. The cells were rapidly washed with 5 ml of ice-cold PBS, and then extraction was performed by incubation of cells for 5 min in 5 ml of ethanol (80%). The suspension was then transferred into a glass tube and incubated for 3 min at 80 °C. Samples were evaporated using a rotavapor device. The residue was suspended in sterile water (100 μl/107cells), and insoluble particles were eliminated by centrifugation (1 h, 4 °C, 21,000 × g). For both yeast and mammalian cells, metabolite separation was performed on an ICS3000 chromatography station (Dionex, Sunnyvale, CA) using a CarboPac PA1 column (250 × 2 mm; Dionex) with the 50–800 mm acetate gradient in 50 mm NaOH described in Ref. 1. Peaks were identified by their retention time as well as by co-injection with standards and/or their UV spectrum signature. Of note, intracellular AICAr concentration is hardly measurable when adenosine kinase is present, and furthermore it is often contaminated by external AICAr (especially true for mammalian cells), so we routinely used AICAR accumulation as readout of AICAr entry.
AICAr Uptake
Yeast cells were exponentially grown (<2 × 107 cells/ml) for 24 h before being harvested by centrifugation (3,200 × g, 2 min, 30 °C). The cells were then suspended (4–5 × 107 cells/ml) in 1 ml of uptake buffer (50 mm sodium citrate, pH 4.5, containing 2% glucose) prewarmed at 30 °C and were kept under agitation at this temperature. Uptake was started by the addition of [3H]AICAr (100 μm final concentration; 925 MBq/mmol; Hartmann analytic). As a function of time (up to 15 min), aliquots (300 μl of cell suspension) were harvested by filtration (nitrocellulose membranes, 0.8 μm; Millipore), filters were then washed twice with 3 ml of cold water and dried, and the retained radioactivity was determined by scintillation counting. AICAr uptake (pmol/min/107 cells) was calculated by linear regression of the [3H]AICAr incorporated as function of time.
Miscellaneous Methods
Yeast growth test and βGAL assays were performed as described in Refs. 24 and 25, respectively. Nicotinamide riboside (NmR) was prepared by dephosphorylation of nicotinamide mononucleotide (Sigma-Aldrich) as described (26).
RESULTS
Accumulation of Intracellular AICAR in Yeast
We first aimed at identifying experimental conditions allowing AICAR accumulation in yeast. Clearly, addition of AICAr (the riboside form) to the growth medium did not result in massive intracellular AICAR (the monophosphate form) accumulation in a wild-type yeast strain (Fig. 1A). This could be due to poor uptake of AICAr, inefficient metabolization to the monophosphate form by Ado1 and/or to highly active metabolization of AICAR to IMP by ATIC (AICAR transformylase IMP cyclohydrolase) (Fig. 1B). The latter possibility was evaluated by monitoring AICAR accumulation in a quadruple ade16 ade17 ade8 his1 mutant unable to synthesize AICAR and to further metabolize it to IMP, because it is blocked upstream and downstream in the pathway (Fig. 1B). By contrast to the wild-type strain, in the quadruple ade16 ade17 ade8 his1 mutant AICAr feeding led to robust AICAR accumulation (Fig. 1C). This result shows that yeast cells can take up external AICAr and accumulate AICAR in strains lacking ATIC activity. Accordingly, AICAr was toxic at high concentration in the ade16 ade17 ade8 his1 mutant but not in the ade8 his1 mutant that has wild-type ATIC activity (Fig. 1D). Of note, for an unknown reason, AICAr was significantly and reproducibly more toxic at 37 °C than at 30 °C (Fig. 1D). At low concentrations (up to 2 mm at 30 °C and 1 mm at 37 °C), the quadruple ade16 ade17 ade8 his1 mutant was resistant to AICAr (Fig. 1D). These subtoxic concentrations were hereafter used to isolate and characterize yeast mutants that are hypersensitive to AICAr. The ade16 ade17 ade8 his1 yeast strain, which can accumulate AICAR, was mutagenized, and several AICAr hypersensitive mutants that grew poorly in the presence of low concentrations of AICAr were isolated (see “Experimental Procedures” for details). Three of these mutants, corresponding to strains Y7314, Y7321, and Y7506 (Table 1), were further studied and are presented below.
FIGURE 1.

AICAR accumulation and toxicity in yeast. A, addition of extracellular AICAr does not result in a massive intracellular AICAR accumulation in yeast. After an overnight preculture, wild-type cells (Y175) were diluted and kept in exponential phase for 24 h in SDcasaWAU medium. External AICAr (5 mm) was then added (red line) or not (blue line) for 20 min, and metabolites were extracted and separated by liquid chromatography (only the 60–65-min elution time region of the chromatogram is presented). AICAR intracellular concentration was determined by measuring yeast cell volume as described (1). The inset corresponds to a zoom of the indicated region. Internal AICAR values correspond to three independent extractions, and standard deviation is indicated. B, schematic representation of purine and histidine pathways in yeast. Ade, adenine; Hypox, hypoxanthine; PM, plasma membrane; PRPP, 5-phosphoribosyl-1-pyrophosphate. Only the enzymes mentioned in the text are shown (in blue). C, intracellular AICAR accumulation is strongly increased in the ade16 ade17 ade8 his1 mutant. The cells (Y6986) were grown in SDcasaWAU medium, treated (green line) or not (orange line) with AICAr (5 mm, 20 min), and the metabolites were extracted and separated as in Fig. 1A. Internal AICAR values correspond to three independent extractions, and standard deviation is indicated. N.D., not detectable. D, effect of external AICAr on growth of the ade8 his1 (Y2660) or ade16 ade17 ade8 his1 (Quadruple; Y6986) strains. The cells were grown overnight, serially diluted, and spotted on SDcasaWAU medium containing external AICAr. Plates were imaged after an incubation of 2 days at the indicated temperature.
AICAr-hypersensitive Mutants Are Affected in Thiamine Metabolism
The Y7506 mutant strain, presenting a severe growth defect in the presence of AICAr (Fig. 2A) was backcrossed to an ade16 ade17 ade8 his1 strain. Among the spores resulting from meiosis, AICAr hypersensitivity segregated 2:2 in nine tetrads, indicating that AICAr sensitivity of this mutant is a monogenic character. Genomic DNA extracted from 10 AICAR hypersensitive spores was mixed and sequenced. In parallel, the genomes of a mix of 7 spores showing wild-type AICAr sensitivity were sequenced. Comparison of these genome sequences to the S288c reference genome allowed us to identify one single-nucleotide polymorphism only found in the genome mix from AICAr sensitive spores. This single-nucleotide polymorphism corresponded to a T269C mutation in the THI80 gene, resulting in a leucine to proline substitution at position 90 in the Thi80 protein. Sequencing of the THI80 gene in the parental strain and in the AICAr-sensitive mutant confirmed that this mutation was indeed present in the mutant and absent in the control and was the only mutation in this open reading frame. This L90P substitution was found in all 10 hypersensitive AICAr segregants and in none of the 7 spores showing wild-type resistance to AICAr (see examples spores 1–6 in Fig. 2A). The mutation was clearly recessive (Fig. 2B), and AICAr hypersensitivity could be complemented by the wild-type THI80 gene carried on a centromeric plasmid (Fig. 2C). We conclude that the L90P substitution in Thi80 results in hypersensitivity to AICAr in yeast cells. THI80 is an essential gene encoding thiamine pyrophosphokinase the enzyme synthesizing thiamine pyrophosphate from thiamine (Fig. 2D). Because THI80 is essential for viability and because the mutation is recessive, the L90P substitution most probably results in a partial loss of function that confers hypersensitivity to AICAr.
FIGURE 2.

The S90P recessive mutation in the THI80 gene is responsible for AICAr hypersensitivity of the Y7506 mutant strain. A, the thi80 mutation is genetically linked to AICAr sensitivity. The cells were grown overnight, serially diluted, and spotted on SDcasaWAU medium containing external AICAr. Parental strains ade16 ade17 ade8 his1 either mutated (Y7506) or not (control, Y7242) in the THI80 gene, and six derived spores are shown. The plates were imaged after 3 days at 37 °C. WT and mut, respectively, stand for wild-type (THI80) and mutated (thi80) versions of thiamine pyrophosphate kinase gene, as determined by sequencing. B, the thi80 (S90P) mutation leading to AICAr sensitivity is recessive. Diploid ade16 ade17 ade8 his1 strains mutated (thi80) or not (THI80) in thiamine pyrophosphate kinase gene were grown, diluted, and spotted on SDcasaWAU medium as in Fig. 2A. C, complementation by the wild-type THI80 gene. Haploid ade16 ade17 ade8 his1 strains (WT, Y7242; or thi80, Y7506) were transformed with a centromeric plasmid expressing (THI80) or not (vector) a wild-type copy of THI80. Transformants were grown, diluted, and spotted on SDcasaWA medium as in Fig. 2A. In A–C, the plates were imaged after 3 days at 37 °C. D, schematic representation of thiamine metabolism and resulting regulation of THI gene expression.
Two additional AICAr hypersensitive mutants were characterized by analogous methods. Genome sequencing revealed that the Y7321 mutant strain carried a C1205T mutation located in the THI3 gene that co-segregated perfectly with AICAr hypersensitivity in 14 spores and in none of the 13 AICAr-insensitive spores (Fig. 3A). This mutation resulted in a S402F substitution in Thi3, a regulatory protein that binds and activates the Pdc2p and Thi2p transcription factors (Fig. 2D). Finally, the Y7314 mutant strain was found to carry a T1901C mutation in the PDC2 gene co-segregating with AICAr hypersensitivity in 7 spores and in none of the 7 AICAr-insensitive spores (Fig. 3B). This mutation resulted in a L634S replacement in Pdc2, a transcriptional factor activating the thiamine metabolism regulon when thiamine pyrophosphate (the product of Thi80) is scarce (Fig. 2D). Importantly the thi3 and pdc2 heterozygote strains were still partially sensitive to AICAr, suggesting haploinsufficiency or semidominance (Fig. 3, C and D). Identification of these three mutations thus revealed a strong connection between thiamine metabolism and AICAr sensitivity.
FIGURE 3.

Mutations in THI3 and PDC2 lead to AICAr sensitivity. A and B, mutations in THI3 (A) and PDC2 (B) are linked to AICAr hypersensitivity. The cells were grown overnight, serially diluted, and spotted on SDcasaWAU medium containing external AICAr. Strains correspond to ade16 ade17 ade8 his1 parental cells mutated (thi3, Y7321; or pdc2, Y7314) or not (control, Y6986) and to derived spores. The plates were imaged after 3 days at 30 °C. WT and mut, respectively, stand for wild-type and mutated versions of either THI3 or PDC2 gene as determined by sequencing of the corresponding gene. C and D, mutations in THI3 (C) and PDC2 (D) are haploinsufficient or semidominant. Diploid ade16 ade17 ade8 his1 strains each carrying the indicated combination of mutated (thi3 or pdc2) and wild-type (THI3 or PDC2) alleles were grown, diluted, and spotted on SDcasaWAU medium. The plates were imaged after 48 h at 30 °C.
Accumulation of AICAR and Thiamine in the AICAr-hypersensitive Mutants
To get a better understanding of the mechanisms leading to AICAr hypersensitivity in these mutants, the effects of AICAr on the thi80, thi3, and pdc2 mutant strains were further examined by analytical chromatography. Mutant and control strains showed similar metabolic profiles except for intracellular content of AICAR and thiamine derivatives (Fig. 4, A and B). The major effect associated with AICAr addition in the mutants compared with the control strain was accumulation of AICAR monophosphate (Fig. 4, A and B). This result indicates that the hypersensitivity of the three mutants to AICAr is likely to be due to increased accumulation of the monophosphate derivative, which is likely to be the toxic form, as previously suggested by the absence of toxicity of AICAr in an adenosine kinase mutant (1). Remarkably, the three mutants accumulated thiamine (Fig. 4, A and B), whereas the thiamine pyrophosphate level was reduced in the thi80 mutant in good consistence with the role of Thi80 in thiamine metabolism (Fig. 2D). Together, these results indicated that both thiamine and AICAr uptake could be enhanced in these mutants. Thiamine is known to strongly interfere with adenosine uptake in yeast (27), and AICAr is structurally close to adenosine (Fig. 4C) and is metabolized by adenosine kinase (1). Hence, AICAr-hypersensitive mutations could somehow increase AICAr uptake, thereby exacerbating sensitivity to AICAr. Indeed, AICAr uptake was strongly enhanced in the three mutant strains (Fig. 4D). We thus further examined how mutations affecting Thi80, Thi3, and Pdc2 could impact on this process.
FIGURE 4.
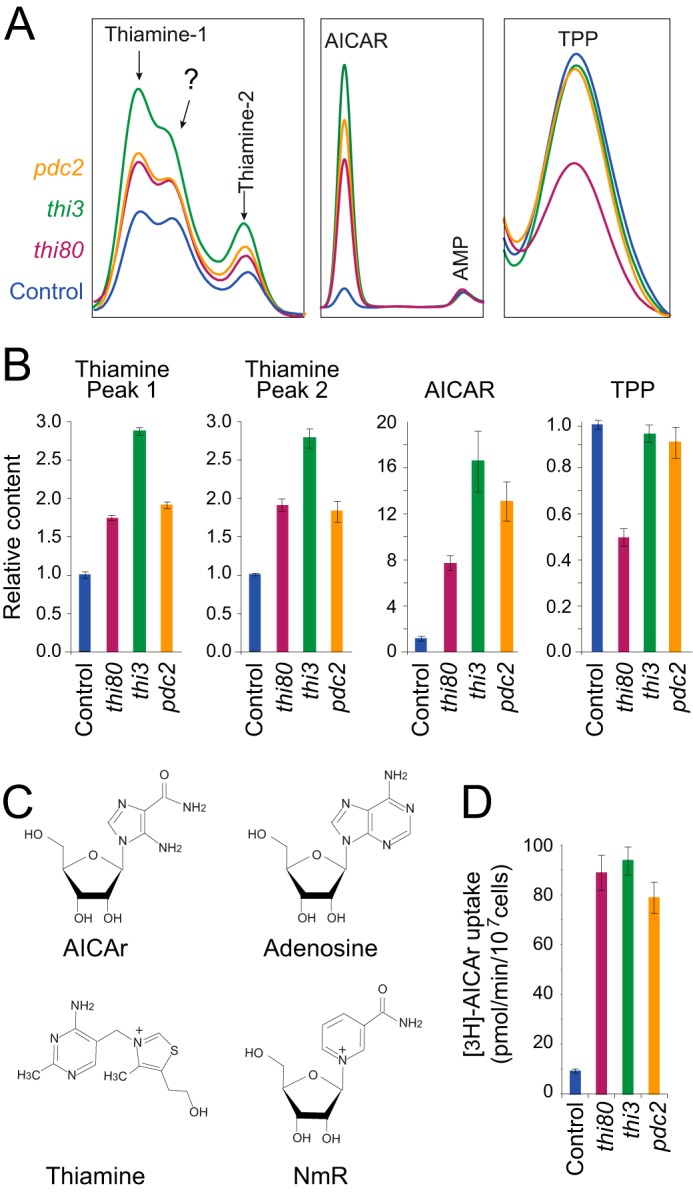
Intracellular concentration of thiamine and AICAR are higher in the AICAr-hypersensitive mutants. A, metabolic analysis of AICAR and thiamine derivatives in the AICAr-sensitive mutants. After an overnight preculture, cells were diluted and kept in exponential phase for 24 h in SDcasaWAU medium containing AICAr (0.5 mm). Metabolites were then extracted and separated by liquid chromatography. TPP, thiamine pyrophosphate. The question mark corresponds to an unidentified peak. B, relative intracellular content of thiamine and AICAR derivatives. Quantifications were determined from at least three independent metabolite extractions and separations for each strain (control, Y6986; thi80, Y7506; thi3, Y7321; pdc2, Y7314), and error bars indicate variations to the mean. For each metabolite, content of the control strain (blue bar) was set at 1. C, chemical structure of adenosine, AICAr, NmR, and thiamine. D, AICAr uptake is strongly enhanced in the AICAr-hypersensitive mutants. the cells were exponentially grown for 24 h in SDcasaWAU medium, and [3H]AICAr uptake was determined from at least two independent kinetics, as described under “Experimental Procedures.” The error bars indicate variations to the mean. Control, Y6986; thi80, Y8845; thi3, Y7321; pdc2, Y7314.
Overexpression of Thiamine Transporters Results in AICAr Hypersensitivity
It should be stressed that thiamine pyrophosphate (the product of Thi80), Thi3, and Pdc2 play a major regulating role on the transcriptional expression of the thiamine regulon (Fig. 2D), including the THI7 gene encoding the major thiamine transporter (28). Mutations isolated in our AICAr-hypersensitive mutant screen could therefore affect expression of the thiamine regulon. Indeed, a lacZ fusion driven by the THI7 promoter was found up-regulated in all three thi80, thi3, and pdc2 AICAr-hypersensitive mutants (Fig. 5A). Together our results showing stimulation of Thi7 expression and concomitant accumulation of AICAR and thiamine in the mutant cells pointed to a connection between thiamine and AICAr uptake by Thi7. Accordingly, constitutive overexpression of the thiamine transporter Thi7 enhanced AICAr sensitivity of the ade16 ade17 ade8 his1 yeast strain (Fig. 5B) and AICAR monophosphate accumulation (Fig. 5C). Similar results were obtained, although to a lesser extent, by overexpressing Thi72, a paralog of Thi7 (Fig. 5, B and C). Finally, AICAR accumulation was severely reduced in yeast cells treated with thiamine uptake inhibitors (chloroquine and amodiaquine) (29) (Fig. 5D), and AICAR toxicity in vivo was abolished by chloroquine (Fig. 5E). Together, our data show that increased expression of the thiamine transporters Thi7 and its paralog Thi72 results in robust stimulation of AICAr uptake in yeast cells. In addition, our results establish that AICAr uptake is limiting for AICAR toxicity in yeast cells and that the thiamine carriers are involved in AICAr uptake in the AICAr-hypersensitive mutants.
FIGURE 5.
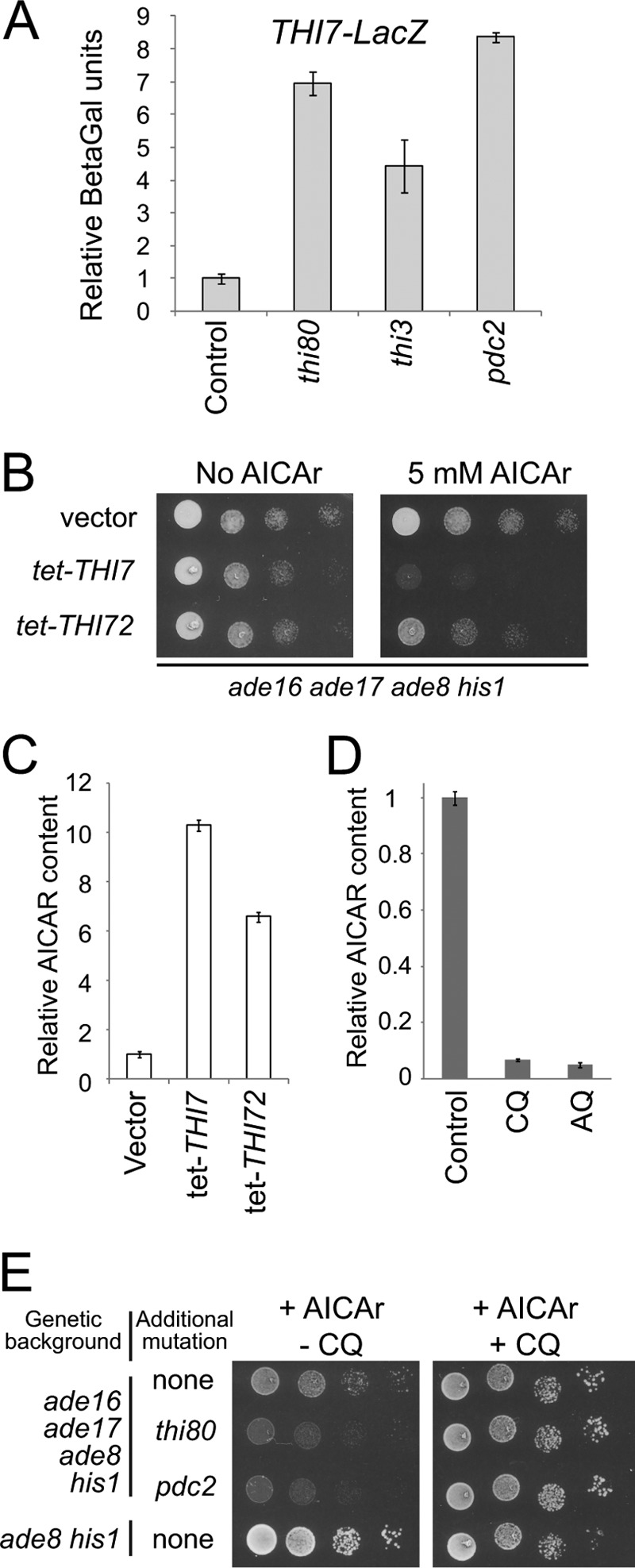
AICAR accumulation and toxicity correlates with thiamine transporter expression and activity. A, THI7 expression is increased in AICAr-sensitive mutants. The ade16 ade17 ade8 his1 cells (control, Y7242; thi80, Y8845; thi3, Y8848; pdc2, Y8843) transformed with a THI7-LacZ fusion plasmid (p4884) were grown in SC-U-L medium to A600 nm = 1. Relative β-Gal activities were measured as described under experimental procedures and are given using the Y7242 strain as reference (set up at 1). B, overexpression of thiamine transporter genes leads to AICAr hypersensitivity. The ade16 ade17 ade8 his1 (Y6986) strain was transformed with a plasmid allowing or not (vector) the overexpression of either THI7 (p4678) or THI72 (p4680) gene. Transformants were diluted and spotted on SDcasaWA medium containing or not AICAr. The plates were imaged after 2 days at 30 °C. C, intracellular AICAR accumulation is enhanced by overexpression of thiamine carriers. Transformants from Fig. 5B were grown and treated with AICAr (5 mm) for 20 min, and metabolites were extracted and separated as in Fig. 1A. Internal AICAR values correspond to three independent determinations, and error bars indicate variations to the mean. AICAR content in the strain containing the empty vector was set up at 1. D, treatment with inhibitors of thiamine transporters severely affects AICAR accumulation. Intracellular AICAR content was determined on metabolites extracts from ade16 ade17 ade8 his1 cells (Y6986) grown in SDcasaWAU medium and incubated for 20 min with external AICAr (5 mm) as in Fig. 1A. Chloroquine (CQ, 2 mm) or amodiaquine (AQ, 100 μm) were added 1 min before AICAr addition. The results correspond to at least four independent metabolite extractions, and error bars indicate variations to the mean. AICAR content found in absence of inhibitor was set up at 1. E, chloroquine abolishes AICAr sensitivity of thi80 and pdc2 mutants. The cells were grown overnight, serially diluted, and spotted on SDcasaWAU medium containing external AICAr (5 mm) and supplemented or not with chloroquine (2 mm). The plates were imaged after 2 days at 30 °C.
The Nicotinamide Riboside Transporter Nrt1 Is the Major AICAr Transporter in Yeast
Because our results indicated that AICAr sensitivity of the thi80, pdc2, and thi3 mutants was linked to enhanced AICAr uptake through Thi7 and Thi72, we evaluated the effect on AICAr toxicity of mutations in the thiamine transporter genes. Surprisingly, knock-out of thi7 or thi72 only very slightly increased resistance to AICAr (Fig. 6A), thus suggesting that AICAr could be taken up by other means. We hence explored the possible involvement of Nrt1 (previously identified as Thi71), a third transporter structurally related to Thi7, capable of taking up thiamine with low affinity (30) and known to be sensitive to chloroquine (29). Nrt1 is the major transporter of NmR (31), a precursor of NAD(H). Knock-out of nrt1 efficiently protected ade16 ade17 ade8 his1 cells from toxic effects of AICAr (Fig. 6A), thus suggesting that Nrt1 is responsible for most AICAr uptake under thiamine replete conditions (classical yeast growth media). This prediction was directly assayed by comparing AICAR accumulation in mutants lacking Thi7, Thi72, Nrt1, or all three transporters. Although AICAR accumulation was only slightly affected by the thi7 or thi72 mutations, it was strongly diminished in the nrt1 mutant and almost fully abolished in the triple mutant (Fig. 6B), thus pointing to a major role for Nrt1 in AICAr uptake.
FIGURE 6.

Nrt1 is the major AICAr transporter in yeast. A, deletion of NRT1 alleviates the growth defect of an ade16 ade17 ade8 his1 strain on AICAr. The cells were grown overnight, serially diluted, and spotted on SDcasaWAU medium containing or not external AICAr (5 mm). All strains are in an ade16 ade17 ade8 his1 genetic background and carry the indicated additional knock-out (none, Y2950; thi7, Y8750; thi72, Y8755; nrt1, Y9116). The plates were imaged after 2 days at 37 °C. B, AICAR accumulation is severely decreased in the absence of NRT1. Relative intracellular AICAR contents were determined on metabolite extracts from strains grown in SD casaWAU medium and treated with external AICAr as in Fig. 1A. The amounts of AICAR found in the ade16 ade17 ade8 his1 strain (control, Y2950; content set up at 1) and isogenic mutant strains nrt1 (Y9116), thi7 (Y8750), thi72 (Y8755), and thi7 thi72 nrt1 (Y9188) are presented. C, AICAr uptake correlates with Nrt1 expression level. Triple mutant cells (thi7 thi72 nrt1; Y9188) were transformed with vectors allowing or not (−) expression of the NRT1 gene (CEN, centromeric plasmid: p4980; 2μ, multicopy plasmid: p4926). Transformants were exponentially grown for 24 h in SDcasaWA medium, and [3H]AICAr uptake was calculated from at least two independent kinetics. The error bars indicate variations to the mean. D, overexpression of NRT1 increases AICAr sensitivity. The ade16 ade17 ade8 his1 strain (Y2950) was transformed with a multicopy plasmid (p4926) allowing overexpression of NRT1. Transformants were grown overnight, serially diluted, and spotted on SDcasaWA medium containing external AICAr (5 mm) and in the presence or not of amodiaquine (AQ, 100 μm). The plates were imaged after 2 days at 37 °C. E, AICAr hypersensitivity of thi80 mutant is independent to the presence of NRT1 gene. The cells were grown overnight, serially diluted, and spotted on SDcasaWAU medium containing or not external AICAr (2 mm). All strains are in an ade16 ade17 ade8 his1 genetic background and carry the indicated additional knock-out (thi80, Y7506; nrt1, Y9116; thi80 nrt1, Y9437). The plates were imaged after 2 days at 37 °C.
The role of Nrt1 in AICAr uptake was established by showing that reintroduction of the NRT1 gene carried on a centromeric plasmid in the septuple ade16 ade17 ade8 his1 thi7 thi72 nrt1 mutant resulted in a robust enhancement of AICAr uptake, which was even further stimulated by overexpression of NRT1 on a multicopy plasmid (Fig. 6C). Consistently, increased AICAr sensitivity was associated with overexpression of NRT1 (Fig. 6D), an effect that was fully reversed by amodiaquine (Fig. 6D). Finally, we established that the hypersensitivity of the thi3, thi80, and pdc2 mutants was not dependent on Nrt1, because these mutants were still sensitive to AICAr even in the absence of the NRT1 gene (Fig. 6E and data not shown). Together, these experiments establish that Nrt1 is the major AICAr transporter in yeast.
AICAr Uptake in Mammalian Cells
Based on our results obtained in yeast, we wished to investigate AICAr uptake in human cells. We first showed that addition of AICAr was sufficient to trigger AICAR monophosphate accumulation in several cells lines, although substantial disparities among them were observed (Fig. 7A). We then used SF188 cells, in which AICAr addition resulted in a 60-fold increase in intracellular AICAR level (Fig. 7B), to examine whether AICAR accumulation was competed by addition of thiamine or NmR, which are the substrates of yeast AICAr transporters. Clearly, neither thiamine nor NmR affected AICAR accumulation in SF188 cells (Fig. 7C). By contrast, nucleosides and in particular adenosine strongly affected AICAR accumulation (Fig. 7C), suggesting that nucleoside transporters could be involved in AICAr uptake in human cells. Previous work showed that dipyridamole, an inhibitor of nucleoside uptake, could abolish the effects of AICAr addition on neuroblastoma cells (8, 9). Indeed, we found that dipyridamole strongly inhibited AICAR accumulation (Fig. 7D). Accordingly, dipyridamole alleviates AICAr-induced cytotoxic effect on SF188 cells in a dose-dependent manner (Fig. 7E). Yeast strains each expressing one single type of human nucleoside transporter were then used to evaluate the ability of individual transporters to take up AICAr. Clearly, hENT1, hENT2, and hCNT3 could efficiently take up AICAr (Fig. 7F). These results strongly substantiate the conclusion that AICAr uptake is mediated by nucleoside transporters in mammalian cells.
FIGURE 7.

AICAr uptake occurs through adenosine transport systems in human cells. A, AICAR accumulation in various human cell lines. The cells grown in complete medium were treated with external AICAr (250 μm) for 12 h, and metabolites were extracted and separated by liquid chromatography as described under “Experimental Procedures.” The tissular origin of the different human cell lines is as follows: SF188, pediatric glioblastoma; U87 and SF126, adult glioblastomas; NHA/TS, immortalized astrocytes; A549, lung carcinoma; Huh7, hepatocellular carcinoma. B, SF188 cells accumulate AICAR when incubated with AICAr. The cells were treated (red line) or not (blue line) with external AICAr (500 μm) for 20 min, and metabolites were extracted and separated by liquid chromatography. The inset corresponds to a zoom of the indicated region. AICAR content in the absence of external AICAr was set up at 1. C, effect of various compounds on intracellular AICAR accumulation. Relative AICAR content was determined in SF188 cells grown and treated with external AICAr as in Fig. 7B and in the presence or the absence (control, set up at 1) of potential AICAr uptake competitors (100 μm each) added 1 min before AICAr treatment (500 μm, 20 min). D, dipyridamole alleviates AICAr accumulation in SF188 cells. Relative AICAR content was determined after metabolite extractions of cells grown and treated with AICAr (500 μm, 20 min) as in Fig. 7B, but in the absence (control, set up at 1) or presence of increasing concentrations of the nucleoside transporter inhibitors dipyridamole (DP) added 1 min before AICAr. E, dipyridamole protects SF188 cells against AICAr toxicity. Cell proliferation was determined by using the WST-1 colorimetric assay on SF188 cells grown for 3 days in the absence (blue triangles) or in the presence of 0.3 mm (orange circles) or 1 mm (red circles) of external AICAr and in the presence or not of dipyridamole. WST-1 signal obtained in the absence of both AICAR and inhibitors was set up at 1. F, expression of the human nucleoside transporters hENT1, hENT2, and hCNT3 enhances AICAr uptake in yeast. Yeast cells (KY114 (38) expressing or not (control) the various human nucleoside transporters (hENT1, SLC29A1; hENT2, SLC29A2; hCNT2, SLC28A2; hCNT3, SLC28A3)) were exponentially grown for 24 h in SDcasaWA medium prior to [3H]AICAr uptake measurements determined as in Fig. 4D. A–F, in all panels, values correspond to at least three independent determinations, and error bars indicate variations to the mean.
Importantly, neither AICAR accumulation (Fig. 8A) nor AICAR effects on cell proliferation (Fig. 8B) were affected in AMPKα1/α2 double knock-out murine embryonic fibroblasts, when compared with wild-type cells. Accordingly, AICAR toxicity in these cell lines was comparable and was similarly suppressed by dipyridamole treatment (Fig. 8, C and D). Together, these results demonstrate that the anti-proliferative effects of AICAR are not AMPK-dependent.
FIGURE 8.
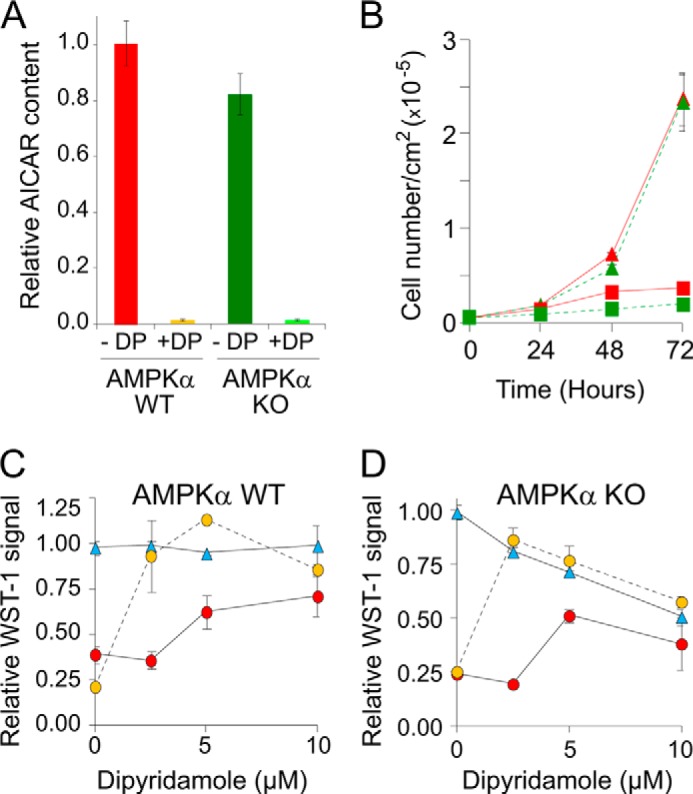
AICAr uptake and AICAR toxicity are AMPK-independent in mouse fibroblasts. A, the AMPK catalytic subunits α1 and α2 are not required for AICAR accumulation in MEFs. Relative AICAR content was determined in MEF double knock-out (AMPKα KO stands for AMPKα1/α2 KO) or not (AMPKα WT) in AMPKα genes. The cells were grown and treated with external AICAr as in Fig. 7B and in the presence (+DP) or the absence (−DP) of dipyridamole (2.5 μm) added 1 min before AICAr treatment (500 μm, 20 min). AICAR content was set up at 1 for WT MEF in the absence of DP. B, AICAR effect on cell proliferation is not altered in the absence of AMPK-α1 and -α2 catalytic subunits. Cell proliferation was measured using WT cells (red curves) or AMPKα1/α2 KO cells (green curves) MEF grown for 3 days in the presence (squares) or the absence (triangles) of AICAr (1 mm). C and D, dipyridamole protects MEF against AICAr toxicity in an AMPK-independent manner. Cell proliferation was determined using the WST-1 test on wild-type and AMPKα KO MEFs grown for 3 days in the absence (blue triangles) or in the presence of either 0.3 mm (orange circles) or 1 mm (red circles) of external AICAr and in the presence or not of dipyridamole. A–D, in all panels, values correspond to at least three independent determinations, and errors bars indicate variations to the mean.
DISCUSSION
Our screen for AICAr hypersensitive mutants in yeast revealed that three mutations affecting the regulation of thiamine metabolism strongly impact on AICAR accumulation in yeast cells. Importantly, all three mutations result in up-regulation of THI7 encoding the thiamine high affinity transporter. Our results establish that overexpression of THI7 under control of a heterologous promoter indeed mimics these mutants by increasing sensitivity to AICAr and resulting in higher AICAR intracellular concentration, yet we also show that knock-out of THI7 or THI72 only slightly affects AICAr sensitivity in yeast, indicating that, under standard growth conditions (when thiamine is replete), these transporters are most probably poorly expressed and marginally contribute to AICAr uptake. We thus tested a third transporter, named Nrt1, suspected to contribute to thiamine uptake (30) and also identified as sensitive to chloroquine (29), a drug that we have shown to inhibit AICAr uptake in yeast (Fig. 5D). Clearly, deletion of NRT1 resulted in robust resistance to AICAr and impaired AICAR accumulation, suggesting that Nrt1, by opposition to Thi7, is functional under standard growth conditions. We conclude that Nrt1, the main NmR transporter in yeast (31), is also the major AICAr transporter under standard growth conditions.
Accumulation of intracellular AICAR in yeast and mammalian cells was clearly very different. Indeed, by contrast to human SF188 cells (Fig. 7A), no significant intracellular AICAR accumulation was observed in wild-type yeast cells after addition of AICAr to the growth medium (Fig. 1A). This difference between species could be due to poor AICAr uptake in yeast, to its inefficient phosphorylation to AICAR by adenosine kinase, and/or to vigorous AICAR metabolization to IMP by ATIC. Indeed, mutations in the yeast ATIC coding genes resulted in a drastic enhancement of AICAR accumulation (Fig. 1C), thus indicating that the low AICAR accumulation in the wild-type strain was, in part, due to its efficient metabolization to IMP by ATIC. By contrast, this also suggested that AICAR accumulation in mammalian cells results from the fact that ATIC was not active enough to metabolize all the AICAR synthesized from the precursor. The steady state AICAR accumulation, which reflects the balance between AICAr entry and utilization, varied significantly from one cell line to another (Fig. 7A). Importantly, in human SF188 cells, AICAr uptake was not affected by thiamine or NmR but instead was decreased in the presence of nucleosides in the growth medium. Accordingly AICAR accumulation was inhibited by dipyridamole, an inhibitor of adenosine transport, and dipyridamole significantly reduced the effect of AICAr on cell proliferation, as previously reported for undifferentiated human neuroblastoma cells (SH-SY5Y) (9). We identified three nucleoside transporters, namely hENT1, hENT2, and hCNT3, as AICAr transporters. The nucleoside transporters constitute a multiprotein family with two subgroups (32): SLC28A (three members, hCNT1–3) and SLC29A (four members, hENT1–4), with each transporter presenting various substrate specificity and sensitivity to inhibitors (32). The remaining members of the family were not able to stimulate AICAr uptake in yeast (hCNT1 and hCNT2, Fig. 7F) or are unlikely to play a major role in AICAr uptake. Indeed, hENT3 is a lysosomal protein, and hENT4 is highly resistant to dipyridamole (33). By contrast, yeast cells do not have a dedicated adenosine transport system and can only inefficiently take up adenosine (34). Interestingly thiamine is known to strongly interfere with adenosine uptake in yeast (27), as well as with entry of the nucleoside analog cordicepin (27). Thus, yeast thiamine transporters are thought to be responsible for most of adenosine uptake and are also involved in AICAr uptake (Fig. 6B). Importantly, AICAr uptake in yeast is far less efficient than it is in mammalian cells. Indeed, a 10-fold higher concentration of extracellular AICAr was required in yeast cells (5 mm) compared with human and mouse cells (0.5 mm) to achieve comparable levels of intracellular AICAR accumulation. The yeast and human AICAr uptake systems thus appear clearly distinct. In addition, the yeast AICAr transporter Nrt1 does not have any identified ortholog in human cells (31), and neither the SLC19A2 nor the SLC19A3 human thiamine transporters (35, 36) enhanced AICAr uptake when expressed in yeast.4 However, despite these differences, AICAr uptake occurs through adenosine transport systems in both yeast and human cells.
Importantly, our results also demonstrate that AICAR accumulation is normal in AMPKα1/α2 KO embryonic fibroblasts. In addition, the anti-proliferative effects of AICAR are not affected in the same cells. Thus, these effects are not AMPK-dependent. This result is in good agreement with recently published work by others (37) and uncovers the acute necessity to identify the AICAR targets that are critical for its anti-proliferative effects.
Acknowledgments
We thank Drs. Cass, Damaraju, and Sawyer for providing the yeast strain expressing the human nucleoside transporters. We also thank J. E. Gomes for helpful discussions and J. Tissot-Dupont for technical support.
This work was supported by Association de la Recherche contre le Cancer Grant SFI-2012-120-5915 (to B. D.-F.) and a grant from Ligue contre le Cancer du Périgord (to M. M.).
M. Moenner and B. Pinson, unpublished results.
- AICAR
- 5-amino-4-imidazole carboxamide ribonucleotide 5′-phosphate
- AICAr
- 5-amino-4-imidazole carboxamide ribonucleoside
- AMPK
- AMP-activated protein kinase
- ATIC
- AICAR-transformylase IMP-cyclohydrolase
- NmR
- nicotinamide riboside
- MEF
- mouse embryonic fibroblast.
REFERENCES
- 1. Hürlimann H. C., Laloo B., Simon-Kayser B., Saint-Marc C., Coulpier F., Lemoine S., Daignan-Fornier B., Pinson B. (2011) Physiological and toxic effects of purine intermediate 5-amino-4-imidazolecarboxamide ribonucleotide (AICAR) in yeast. J. Biol. Chem. 286, 30994–31002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sullivan J. E., Carey F., Carling D., Beri R. K. (1994) Characterisation of 5′-AMP-activated protein kinase in human liver using specific peptide substrates and the effects of 5′-AMP analogues on enzyme activity. Biochem. Biophys. Res. Commun. 200, 1551–1556 [DOI] [PubMed] [Google Scholar]
- 3. Day P., Sharff A., Parra L., Cleasby A., Williams M., Hörer S., Nar H., Redemann N., Tickle I., Yon J. (2007) Structure of a CBS-domain pair from the regulatory γ1 subunit of human AMPK in complex with AMP and ZMP. Acta Crystallogr. D Biol. Crystallogr. 63, 587–596 [DOI] [PubMed] [Google Scholar]
- 4. Narkar V. A., Downes M., Yu R. T., Embler E., Wang Y. X., Banayo E., Mihaylova M. M., Nelson M. C., Zou Y., Juguilon H., Kang H., Shaw R. J., Evans R. M. (2008) AMPK and PPARδ agonists are exercise mimetics. Cell 134, 405–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rattan R., Giri S., Singh A. K., Singh I. (2005) 5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranoside inhibits cancer cell proliferation in vitro and in vivo via AMP-activated protein kinase. J. Biol. Chem. 280, 39582–39593 [DOI] [PubMed] [Google Scholar]
- 6. Tang Y. C., Williams B. R., Siegel J. J., Amon A. (2011) Identification of aneuploidy-selective antiproliferation compounds. Cell 144, 499–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Corton J. M., Gillespie J. G., Hawley S. A., Hardie D. G. (1995) 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 229, 558–565 [DOI] [PubMed] [Google Scholar]
- 8. Nakamaru K., Matsumoto K., Taguchi T., Suefuji M., Murata Y., Igata M., Kawashima J., Kondo T., Motoshima H., Tsuruzoe K., Miyamura N., Toyonaga T., Araki E. (2005) AICAR, an activator of AMP-activated protein kinase, down-regulates the insulin receptor expression in HepG2 cells. Biochem. Biophys. Res. Commun. 328, 449–454 [DOI] [PubMed] [Google Scholar]
- 9. Garcia-Gil M., Pesi R., Perna S., Allegrini S., Giannecchini M., Camici M., Tozzi M. G. (2003) 5′-aminoimidazole-4-carboxamide riboside induces apoptosis in human neuroblastoma cells. Neuroscience 117, 811–820 [DOI] [PubMed] [Google Scholar]
- 10. Sabina R. L., Patterson D., Holmes E. W. (1985) 5-Amino-4-imidazolecarboxamide riboside (Z-riboside) metabolism in eukaryotic cells. J. Biol. Chem. 260, 6107–6114 [PubMed] [Google Scholar]
- 11. Guigas B., Bertrand L., Taleux N., Foretz M., Wiernsperger N., Vertommen D., Andreelli F., Viollet B., Hue L. (2006) 5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranoside and metformin inhibit hepatic glucose phosphorylation by an AMP-activated protein kinase-independent effect on glucokinase translocation. Diabetes 55, 865–874 [DOI] [PubMed] [Google Scholar]
- 12. Guigas B., Taleux N., Foretz M., Detaille D., Andreelli F., Viollet B., Hue L. (2007) AMP-activated protein kinase-independent inhibition of hepatic mitochondrial oxidative phosphorylation by AICA riboside. Biochem. J. 404, 499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jacobs R. L., Lingrell S., Dyck J. R., Vance D. E. (2007) Inhibition of hepatic phosphatidylcholine synthesis by 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside is independent of AMP-activated protein kinase activation. J. Biol. Chem. 282, 4516–4523 [DOI] [PubMed] [Google Scholar]
- 14. Kuo C. L., Ho F. M., Chang M. Y., Prakash E., Lin W. W. (2008) Inhibition of lipopolysaccharide-induced inducible nitric oxide synthase and cyclooxygenase-2 gene expression by 5-aminoimidazole-4-carboxamide riboside is independent of AMP-activated protein kinase. J. Cell Biochem. 103, 931–940 [DOI] [PubMed] [Google Scholar]
- 15. López J. M., Santidrián A. F., Campàs C., Gil J. (2003) 5-Aminoimidazole-4-carboxamide riboside induces apoptosis in Jurkat cells, but the AMP-activated protein kinase is not involved. Biochem. J. 370, 1027–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pinson B., Vaur S., Sagot I., Coulpier F., Lemoine S., Daignan-Fornier B. (2009) Metabolic intermediates selectively stimulate transcription factor interaction and modulate phosphate and purine pathways. Genes Dev. 23, 1399–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mayer F. V., Heath R., Underwood E., Sanders M. J., Carmena D., McCartney R. R., Leiper F. C., Xiao B., Jing C., Walker P. A., Haire L. F., Ogrodowicz R., Martin S. R., Schmidt M. C., Gamblin S. J., Carling D. (2011) ADP regulates SNF1, the Saccharomyces cerevisiae homolog of AMP-activated protein kinase. Cell Metab. 14, 707–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sherman F., Fink G. R., Hicks J. B. (1986) Methods in Yeast Genetics, Cold Spring Harbor, New York [Google Scholar]
- 19. Sikorski R. S., Hieter P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Garí E., Piedrafita L., Aldea M., Herrero E. (1997) A set of vectors with a tetracycline-regulatable promoter system for modulated gene expression in Saccharomyces cerevisiae. Yeast 13, 837–848 [DOI] [PubMed] [Google Scholar]
- 21. Gietz R. D., Sugino A. (1988) New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74, 527–534 [DOI] [PubMed] [Google Scholar]
- 22. Myers A. M., Tzagoloff A., Kinney D. M., Lusty C. J. (1986) Yeast shuttle and integrative vectors with multiple cloning sites suitable for construction of lacZ fusions. Gene 45, 299–310 [DOI] [PubMed] [Google Scholar]
- 23. Laporte D., Lebaudy A., Sahin A., Pinson B., Ceschin J., Daignan-Fornier B., Sagot I. (2011) Metabolic status rather than cell cycle signals control quiescence entry and exit. J. Cell Biol. 192, 949–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Saint-Marc C., Pinson B., Coulpier F., Jourdren L., Lisova O., Daignan-Fornier B. (2009) Phenotypic consequences of purine nucleotide imbalance in Saccharomyces cerevisiae. Genetics 183, 529–538, 521SI–527SI [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pinson B., Sagot I., Borne F., Gabrielsen O. S., Daignan-Fornier B. (1998) Mutations in the yeast Myb-like protein Bas1p resulting in discrimination between promoters in vivo but not in vitro. Nucleic Acids Res. 26, 3977–3985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bieganowski P., Brenner C. (2004) Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 117, 495–502 [DOI] [PubMed] [Google Scholar]
- 27. Iwashima A., Kawasaki Y., Nosaka K., Nishimura H. (1992) Effect of thiamin on cordycepin sensitivity in Saccharomyces cerevisiae. FEBS Lett. 311, 60–62 [DOI] [PubMed] [Google Scholar]
- 28. Singleton C. K. (1997) Identification and characterization of the thiamine transporter gene of Saccharomyces cerevisiae. Gene 199, 111–121 [DOI] [PubMed] [Google Scholar]
- 29. Huang Z., Srinivasan S., Zhang J., Chen K., Li Y., Li W., Quiocho F. A., Pan X. (2012) Discovering thiamine transporters as targets of chloroquine using a novel functional genomics strategy. PLoS Genet. 8, e1003083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mojzita D., Hohmann S. (2006) Pdc2 coordinates expression of the THI regulon in the yeast Saccharomyces cerevisiae. Mol. Genet. genomics 276, 147–161 [DOI] [PubMed] [Google Scholar]
- 31. Belenky P. A., Moga T. G., Brenner C. (2008) Saccharomyces cerevisiae YOR071C encodes the high affinity nicotinamide riboside transporter Nrt1. J. Biol. Chem. 283, 8075–8079 [DOI] [PubMed] [Google Scholar]
- 32. Young J. D., Yao S. Y., Baldwin J. M., Cass C. E., Baldwin S. A. (2013) The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol. Aspects Med. 34, 529–547 [DOI] [PubMed] [Google Scholar]
- 33. Wang C., Lin W., Playa H., Sun S., Cameron K., Buolamwini J. K. (2013) Dipyridamole analogs as pharmacological inhibitors of equilibrative nucleoside transporters. Identification of novel potent and selective inhibitors of the adenosine transporter function of human equilibrative nucleoside transporter 4 (hENT4). Biochem. Pharmacol. 86, 1531–1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thomas D., Becker A., Surdin-Kerjan Y. (2000) Reverse methionine biosynthesis from S-adenosylmethionine in eukaryotic cells. J. Biol. Chem. 275, 40718–40724 [DOI] [PubMed] [Google Scholar]
- 35. Oishi K., Hofmann S., Diaz G. A., Brown T., Manwani D., Ng L., Young R., Vlassara H., Ioannou Y. A., Forrest D., Gelb B. D. (2002) Targeted disruption of Slc19a2, the gene encoding the high-affinity thiamin transporter Thtr-1, causes diabetes mellitus, sensorineural deafness and megaloblastosis in mice. Hum. Mol. Genet. 11, 2951–2960 [DOI] [PubMed] [Google Scholar]
- 36. Reidling J. C., Lambrecht N., Kassir M., Said H. M. (2010) Impaired intestinal vitamin B1 (thiamin) uptake in thiamin transporter-2-deficient mice. Gastroenterology 138, 1802–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu X., Chhipa R. R., Pooya S., Wortman M., Yachyshin S., Chow L. M., Kumar A., Zhou X., Sun Y., Quinn B., McPherson C., Warnick R. E., Kendler A., Giri S., Poels J., Norga K., Viollet B., Grabowski G. A., Dasgupta B. (2014) Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc. Natl. Acad. Sci. U.S.A. 111, E435–E444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Visser F., Vickers M. F., Ng A. M., Baldwin S. A., Young J. D., Cass C. E. (2002) Mutation of residue 33 of human equilibrative nucleoside transporters 1 and 2 alters sensitivity to inhibition of transport by dilazep and dipyridamole. J. Biol. Chem. 277, 395–401 [DOI] [PubMed] [Google Scholar]