

Background: p27 inhibits cell cycle and regulates autophagy in proliferating cells. However, the role of p27-regulated autophagy in nonproliferating cells was unknown.
Results: A TAT-p27 fusion protein reduced apoptosis in metabolically stressed cardiomyocytes in vitro and in vivo by increasing autophagy.
Conclusion: p27 prevents apoptosis in metabolically stressed cardiomyocytes through autophagy.
Significance: These findings identify TAT-p27 as an autophagy-targeting therapeutic for cardiomyopathy.
Keywords: Apoptosis, Autophagy, Cardiac Metabolism, Cardiovascular Disease, Heart Failure, p27
Abstract
p27Kip1 (p27), a key regulator of cell division, has been implicated in autophagy of cancer cells. However, its role in autophagy, the evolutionarily conserved catabolic process that enables cells to remove unwanted proteins and damaged organelles, had not been examined in the heart. Here we report that ectopic delivery of a p27 fusion protein (TAT-p27) was sufficient to induce autophagy in neonatal rat ventricular cardiomyocytes in vitro, under basal conditions and after glucose deprivation. Conversely, lentivirus-delivered shRNA against p27 successfully reduced p27 levels and suppressed basal and glucose-deprived levels of autophagy in cardiomyocytes in vitro. Glucose deprivation mimics myocardial ischemia and induces apoptosis in cardiomyocytes. During glucose deprivation, TAT-p27 inhibited apoptosis, whereas down-regulation of p27 decreased survival of cardiomyocytes. However, inhibition of autophagy by pharmacological (3-methyladenine, chloroquine, or bafilomycin A1) or genetic approaches (siRNA-mediated knockdown of Atg5) sensitized cardiomyocytes to glucose deprivation-induced apoptosis, even in the presence of TAT-p27. TAT-p27 was also able to provoke greater levels of autophagy in resting and fasting cardiomyocytes in vivo. Further, TAT-p27 enhanced autophagy and repressed cardiomyocytes apoptosis, improved cardiac function, and reduced infarct size following myocardial infarction. Again, these effects were lost when cardiac autophagy in vivo was blocked by chloroquine. Taken together, these data show that p27 positively regulates cardiac autophagy in vitro and in vivo, at rest and after metabolic stress, and that TAT-p27 inhibits apoptosis by promoting autophagy in glucose-deprived cardiomyocytes in vitro and in post-myocardial infarction hearts in vivo.
Introduction
p27Kip1 (p27),2 commonly known as a tumor suppressor, is a cyclin-dependent kinase inhibitor that negatively regulates cell growth and cell division. Reduced tissue p27 levels have been found in various human cancers (1). In addition, decreased p27 levels are found in both acute and end-stage heart failure in humans (2). We showed that p27 is functionally inactivated during cardiac hypertrophy, whereas ectopic delivery of p27 inhibits cardiac hypertrophy (3, 4). We also demonstrated that following myocardial infarction (MI), rats injected with recombinant p27 protein manifest decreased cardiac apoptosis, less cardiomyocyte hypertrophy and fibrosis, less diminished cardiac function, and greater survival (4). However, the mechanisms underlying these observations were not fully determined.
Autophagy is an evolutionarily conserved cell survival mechanism used by stressed cells to degrade unwanted cytoplasmic proteins and organelles and generate energy (5). At first, cytoplasmic materials are sequestered by isolation membranes to form autophagosomes, which then fuse with lysosomes to form autolysosomes leading to the degradation of the substrates. This multistepped autophagy process is dynamically regulated and can be inhibited at different stages using chemical reagents, e.g. 3-methyladenine (3-MA) to inhibit initial sequestration and bafilomycin A1 (Baf-A1) or chloroquine (CQ) to inhibit fusion (5). Autophagy levels are usually low under basal conditions but higher in response to stimuli such as metabolic stress. Dysregulated autophagy has been found in various heart diseases, including heart failure (6). Even in the absence of pressure overload, cardiac-specific loss of autophagy-related protein 5 (ATG5) leads to cardiac hypertrophy and dysfunction (7).
Overexpression of p27 has been shown to induce autophagy in cancer cells (8, 9). However, the role of p27 in the autophagy response to metabolic stress had not been addressed in the heart. Here we examined the role of p27 in regulating autophagy in neonatal rat cardiomyocytes in vitro and in mouse hearts in vivo, under both basal and metabolic stress conditions. We find that ectopic delivery of a TAT-p27 fusion protein was able to induce autophagy in cardiomyocytes with or without glucose deprivation, both in vitro and in vivo, as determined by increased microtubule-associated protein light chain 3 (LC3) levels and puncta of the autophagosome-associated-LC3-II. Lentivirus-delivered shRNA against p27 knocked down p27 levels and suppressed basal and glucose deprivation-induced levels of autophagy. Meanwhile, TAT-p27 was able to inhibit apoptosis induced by glucose deprivation as identified by cleaved caspase 3 levels and TUNEL assay. This effect was abolished when autophagy was blocked by either chemical inhibitors of autophagy or siRNA against Atg5. We also demonstrate that TAT-p27 promoted higher levels of autophagy, inhibited apoptosis, reduced infarct scar size, and improved cardiac function in a mouse permanent left anterior descending (LAD) ligation model of MI in vivo. This effect was compromised when in vivo autophagy was interfered with by CQ. Together, these results show that p27 prevents apoptosis in metabolically stressed cardiomyocytes by promoting cardiac autophagy.
EXPERIMENTAL PROCEDURES
Primary Culture of Rat Neonatal Cardiomyocytes
Neonatal rat ventricular cardiomyocytes were isolated from 2–3-day-old Wistar rat pups as described previously (3). Hearts were dissected, minced, and enzymatically isolated with collagenase II (0.5 mg/ml; Invitrogen) and pancreatin (1 mg/ml; Sigma). The resultant cell suspension was preplated with culture medium DMEM/F-12 (Invitrogen 11320) containing 1% penicillin/streptomycin, 3 mm sodium pyruvate, 2 mm l-glutamine, 0.2% (v/v) BSA, 0.1 mm ascorbic acid, and 0.5% (v/v) insulin-transferrin-selenium (Invitrogen). Then cardiomyocytes were grown in the presence of 5% horse serum (Invitrogen) and 25 mm arabinosylcytosine (Sigma) for at least 36 h to inhibit noncardiomyocyte proliferation. For starvation experiments, the cells were incubated with glucose-free medium (Invitrogen 11966) for 24 h. TAT-p27 or TAT-β-Gal was added at the beginning of this starvation period. To assay autophagy flux, cells were pretreated with inhibitor Baf-A1 (100 nm; InvivoGen) 4 h prior to harvest for protein isolation or fixation for immunofluorescence staining. In autophagy blocking experiments, cardiomyocytes were incubated in glucose-free medium containing Baf-A1, 3-MA (10 mm; Sigma), or CQ (10 μm; InvivoGen) for 24 h.
Animal Studies
All animal experimental protocols conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (publication 85-23, revised 1996) and were in compliance with approved institutional (University Health Network, Animal Care and Use Committee) and Canadian Council on Animal Care guidelines. All mice were raised in 12-h light (6 a.m.–6 p.m.)/dark (6 p.m.–6 a.m.) cycles.
Mouse Fasting
C57BL/6J mice (Charles River) 7–10 weeks old underwent a 48-h fast from food with water provided ad libitum (10). TAT-p27 and control peptides (10 mg/kg) were injected intraperitoneally twice daily. Heart samples were fixed with fresh 4% paraformaldehyde for immunofluorescence detection or homogenized for Western blot analysis.
Experimental Model of Myocardial Infarction
LAD coronary artery ligation was performed as described (4). Animals were anesthetized with 10 mg/ml ketamine (MTC Pharmaceuticals) and 10% xylazine (10 mg/kg; Bayer), intubated, and ventilated with room air using a pressure control ventilator (Kent Scientific). Thorax and pericardium were opened, and the heart was exposed. With the use of a 7.0 silk suture (Deknatel), the LAD was ligated, the chest was closed, and the animal was allowed to recover. Sham animals underwent the identical procedure except for ligation of the coronary artery. Three days later, mice were randomly divided into four groups and received the following treatments through intraperitoneal injection: 1) TAT-β-Gal, 10 mg/kg twice daily; 2) TAT-p27, 10 mg/kg twice daily; 3) TAT-β-Gal+CQ, 60 mg/kg twice daily; 4) TAT-p27+CQ. Mice were sacrificed by cervical dislocation on day 5 (n = 3/group).
Echocardiography
M-mode echocardiography was performed as described previously (11, 12) in LAD-ligated and sham controls at three time points (see Fig. 8A): 1) day 0, pre-LAD ligation (baseline); 2) 2 days post-MI (before daily treatments with TAT-β-Gal, TAT-p27, or CQ were administered on days 3–5); and 3) 14 day after LAD ligation (9 days after treatment).
FIGURE 8.
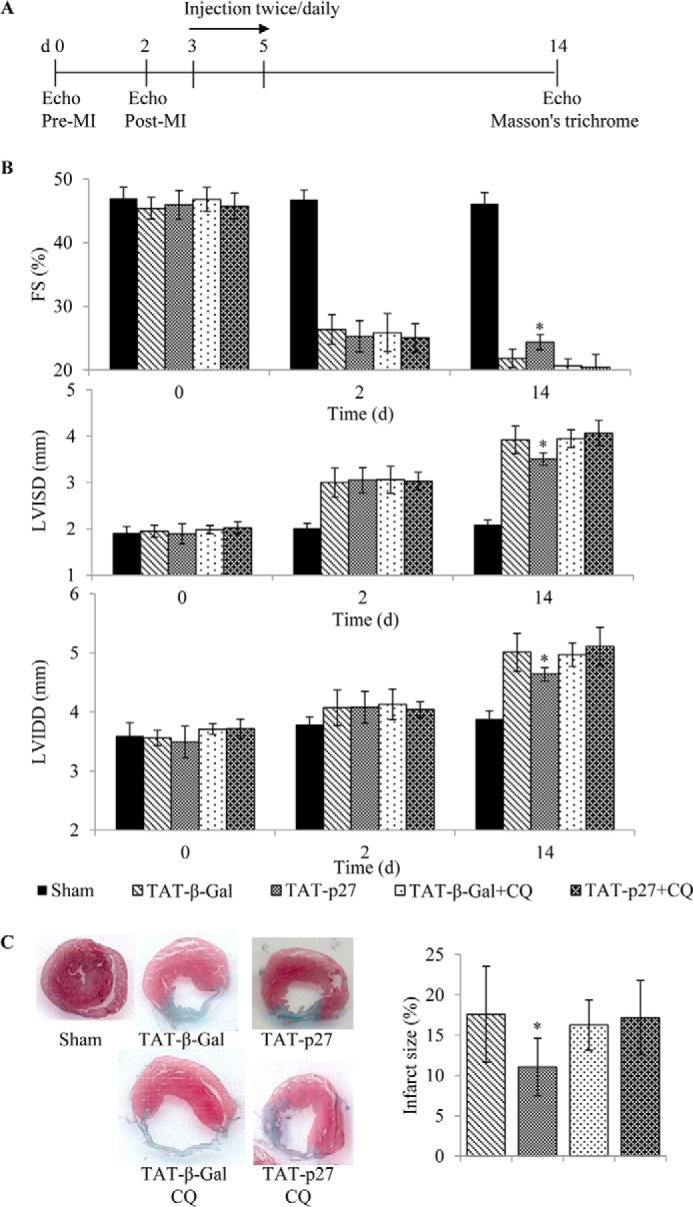
TAT-p27 reduced infarct size, prevented adverse cardiac remodeling, and improved cardiac function post-MI through enhanced autophagy. A, experimental scheme. B, fractional shortening (FS; percentage), LVISD and LVIDD (mm) were determined by M-mode echocardiography 14 days after sham or LAD ligation surgery, with the latter groups receiving treatment with TAT-β-Gal or TAT-p27 in the presence or absence of the autophagy inhibitor CQ. TAT-p27 treatment limited the reduced fractional shortening and increased LVISD and LVIDD observed post-MI in TAT-β-Gal-treated controls. This effect of TAT-p27 was lost in the presence of CQ. C, cardiac sections stained for determination of infarct area. The ability of TAT-p27 to reduce infarct size was also abolished in the presence of CQ. Both analyses have n = 5–7/group; *, p < 0.05 versus TAT-β-Gal-treated controls in the absence of CQ.
Determination of Infarct Size
Myocardial infarct size was quantified as described previously (4, 13). Briefly, 14 days post-MI (i.e. 9 days following the last treatment), mice were sacrificed, and the hearts were excised and fixed. Hearts were sectioned in 5-μm slices every 500 μm from apex to base and stained with Masson's trichrome. Photomicrographs obtained on a digital imaging system were used for morphometry. The myocardium was traced manually and measured in a blinded manner using ImageJ (1.47v). Infarct size (percentage) was calculated by dividing the sum of infarct areas from all sections by the sum of left ventricle (LV) areas from all sections (including those without any infarct).
Immunofluorescence and Confocal Microscopy
Fixed mouse hearts were incubated with 0.3 m glycine for at least 4 h and then in 30% sucrose overnight. Samples were frozen and cut in 5-μm-thick sections (LEICA CM3050S). Sections were treated with TBS containing 2% Triton X-100 and probed with antibodies for immunofluorescence detection. Cardiomyocytes were cultured in collagen-coated chamber slides (BD Biosciences). After treatments, cells were fixed in PBS with 4% formaldehyde and washed in PBSB (1×PBS with 0.2% Triton X-100, 0.5% BSA) before incubation with primary antibodies overnight at 4 °C. Dilutions for primary antibodies were rabbit anti-LC3 (1:200; Cell Signaling), mouse anti-α-actinin (1:50; Sigma), mouse anti-GATA4 (1:50; BD Biosciences). Secondary antibodies were Alexa Fluor 488 and 555 (1:100; Invitrogen). Nuclei were stained with 4′-6-diamidino-2-phenylindole (DAPI) (1 μg/ml). Apoptotic nuclei were detected by in situ terminal transferase (TdT)-mediated fluorescein dUTP nick end labeling (TUNEL assay; Roche Applied Science). The images were captured on a Zeiss Axio Observer Z1 microscope using software Zen 2011 (Zeiss).
Protein Isolation and Western Blotting
Heart homogenates and cultured rat neonatal cardiomyocytes were lysed with ice-cold radioimmuneprecipitation assay buffer (New England Biolabs) and cell lysis buffer (Cell Signaling), respectively, with protease inhibitor mix (Amersham Biosciences). Protein concentration was determined by the BCA kit (Pierce). Protein extracts were subjected to 4–20% gradient SDS-PAGE (Bio-Rad) and then transferred to PVDF membrane. Membranes were blocked with 5% nonfat dry milk in PBS containing 0.1% Tween 20 for 1 h at room temperature and then probed overnight at 4 °C for LC3 (1:1000; Cell Signaling), p27 (1:1500; Cell Signaling), p62 (1:1000; Cell Signaling), ATG5 (1:1000; Cell Signaling), cleaved caspase 3 (1:1500; Cell Signaling), β-Gal (1:5000; Millipore). Probes for GAPDH (1:10,000; Sigma) or β-tubulin (1:1500; Cell Signaling) were used to quantify loading. The corresponding horseradish peroxidase-conjugated goat anti-mouse (1:10,000) or goat anti-rabbit (1:5000) secondary antibodies (Cell Signaling) were used, respectively. The chemoluminescence signal was detected using reagents from Cell Signaling. Bands were quantified with Bio-Rad GS-800 Calibrated Densitometer using software Quantity One (4.6). Data shown represent at least three independent experiments.
siRNA and shRNA Knockdown of Gene Expression
5 nm siRNA against Atg5 or negative control (Invitrogen) was used to transfect cardiomyocytes in the presence of Lipofectamine RNAi Max (Invitrogen) according to the manufacturer's instructions. Following 24 h of siRNA incubation, cardiomyocytes were switched to glucose-free medium for 24 h. Lentiviral shRNA constructs were obtained from Sigma. The selected siRNA sequences were confirmed with BLAST searches of rat and mouse genomes to ensure that only gene p27 was targeted and that the control siRNA did not target any known genes. HEK293FT cells (Invitrogen) were used for lentivirus production and packaging. Lentiviral supernatants were concentrated 100-fold by PEG precipitation (System Biosciences). Virus titers were determined through crystal violet (Bioshop) staining of HT1080 human fibrosarcoma cells (CCL-121, ATCC) using puromycin selection. Cultured cardiomyocytes were transduced with lentiviruses at 100 plaque-forming units/cell in the presence of 4 mg/ml Polybrene (Sigma).
Statistics
Data were expressed as mean ± S.D. One-way analysis of variance followed by post hoc tests was used as appropriate. A value of p < 0.05 was considered significant.
RESULTS
TAT-p27 Induces Autophagy in Isolated Neonatal Rat Cardiomyocytes
As basal levels of autophagy are required for cellular housekeeping in the rodent heart (7), we first investigated how altered p27 levels would affect this. Recombinant TAT-p27 was used to deliver a p27 fusion protein via the HIV-1 TAT protein transduction domain (3, 14). TAT fusion proteins are capable of transducing 100% of targeted cells, including cardiomyocytes, in a concentration-dependent and receptor- and transporter-independent manner (3). These transductions were performed as we tracked autophagy activity with assays including autophagy flux. First, activation of autophagy was examined by immunoblotting for modification of microtubule-associated protein LC3, a marker for the autophagosome membrane, with cytosolic LC3-I being lipidated into active LC3-II during autophagy (15, 16). We found that LC3-II levels, which correlate with autophagosome abundance (17), were increased after addition of TAT-p27 (2.3 ± 0.27-fold) compared with untreated controls (Fig. 1A). Autophagosome abundance, as assessed by immunofluorescence for LC3-stained “dots,” was also increased after TAT-p27 treatment (Fig. 1, D and E). As LC3-II protein is also degraded during autophagy, increases in LC3-II can be due to increased synthesis and/or impaired degradation. To distinguish between these possibilities, autophagy flux was assessed using the lysosomal inhibitor Baf-A1 (17). Directly implicating TAT-p27 in the induction of autophagy, LC3-II levels and LC3-positive dots were increased by TAT-p27 in the presence of Baf-A1 (4.5 ± 0.26-fold versus 2.9 ± 0.26) (Fig. 1, A–C). Levels of p62, a polyubiquitin-binding protein degraded by autophagy, are inversely related to autophagy activity (18, 19). p62 levels were decreased by treatment with TAT-p27 but were increased by Baf-A1 (Fig. 1A). The combination of TAT-p27 and Baf-A1 resulted in an accumulation of p62, supporting the finding that TAT-p27 increased autophagy activity (Fig. 1A). Additionally, a dose-dependent increase in LC3-II and a dose-dependent decrease in p62 were found in cardiomyocytes treated with TAT-p27, but not TAT-β-Gal (Fig. 1C). Together, these results show that p27 is capable of promoting basal levels of autophagy in cardiomyocytes.
FIGURE 1.

TAT-p27 increased basal level of autophagy in rat neonatal cardiomyocytes. Cardiomyocytes were treated with TAT-p27 in the presence or absence of autophagy flux inhibitor Baf-A1. Autophagy, as measured by LC3-II expression and autophagosome formation, was monitored by Western blotting (A and B) and immunocytochemistry (D and E). A and B, densitometry-defined β-tubulin-normalized LC3-II levels were increased whereas p62 levels were decreased in TAT-p27-treated cardiomyocytes. C, dosage effect of TAT-p27 on the levels of LC3 and p62 in TAT protein-treated cardiomyocytes are shown. D, representative photomicrographs show cardiomyocytes fixed and stained for LC3 (green), α-actinin (red), and nuclei (blue). E, histogram shows the number of LC3+ dots/cell. *, p < 0.05 versus control (1); #, p < 0.05 versus Baf-A1-only group (2).
TAT-p27 Inhibits Apoptosis in Cardiomyocytes through Autophagy
Nutrient starvation is a potent inducer of autophagy in cells (20), including cardiomyocytes (6). Under the condition of glucose deprivation, delivery of TAT-p27 increased LC3-II levels compared with TAT-β-Gal-treated controls (1.5 ± 0.1-fold, p < 0.05) (Fig. 2, A and B, lanes 3 versus 1), suggesting increased autophagosome formation. These results show that p27 also enhances autophagy under metabolic stress.
FIGURE 2.

TAT-p27-reduced cardiomyocyte apoptosis following glucose deprivation is autophagy-dependent. Glucose-deprived (GD) cardiomyocytes were treated with TAT-p27 in the presence or absence of autophagy flux inhibitor Baf-A1. A and B, TAT-p27-treated glucose-deprived cardiomyocytes showed increased LC3-II levels and reduced p62 and caspase 3 cleavage compared with TAT-β-Gal (lanes 3 versus 1). However, addition of Baf-A1 increased apoptosis. *, p < 0.05 versus GD (1); #, p < 0.05 versus Baf-A1-treated (2). C, the rate of apoptosis was also determined by TUNEL staining (green) with DAPI nuclear staining (blue). GATA4 (red) is a cardiomyocyte-specific nuclear marker. Scale bar, 50 μm. D, TUNEL+ nuclei are quantified.
Glucose deprivation is also known to result in apoptosis in cardiomyocytes (21), and we consistently detected caspase 3 cleavage (Fig. 2, A and B, lanes 1) and TUNEL staining (Fig. 2, C and D) in cardiomyocytes deprived of glucose for 24 h. Treatment with TAT-p27 reduced both caspase 3 cleavage (Fig. 2, A and B, lanes 3 versus 1) and the percentage of apoptotic nuclei (Fig. 2, C and D), suggesting that TAT-p27 could repress glucose deprivation-induced apoptosis. To define whether this effect derived from TAT-p27-promoted autophagy, we used Baf-A1 to block autophagy in TAT-p27-treated cardiomyocytes during glucose deprivation. We found that inhibition of autophagy by Baf-A1 increased the rate of apoptosis, whether or not cells were treated with TAT-p27 (Fig. 2), suggesting that TAT-p27 inhibits apoptosis through autophagy.
To identify the stages of autophagy affected by TAT-p27, two more chemical inhibitors of autophagy, namely 3-MA and CQ, were also used. 3-MA is commonly used as an inhibitor for starvation-induced autophagy sequestration (17). In 3-MA-treated cardiomyocytes, the addition of TAT-p27 failed to increase LC3-II levels during glucose deprivation (Fig. 3), suggesting that TAT-p27-promoted autophagy was abolished when autophagy sequestration was blocked. In cardiomyocytes treated with CQ, which blocks the final steps of autophagy (17), we observed increased levels of LC3-II (Fig. 3C). Moreover, delivery of TAT-p27 increased LC3-II levels more than TAT-β-Gal in the presence of CQ (Fig. 3C). Both 3-MA and CQ abolished the TAT-p27 effect on apoptosis in cardiomyocytes undergoing glucose deprivation (Fig. 3), consistent with data obtained with Baf-A1 treatment (Fig. 2). Therefore, perturbations of autophagy at either early or late stages were shown to increase the rate of apoptosis in cardiomyocytes exposed to glucose deprivation, suggesting that the completion of all stages of autophagy is essential for cardiomyocytes to survive metabolic stress. As pharmacological inhibition of autophagy abolished TAT-p27-repressed cardiomyocyte apoptosis following glucose deprivation, our data suggest that TAT-p27 inhibits apoptosis by promoting cardiac autophagy and that TAT-p27 is an upstream regulator of autophagy.
FIGURE 3.

TAT-p27 reduced glucose deprivation-induced apoptosis by promoting autophagy. TAT-p27-reduced apoptosis in glucose-deprived cardiomyocytes was abolished by 3-MA or CQ. A and B, 3-MA blocks the initial autophagy step, reducing LC3-II activation and increasing cleaved caspase 3 levels even in the presence of TAT-p27. *, p < 0.05 versus glucose-deprived (1); #, p < 0.05 versus 3-MA-treated (2). C and D, CQ, as an autophagosome fusion blocker, prevents maturation of autophagy and increased LC3-II levels and caspase 3 cleavage in glucose-deprived cardiomyocytes. *, p < 0.05 versus glucose-deprived (1); #, p < 0.05 versus CQ-treated (2).
Given the possibility that chemical inhibitors of autophagy may not be target-specific (17), we also blocked autophagy using a genetic approach. siRNA knockdown of Atg5 gene products enhanced glucose deprivation-induced apoptosis and abolished the antiapoptotic effect of TAT-p27 compared with nontargeted control (Fig. 4). Together, these data indicate that TAT-p27 protects glucose-deprived cardiomyocytes from apoptosis by promoting autophagy in vitro.
FIGURE 4.

TAT-p27 fails to promote autophagy and reduce apoptosis following Atg5 knockdown. Cardiomyocytes were transfected with 5 nm Atg5 siRNA or nontargeting control siRNA (N/C) for 24 h and then deprived of glucose for a further 24 h. siRNA knockdown of Atg5 reduces glucose deprivation-induced LC3 processing (A and B) and cardiomyocytes survival (A–C) even in the presence of TAT-p27.
Endogenous p27 Is Required for Cardiomyocytes to Survive Glucose Deprivation
Reduced cardiac p27 levels have been found in both acute and end-stage heart failure in humans (2). Thus, we next investigated whether decreasing endogenous p27 affects cardiomyocyte responses to glucose deprivation. To knock down p27, we used a lentivirus-delivered shRNA against p27 (shRNAp27) with a lentiviral nontarget (NT) negative control. With shRNAp27, levels of p27 protein in cardiomyocytes were reduced by 67 ± 10% of the levels observed in NT shRNA-treated controls (p < 0.05) (Fig. 5, A and B). Knockdown of p27 was sufficient to decrease LC3-II levels by 30 ± 10% (0.7 ± 0.1-fold) compared with NT-treated cardiomyocytes (p < 0.05) (Fig. 5, A and B), suggesting that LC3-II generation is decreased in conditions of reduced p27 expression. Similar treatments in glucose-deprived cardiomyocytes resulted in similar reductions in LC3-II levels (0.4 ± 0.1-fold, p < 0.05) and dots (Fig. 5, A–D). These results suggest that p27 is required to achieve basal (and metabolically stressed) levels of autophagy in cardiomyocytes. Furthermore, we found that treatment of cardiomyocytes with shRNAp27 increased their levels of cleaved caspase 3 in response to glucose deprivation (2.0 ± 0.1 versus 1.3 ± 0.1 NT, p < 0.05) (Fig. 5, A and B), suggesting that endogenous p27 is required for cardiomyocytes to maintain autophagy and survive nutrient stress.
FIGURE 5.

Knockdown of p27 suppresses autophagy and increases apoptosis in glucose-deprived (GD) cardiomyocytes. A, Western blot shows lysates from cardiomyocytes treated with and without shRNA to knock down p27 versus control shRNA (NT) in the presence or absence of glucose. B, shRNAp27 reduced p27 and LC3-II levels and increased p62 levels. Apoptosis marker cleaved caspase 3 was increased in cardiomyocytes undergoing p27 knockdown. C, representative photomicrographs show cardiomyocytes fixed and stained for LC3 (green), α-actinin (red), and nuclei (blue). D, histogram shows the number of LC3+ dots/cell. E, TUNEL-positive nuclei are quantified. *, p < 0.05 versus control (1); #, p < 0.05 versus glucose-deprived (2).
Effects of TAT-p27 on Cardiac Autophagy in Vivo
Our in vitro data suggested that a perturbation at either early or late stages of autophagy abolished TAT-p27-inhibited apoptosis in glucose-deprived cardiomyocytes. We next examined whether TAT-p27 could protect metabolically stressed cardiomyocytes in a similar manner in vivo. First, to address whether TAT-p27 is able to induce autophagy in normal hearts in vivo, we used TAT-p27 to increase p27 levels in the heart (3). Western blotting revealed that cardiac LC3-II level increased after TAT-p27 injection (1.6 ± 0.1-fold, p < 0.05; saline: 1.0 ± 0.1, p = NS; TAT-β-Gal: 1.0 ± 0.1, p = NS) (Fig. 6A, lanes 1–4). Consistently, frozen sections showed no detectable LC3-stained dots in nontreated, saline-, or TAT-β-Gal-treated groups, with spontaneous appearance of LC3-positive dots following treatment with TAT-p27 (Fig. 6, B and C).
FIGURE 6.

TAT-p27 induces autophagy in mouse hearts in vivo. A, Western blotting for LC3 in heart homogenates revealed increased LC3-II levels in fasted versus nonfasted groups. TAT-p27 treatment further increased LC3-II levels with and without fasting. B and D, representative photomicrographs show frozen sections from nonfasted (B) or fasted (D) hearts stained for LC3 (green), α-actinin (red), and nuclei (blue). Scale bar, 20 μm. C and E, histograms show numbers of LC3+ dots/cell under nonfasted (C) and fasted (E) conditions. *, p < 0.05 versus control (1); #, p < 0.05 versus fasted (5).
A water-only diet (48 h) is known to induce autophagy in mouse hearts without any appreciable apoptosis (10, 22). This may reflect the importance of autophagy as a means for cells to survive food scarcity. In this context, we found that TAT-p27 provoked a greater induction of cardiac autophagy in mice undergoing caloric restriction for 48 h (3.0 ± 0.1-fold, p < 0.05) (Fig. 6, A, D, and E), compared with nontreated (2.3 ± 0.12-fold), saline-treated (2.2 ± 0.1, p = not significant), or TAT-β-Gal-treated groups (2.3 ± 0.2, p = not significant) (Fig. 6A, lanes 5-8). These results suggest that TAT-p27 is sufficient to induce cardiac autophagy in vivo under both basal and metabolically stressed conditions, providing a feasible way to enhance p27-promoted autophagy in vivo.
TAT-p27 Protects Post-MI Hearts by Increasing Autophagy
Fasting for 48 h produces remarkably low rates of apoptosis in the mouse heart, and most affected cells are not cardiomyocytes (22). Glucose deprivation mimics myocardial ischemia and induces apoptosis in cardiomyocytes (Figs. 2–4) (21). TAT-p27 has also been shown to provide protection in a permanent LAD ligation model of myocardial infarction (4). Therefore, we next investigated whether TAT-p27 prevented apoptosis in post-MI hearts by enhancing cardiac autophagy. We treated LAD-ligated mice with TAT-p27 and monitored autophagy levels. We found that compared with TAT-β-Gal-treated control groups, TAT-p27 increased LC3-II levels (Fig. 7A). Consistently, the levels of p62, which is degraded through autophagy and accumulates during defective autophagy (23), were decreased in TAT-p27-treated animals compared with control groups (Fig. 7A), indicating higher levels of autophagy. Correspondingly, apoptosis in TAT-p27-treated post-MI hearts was significantly lower than in control groups (Fig. 7A). To determine whether such antiapoptotic effects of TAT-p27 were dependent on autophagy, cardiac autophagy in vivo was interfered with by treatment with CQ. In the presence of CQ, no significant difference in autophagy activity was observed in TAT-p27-treated versus control-treated groups (Fig. 7B). Moreover, there was no significant difference in apoptosis between TAT-p27- and control-treated groups (Fig. 7B). Together, these data indicate that TAT-p27-inhibited apoptosis post-MI is dependent on cardiac autophagy.
FIGURE 7.

TAT-p27 reduced apoptosis post-MI by promoting autophagy. A and B, homogenates from mice undergoing sham and LAD ligation surgery, with the latter receiving treatments with TAT-p27 or TAT-β-Gal alone or in combination with the autophagy inhibitor CQ, were probed for LC3-II, p62, and cleaved caspase 3. β-Tubulin served as loading control. Compared with sham-operated controls, post-MI groups revealed higher levels of LC3-II and cleaved caspase 3 levels (*, p < 0.05 versus sham). Levels of p62 were more varied. Compared with TAT-β-Gal-treated controls, TAT-p27-treated hearts revealed higher levels of LC3-II and lower levels of p62, consistent with increased autophagy. TAT-p27-treated hearts also revealed lower levels of cleaved caspase 3 than TAT-β-Gal-treated controls, consistent with reduced apoptosis (#, p < 0.05 versus TAT-β-Gal). In the presence of CQ, treatment with TAT-p27 failed to reduce expression of the apoptosis marker cleaved caspase 3 versus TAT-β-Gal-treated controls (p = not significant).
To investigate the pathophysiological significance of these findings, we next assessed cardiac structure and function with M-mode echocardiography at 14 days post-MI (Fig. 8A). TAT-p27-treated post-MI hearts (n = 7) showed significantly greater fractional shortening than TAT-β-Gal-treated controls (n = 7) (p < 0.05, Fig. 8B), consistent with our previous report (4). However, this improvement in cardiac function attributable to TAT-p27 was lost in the presence of CQ (Fig. 8B). Treatment with TAT-p27 also prevented the increase in LV internal systolic diameter (LVISD) and LV internal diastolic diameter (LVIDD) observed in TAT-β-Gal-treated controls (Fig. 8B). This beneficial effect of TAT-p27 on LV remodeling post-MI was also abolished in the presence of the autophagy inhibitor CQ. Finally, infarct size was determined by morphometric analysis of Masson trichrome-stained cardiac tissue sections. Histologically defined infarct scar area was smaller in TAT-p27-treated animals (n = 7) than TAT-β-Gal-treated controls (n = 7) (p < 0.05, Fig. 8C). Again, this infarct-sparing effect was not seen in the presence of CQ (Fig. 8C). Together, these data suggest that TAT-p27 reduced infarct size, prevented adverse cardiac remodeling, and improved cardiac function post-MI via its ability to enhance autophagy.
Although comparisons between TAT-β-Gal versus TAT-β-Gal+CQ groups on day 5 post-MI suggested more LC3-II and cleaved caspase 3 abundance in the presence of autophagy inhibitor CQ (Fig. 7), this did not translate into larger infarct size or worse cardiac function at day 14 (Fig. 8). In other words, adverse pathophysiological consequences of blocking autophagy are not as apparent in TAT-β-Gal controls as they are in TAT-p27-treated animals.
DISCUSSION
p27 is a pleiotropic regulator of cell cycle, motility, and apoptosis in diverse cell types (24). However, prior to the present study, knowledge of its role in autophagy was confined to proliferating cell types (8, 9). Liang et al. showed in a variety of proliferating cells that 1) forced overexpression of fluorescent-tagged wild-type p27 and phosphomimetic Thr-198 to Asp-198 (T198D) p27, but not unstable Thr-198 to Ala-198 p27, is sufficient to induce autophagy; 2) that nutrient deprivation following siRNA-mediated knockdown of p27 results in apoptosis; and 3) that the nutrient-sensing LKB1-AMPK pathway stabilizes p27 through Thr-198 phosphorylation (9). However, the specific mechanisms identified by this study were examined in a diverse number of transformed or embryonic cell types in vitro and never comprehensively assessed in a single cell type or in a nonproliferating cell. Moreover, whether the observed p27-induced increases in autophagosome abundance were due to increased autophagosome formation, or decreased clearance, i.e. autophagy flux, was not assessed. Here we show, for the first time in vitro and in vivo, that p27 promotes autophagy in differentiated cardiomyocytes, a cell type with very limited if any proliferative capacity (25), with or without nutrient withdrawal, by directly increasing autophagy flux.
Having said this, p27 is not essential for autophagy. Knock-out (KO) of essential autophagy proteins ATG5, Beclin 1, and ATG7 results in embryonic lethality or death shortly after birth, a known phase of metabolic stress (i.e. neonatal starvation) (5). By contrast, p27 KO mice are viable (24) and thus capable of surviving this metabolically vulnerable phase of development. Also, compared with absent autophagy in Atg5 KO mouse embryonic fibroblasts (MEFs) (26), diminished autophagy is still evident in p27 KO MEFs (9) and in p27 knockdown cardiomyocytes (Fig. 5). Finally, glucose withdrawal was known to induce autophagy in wild-type MEFs, but not p27 KO MEFs (9), and similar levels of metabolic stress have now been shown to induce autophagy in a similar p27-dependent manner in cardiomyocytes (Fig. 5). Together, these findings demonstrate a central role for p27 in modulating autophagy in cardiomyocytes under both basal conditions and following metabolic stress.
It remains unclear whether p27 exerts these effects directly on autophagy machinery or through indirect effects via other regulators of autophagy, for example cell cycle-dependent kinases (Cdks) which are known targets of p27. Liang et al. found that siRNA-mediated depletion of Cdk2 or Cdk4 promoted autophagy in 3T3-L1 fibroblasts, but that this effect was independent of p27 (9). Given the low or absent levels of Cdks in the heart (27), the effects of p27 on cardiomyocyte autophagy are unlikely to be mediated through Cdks. As the ability of TAT-p27 to promote autophagy was blocked by both early-stage (3-MA; siRNA Atg5)- and late-stage (Baf-A1; CQ) autophagy inhibitors, p27 may be an upstream regulator of this pathway. Indeed, this interpretation finds support from a screen for direct p27 interactions performed in our laboratory, which has failed to reveal any interactions with known autophagy proteins.3 As such, future studies will be needed to identify the precise pathways that link p27 with distinct steps of the autophagy process.
Our study confirms the importance of regulating endogenous p27 levels as a means of controlling autophagy. Given that RNAi may fail to implicate genes whose activities need to be completely eliminated to show a phenotype (26), our study reveals that a ∼70% reduction of p27 expression (achieved by shRNAp27) is sufficient to reduce autophagy and increase apoptosis in glucose-deprived cardiomyocytes (Fig. 5). The importance of precise control over p27 abundance is also evident in other biological processes. Reduced p27 protein levels are believed to play a role in various human cancers (1), with p27 KO mice developing pituitary tumors and multiple organ hyperplasia (28, 29) and p27 heterozygous mice being hypersensitive to carcinogens (30). Consistently, p27 KO mice have also been shown to have increased numbers of cardiomyocytes, albeit of reduced cell size (31). Of further interest, p27 KO mice develop age-dependent cardiac hypertrophy and are hypersensitive to thoracic aortic banding (3). How much of these (cardiac) phenotypes can now be attributed to the disturbed autophagy caused by absent or insufficient p27 is a question raised by our study.
Similarly, load- and ischemia-induced cardiac remodeling are common precedents to heart failure, and autophagy has been implicated in both (6). Having previously shown that TAT-p27 prevents apoptosis and adverse cardiac remodeling in rodent models of pressure overload and ischemia (3, 4), our current study suggests that these effects depend on the ability of TAT-p27 to promote autophagy. Having said this, it remains controversial whether activation of autophagy is beneficial or detrimental in load-induced remodeling (7, 32), whereas augmenting autophagy has been more consistent in protecting against postischemic remodeling (33–35). Nakai et al. demonstrated that the cardiac-specific knock-out of ATG5 in adult mice led to cardiac hypertrophy, LV dilatation, and contractile dysfunction (7). Of interest, these phenotypes were not found early in cardiogenesis under baseline conditions, whereas these mice manifest rapid cardiac dysfunction following pressure overload (7). However, Zhu et al. reported that cardiac autophagy and pathologic remodeling due to severe thoracic aortic banding were diminished in mice haploinsufficent for Beclin 1, a protein required for autophagosome formation (32). These conflicting reports regarding the protective versus maladaptive effects of specific autophagy regulators stand in contrast to data from mouse and rat LAD occlusion model of MI. In this model, Kanamori et al. found in mouse that levels of autophagy were greatest in the infarct border zone during the first week post-MI, then greatest in remote areas of the heart during the chronic stage (3 weeks post-MI) and that blocking autophagy (with Baf-A1) exacerbated cardiac dysfunction, although enhancing autophagy (with rapamycin) reduced cardiac dysfunction and remodeling (33, 34). Consistent with this, Buss et al. reported in rat that the mTOR inhibitor everolimus also increased autophagy and prevented adverse cardiac remodeling post-MI (35).
Together, these studies support the potential for TAT-p27 (or other agents that promote cardiac autophagy) to have meaningful effects in select patient populations at risk for adverse cardiac remodeling. However, several limitations to such an approach deserve discussion. First, following MI, large numbers of cardiomyocytes rapidly undergo apoptosis, and any attempt to limit this by enhancing autophagy will require immediate delivery of such an agent. Second, to limit the possibility that antiapoptotic and proautophagic effects might increase the survival of specific tumors (36), the duration of any such therapy would have to be short. Autophagy has also been shown to promote tumorigenesis in oncogene-induced tumors (37–40). Indeed, the need to repeatedly or chronically treat subjects with such agents may only be possible if means are developed to specifically localize the therapy to the heart. Third, the ability of p27-regulated cardiac autophagy to effect whatever endogenous capacity exists to regenerate cardiomyocytes has yet to be explored. Reports of limited cell cycle re-entry in the adult human heart (41), as well as the existence of cardiac progenitor cells (42, 43), suggest the need to specifically examine the effects of an autophagy-enhancing strategy in these cell types. Finally, cardiomyocytes account for <60% of the cell types constituting the adult mouse heart (44). Whether TAT-p27 or similar agents modulate endogenous autophagy levels in nonmyocytes and to what extent this might account for their overall effects on cardiac remodeling are not currently known. Future studies aimed at overcoming the challenges of co-localizing exogenous TAT-p27 versus known fluctuations in the levels of endogenous p27 post-MI (4) with autophagy and/or apoptosis markers in specific cell types at different stages of the post-MI remodeling response will be required to determine whether the in vivo effects of TAT-p27 are specific to cardiomyocyte uptake or potentially cell autonomous. Having said that, our in vitro studies unequivocally demonstrate that p27 exerts autophagy-dependent effects on cell survival in isolated cardiomyocytes.
In summary, we have shown that TAT-p27 regulates autophagy in cardiomyocytes in vitro and in vivo, under both basal and nutrient-deprived conditions, in an autophagy-dependent manner. Together with our earlier work (3, 4), we speculate that treatment with TAT-p27 may facilitate beneficial levels of autophagy in a variety of cardiomyopathic processes. Having said this, therapeutic modulation of autophagy has not been adequately studied and may have context-dependent effects. In cardiomyocytes, both inhibition and activation of autophagy have been reported to lead to hypertrophy, with effects depending either on the models employed or the manner in which autophagy was manipulated (6). The precise mechanism(s) by which p27 activates cardiac autophagy remain unknown and require further investigations.
Acknowledgments
We thank Daniela Grothe and Jeremy Johns for technical assistance.
This work was supported by Canadian Institutes for Health Research Operating Grant MOP-89959 (to R. v. H.).
C. Antony and R. von Harsdorf, unpublished data.
- p27
- p27Kip1
- Atg5
- autophagy-related protein 5
- Baf-A1
- bafilomycin A1
- Cdk
- cell cycle-dependent kinase
- CQ
- chloroquine
- LAD
- left anterior descending artery
- LC3
- microtubule-associated protein light chain 3
- LV
- left ventricle
- LVIDD
- LV internal diastolic diameter
- LVISD
- LV internal systolic diameter
- 3-MA
- 3-methyladenine
- MEF
- mouse embryonic fibroblast
- MI
- myocardial infarction
- NT
- nontarget.
REFERENCES
- 1. Wander S. A., Zhao D., Slingerland J. M. (2011) p27: a barometer of signaling deregulation and potential predictor of response to targeted therapies. Clin. Cancer Res. 17, 12–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Burton P. B., Yacoub M. H., Barton P. J. (1999) Cyclin-dependent kinase inhibitor expression in human heart failure: a comparison with fetal development. Eur. Heart J. 20, 604–611 [DOI] [PubMed] [Google Scholar]
- 3. Hauck L., Harms C., An J., Rohne J., Gertz K., Dietz R., Endres M., von Harsdorf R. (2008) Protein kinase CK2 links extracellular growth factor signaling with the control of p27Kip1 stability in the heart. Nat. Med. 14, 315–324 [DOI] [PubMed] [Google Scholar]
- 4. Konecny F., Zou J., Husain M., von Harsdorf R. (2012) Post-myocardial infarct p27 fusion protein intravenous delivery averts adverse remodelling and improves heart function and survival in rodents. Cardiovasc. Res. 94, 492–500 [DOI] [PubMed] [Google Scholar]
- 5. Mizushima N., Levine B., Cuervo A. M., Klionsky D. J. (2008) Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chiong M., Wang Z. V., Pedrozo Z., Cao D. J., Troncoso R., Ibacache M., Criollo A., Nemchenko A., Hill J. A., Lavandero S. (2011) Cardiomyocyte death: mechanisms and translational implications. Cell Death Dis. 2, e244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nakai A., Yamaguchi O., Takeda T., Higuchi Y., Hikoso S., Taniike M., Omiya S., Mizote I., Matsumura Y., Asahi M., Nishida K., Hori M., Mizushima N., Otsu K. (2007) The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 13, 619–624 [DOI] [PubMed] [Google Scholar]
- 8. Komata T., Kanzawa T., Takeuchi H., Germano I. M., Schreiber M., Kondo Y., Kondo S. (2003) Antitumour effect of cyclin-dependent kinase inhibitors (p16INK4A), p18INK4C, p19INK4D, p21WAF1/CIP1 and p27KIP1) on malignant glioma cells. Br. J. Cancer 88, 1277–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liang J., Shao S. H., Xu Z. X., Hennessy B., Ding Z., Larrea M., Kondo S., Dumont D. J., Gutterman J. U., Walker C. L., Slingerland J. M., Mills G. B. (2007) The energy sensing LKB1-AMPK pathway regulates p27Kip1 phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 9, 218–224 [DOI] [PubMed] [Google Scholar]
- 10. Mizushima N., Yamamoto A., Matsui M., Yoshimori T., Ohsumi Y. (2004) In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 15, 1101–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Noyan-Ashraf M. H., Shikatani E. A., Schuiki I., Mukovozov I., Wu J., Li R. K., Volchuk A., Robinson L. A., Billia F., Drucker D. J., Husain M. (2013) A glucagon-like peptide-1 analog reverses the molecular pathology and cardiac dysfunction of a mouse model of obesity. Circulation 127, 74–85 [DOI] [PubMed] [Google Scholar]
- 12. Hoefer J., Azam M. A., Kroetsch J. T., Leong-Poi H., Momen M. A., Voigtlaender-Bolz J., Scherer E. Q., Meissner A., Bolz S. S., Husain M. (2010) Sphingosine 1-phosphate-dependent activation of p38 MAPK maintains elevated peripheral resistance in heart failure through increased myogenic vasoconstriction. Circ. Res. 107, 923–933 [DOI] [PubMed] [Google Scholar]
- 13. Zaidi S. H., Huang Q., Momen A., Riazi A., Husain M. (2010) Growth differentiation factor 5 regulates cardiac repair after myocardial infarction. J. Am. Coll. Cardiol. 55, 135–143 [DOI] [PubMed] [Google Scholar]
- 14. Nagahara H., Vocero-Akbani A. M., Snyder E. L., Ho A., Latham D. G., Lissy N. A., Becker-Hapak M., Ezhevsky S. A., Dowdy S. F. (1998) Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat. Med. 4, 1449–1452 [DOI] [PubMed] [Google Scholar]
- 15. Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ichimura Y., Kirisako T., Takao T., Satomi Y., Shimonishi Y., Ishihara N., Mizushima N., Tanida I., Kominami E., Ohsumi M., Noda T., Ohsumi Y. (2000) A ubiquitin-like system mediates protein lipidation. Nature 408, 488–492 [DOI] [PubMed] [Google Scholar]
- 17. Klionsky D. J., Abeliovich H., Agostinis P., Agrawal D. K., Aliev G., Askew D. S., Baba M., Baehrecke E. H., Bahr B. A., Ballabio A., Bamber B. A., Bassham D. C., Bergamini E., Bi X., Biard-Piechaczyk M., Blum J. S., Bredesen D. E., Brodsky J. L., Brumell J. H., Brunk U. T., Bursch W., Camougrand N., Cebollero E., Cecconi F., Chen Y., Chin L. S., Choi A., Chu C. T., Chung J., Clarke P. G., Clark R. S., Clarke S. G., Clavé C., Cleveland J. L., Codogno P., Colombo M. I., Coto-Montes A., Cregg J. M., Cuervo A. M., Debnath J., Demarchi F., Dennis P. B., Dennis P. A., Deretic V., Devenish R. J., Di Sano F., Dice J. F., Difiglia M., Dinesh-Kumar S., Distelhorst C. W., Djavaheri-Mergny M. (2008) Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4, 151–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bjørkoy G., Lamark T., Brech A., Outzen H., Perander M., Overvatn A., Stenmark H., Johansen T. (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171, 603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Komatsu M., Waguri S., Koike M., Sou Y. S., Ueno T., Hara T., Mizushima N., Iwata J., Ezaki J., Murata S., Hamazaki J., Nishito Y., Iemura S., Natsume T., Yanagawa T., Uwayama J., Warabi E., Yoshida H., Ishii T., Kobayashi A., Yamamoto M., Yue Z., Uchiyama Y., Kominami E., Tanaka K. (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131, 1149–1163 [DOI] [PubMed] [Google Scholar]
- 20. Mizushima N., Yoshimori T., Levine B. (2010) Methods in mammalian autophagy research. Cell 140, 313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Umansky S. R., Shapiro J. P., Cuenco G. M., Foehr M. W., Bathurst I. C., Tomei L. D. (1997) Prevention of rat neonatal cardiomyocyte apoptosis induced by simulated in vitro ischemia and reperfusion. Cell Death Differ. 4, 608–616 [DOI] [PubMed] [Google Scholar]
- 22. Kanamori H., Takemura G., Maruyama R., Goto K., Tsujimoto A., Ogino A., Li L., Kawamura I., Takeyama T., Kawaguchi T., Nagashima K., Fujiwara T., Fujiwara H., Seishima M., Minatoguchi S. (2009) Functional significance and morphological characterization of starvation-induced autophagy in the adult heart. Am. J. Pathol. 174, 1705–1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bjørkoy G., Lamark T., Pankiv S., Øvervatn A., Brech A., Johansen T. (2009) Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 452, 181–197 [DOI] [PubMed] [Google Scholar]
- 24. Borriello A., Bencivenga D., Criscuolo M., Caldarelli I., Cucciolla V., Tramontano A., Borgia A., Spina A., Oliva A., Naviglio S., Della Ragione F. (2011) Targeting p27Kip1 protein: its relevance in the therapy of human cancer. Expert. Opin. Ther. Targets 15, 677–693 [DOI] [PubMed] [Google Scholar]
- 25. Bergmann O., Bhardwaj R. D., Bernard S., Zdunek S., Barnabé-Heider F., Walsh S., Zupicich J., Alkass K., Buchholz B. A., Druid H., Jovinge S., Frisén J. (2009) Evidence for cardiomyocyte renewal in humans. Science 324, 98–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hosokawa N., Hara Y., Mizushima N. (2006) Generation of cell lines with tetracycline-regulated autophagy and a role for autophagy in controlling cell size. FEBS Lett. 580, 2623–2629 [DOI] [PubMed] [Google Scholar]
- 27. Ahuja P., Sdek P., MacLellan W. R. (2007) Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol. Rev. 87, 521–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fero M. L., Rivkin M., Tasch M., Porter P., Carow C. E., Firpo E., Polyak K., Tsai L. H., Broudy V., Perlmutter R. M., Kaushansky K., Roberts J. M. (1996) A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27Kip1-deficient mice. Cell 85, 733–744 [DOI] [PubMed] [Google Scholar]
- 29. Nakayama K., Ishida N., Shirane M., Inomata A., Inoue T., Shishido N., Horii I., Loh D. Y., Nakayama K. (1996) Mice lacking p27Kip1 display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell 85, 707–720 [DOI] [PubMed] [Google Scholar]
- 30. Fero M. L., Randel E., Gurley K. E., Roberts J. M., Kemp C. J. (1998) The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature 396, 177–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Poolman R. A., Li J. M., Durand B., Brooks G. (1999) Altered expression of cell cycle proteins and prolonged duration of cardiac myocyte hyperplasia in p27KIP1 knockout mice. Circ. Res. 85, 117–127 [DOI] [PubMed] [Google Scholar]
- 32. Zhu H., Tannous P., Johnstone J. L., Kong Y., Shelton J. M., Richardson J. A., Le V., Levine B., Rothermel B. A., Hill J. A. (2007) Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Invest. 117, 1782–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kanamori H., Takemura G., Goto K., Maruyama R., Ono K., Nagao K., Tsujimoto A., Ogino A., Takeyama T., Kawaguchi T., Watanabe T., Kawasaki M., Fujiwara T., Fujiwara H., Seishima M., Minatoguchi S. (2011) Autophagy limits acute myocardial infarction induced by permanent coronary artery occlusion. Am. J. Physiol. Heart Circ. Physiol. 300, H2261–2271 [DOI] [PubMed] [Google Scholar]
- 34. Kanamori H., Takemura G., Goto K., Maruyama R., Tsujimoto A., Ogino A., Takeyama T., Kawaguchi T., Watanabe T., Fujiwara T., Fujiwara H., Seishima M., Minatoguchi S. (2011) The role of autophagy emerging in postinfarction cardiac remodelling. Cardiovasc. Res. 91, 330–339 [DOI] [PubMed] [Google Scholar]
- 35. Buss S. J., Muenz S., Riffel J. H., Malekar P., Hagenmueller M., Weiss C. S., Bea F., Bekeredjian R., Schinke-Braun M., Izumo S., Katus H. A., Hardt S. E. (2009) Beneficial effects of mammalian target of rapamycin inhibition on left ventricular remodeling after myocardial infarction. J. Am. Coll. Cardiol. 54, 2435–2446 [DOI] [PubMed] [Google Scholar]
- 36. Degenhardt K., Mathew R., Beaudoin B., Bray K., Anderson D., Chen G., Mukherjee C., Shi Y., Gélinas C., Fan Y., Nelson D. A., Jin S., White E. (2006) Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 10, 51–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wei H., Wei S., Gan B., Peng X., Zou W., Guan J. L. (2011) Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 25, 1510–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lock R., Roy S., Kenific C. M., Su J. S., Salas E., Ronen S. M., Debnath J. (2011) Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 22, 165–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim M. J., Woo S. J., Yoon C. H., Lee J. S., An S., Choi Y. H., Hwang S. G., Yoon G., Lee S. J. (2011) Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J. Biol. Chem. 286, 12924–12932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guo J. Y., Chen H. Y., Mathew R., Fan J., Strohecker A. M., Karsli-Uzunbas G., Kamphorst J. J., Chen G., Lemons J. M., Karantza V., Coller H. A., Dipaola R. S., Gelinas C., Rabinowitz J. D., White E. (2011) Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 25, 460–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Beltrami A. P., Urbanek K., Kajstura J., Yan S. M., Finato N., Bussani R., Nadal-Ginard B., Silvestri F., Leri A., Beltrami C. A., Anversa P. (2001) Evidence that human cardiac myocytes divide after myocardial infarction. N. Engl. J. Med. 344, 1750–1757 [DOI] [PubMed] [Google Scholar]
- 42. Beltrami A. P., Barlucchi L., Torella D., Baker M., Limana F., Chimenti S., Kasahara H., Rota M., Musso E., Urbanek K., Leri A., Kajstura J., Nadal-Ginard B., Anversa P. (2003) Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 114, 763–776 [DOI] [PubMed] [Google Scholar]
- 43. Oh H., Bradfute S. B., Gallardo T. D., Nakamura T., Gaussin V., Mishina Y., Pocius J., Michael L. H., Behringer R. R., Garry D. J., Entman M. L., Schneider M. D. (2003) Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc. Natl. Acad. Sci. U.S.A. 100, 12313–12318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Banerjee I., Fuseler J. W., Price R. L., Borg T. K., Baudino T. A. (2007) Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am. J. Physiol. Heart Circ. Physiol. 293, H1883–1891 [DOI] [PubMed] [Google Scholar]