

Background: A subgroup of the NLR (nucleotide binding domain, leucine-rich repeat-containing) proteins and a non-NLR protein AIM2 are key mediators of the inflammasome.
Results: Cells lacking Nrf2 are defective in the activation of the NLRP3 and AIM2 inflammasome but not the NLRC4 inflammasome.
Conclusion: Nrf2 is an essential mediator of NLRP3 and AIM2 inflammasome activation.
Significance: Nrf2 plays a proinflammatory role by enhancing inflammasome activation.
Abstract
Despite the number of extensive studies on the immune function and signaling of inflammasomes in various diseases, the activating mechanism of inflammasome, especially the NLRP3 inflammasome, is not fully understood. Nuclear factor E2-related Factor-2 (Nrf2), a key transcription factor that regulates cellular redox homeostasis, has been reported to play both protective and pathogenic roles depending on the disease conditions through undefined mechanism. This study reveals an essential role of Nrf2 in inflammasome activation. LPS stimulation increased Nrf2 protein levels in a Myd88-dependent manner. When compared with wild-type controls, Nrf2-deficient (Nrf2−/−) macrophages showed decreased maturation and secretion of caspase-1 and IL-1β and reduced apoptosis-associated speck-like protein containing CARD (ASC) speck formation in response to various NLRP3 and AIM2 inflammasome stimuli. In contrast, NLRC4 inflammasome activation was not controlled by Nrf2. Biochemical analysis revealed that Nrf2 appeared in the ASC-enriched cytosolic compartment after NLRP3 inflammasome activation. Furthermore, mitochondrial reactive oxygen species-induced NLRP3 activation also required Nrf2. Nrf2−/− mice showed a dramatic decrease in immune cell recruitment and IL-1β generation in alum-induced peritonitis, which is a typical IL-1 signaling-dependent inflammation animal model. This work discovered a critical proinflammatory effect of Nrf2 by mediating inflammasome activation.
Introduction
The activation of inflammasome, a multiprotein cytosolic complex, leads to caspase-1 activation, which causes the maturation and secretion of inflammatory cytokines IL-1β and IL-18. A subgroup of NLR2 (nucleotide binding domain, leucine-rich repeat containing) proteins or non-NLR protein AIM2 together with adaptor protein ASC, assemble to form individual inflammasome complexes (1–6). Although the activating ligands and mechanisms for several inflammasomes (NLRP1, NLRC4, and AIM2 inflammasome) have been defined, it remains unclear how other inflammasomes, especially NLRP3 inflammasome, are activated at the molecular level (7). A diverse series of exogenous and endogenous agonists can activate the NLRP3 inflammasome; however, direct binding of these ligands to NLRP3 has not been clearly demonstrated. Instead, it is generally accepted that NLRP3 senses a multitude of disturbances in cellular homeostasis. Consistent with this notion, recent studies reveal several cell stress-related signaling pathways that cause NLRP3 activation (7). These signals include endoplasmic reticulum stress (8), calcium signaling (9, 10), and mitochondria-associated signals such as mitochondrial ROS (11, 12), phospholipid cardiolipin (13), and mitochondrial DNA (14, 15). Because it has been well documented that the inflammasome, which leads to caspase-1 protease activation, has broad biologic and clinical impact, further identification of new mediators of inflammasome activation will provide potential therapeutic targets toward numerous disease conditions.
Oxidative stress is one established mechanism of inflammation and tissue damage. Nuclear factor E2-related factor-2 (Nrf2) is an essential transcription factor that mediates cellular antioxidant responses by regulating the expression of genes encoding a wide range of phase II detoxifying enzymes and antioxidants (16–19). Under non-stressed conditions, Nrf2 is maintained at low protein levels through a ubiquitin-proteasome mechanism mediated by cullin-3, an E3 ubiquitin ligase, and an adaptor protein kelch-like ECH-associated protein 1 (Keap1). During cellular stress conditions, Nrf2 is released from Keap1 sequestration, and the accumulated Nrf2 is translocated into the nucleus to initiate gene transcription via the antioxidant response element (ARE). The target genes of Nrf2 include those encoding glutathione S-transferases (Gst), NAD(P)H dehydrogenase, quinone 1 (Nqo1), and heme oxygenase 1 (Hmox1) (16–19).
Recent studies indicate a protective role of Nrf2 in multiple inflammatory diseases, such as sepsis (20, 21), asthma (22), infectious diseases (23–25), and cigarette smoke-induced emphysema (26). Paradoxically, Nrf2 has also been shown to play a pathogenic role in metabolic disorders including atherosclerosis (27–29), obesity (30), and type 2 diabetes (31), all of which are typically associated with chronic inflammation. The seemingly conflicting findings suggest an undefined effect Nrf2 may play in various disease conditions. Here we show an essential role of Nrf2 in mediating inflammasome activation, which has been recently implicated in the pathogenesis of metabolic disorders (12, 32–36). Nrf2 deficiency resulted in defective activation of the NLRP3 and AIM2 inflammasome but not the NLRC4 inflammasome. An alum-induced peritonitis animal model confirmed the critical effect of Nrf2 in IL-1 signaling-associated inflammation. Nrf2 is present in the ASC-enriched cytosolic compartment after NLRP3 activation, suggesting a possibility that Nrf2 may directly contribute to the assembly of ASC speck that is an essential step for inflammasome activation.
EXPERIMENTAL PROCEDURES
Mice
Nlrp3−/−, Nlrc4−/−, Nrf2−/−, Myd88−/− mice have been described previously (37–39). C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME). All mouse studies were used in accordance with the National Institutes of Health guide for the Care and Use of Laboratory Animals and Institutional Animal Care and Use Committees guidelines of the University of North Carolina Chapel Hill.
Reagents and Antibodies
Rotenone, antimycin A, N-acetyl-l-cysteine (NAC) and ATP were from Sigma. Ultrapure lipopolysaccharide (LPS), flagellin, nigericin, poly(dA:dT), alhydrogel, and monosodium urate were purchased from InvivoGen. Imject Alum was from Pierce. Silica (MIN-U-SIL 15) was from U. S. Silica. Diphenyleneiodonium (DPI) was from Enzo Life Sciences. TRIzol reagent was from Invitrogen. X-tremeGENE HP DNA Transfection Reagent was from Roche Applied Science. Transfection reagent Profect P1 was from Targeting Systems. Disuccinimidyl substrate was from Thermo Scientific. Antibodies for immunoblotting include anti-caspase-1 (sc-514), anti-β-actin (sc-1615 HRP) (Santa Cruz Biotechnology), anti-NLRP3 (Cryo-2), anti-Asc (AL177) (Adipogen), anti-IL-1β (AF-401-NA) (R&D Systems), and anti-Nrf2 (ab62352) and anti-HMGB1 (ab18256) (Abcam). Antibodies for flow cytometry include CD11b (M1/70; BioLegend), Ly-6C (HK1.4; Abcam), and Ly-6G (1A8; Pharmingen).
Cell Culture and Stimulation
Bone marrow-derived macrophages (BMMs) were generated in the presence of L-929 conditional medium. Cells were cultured in DMEM complemented with 10% FCS, 1 mm sodium pyruvate, 1 mm nonessential amino acid, 100 IU/ml penicillin/streptomycin, and 2 mm l-glutamine. After pretreatment with ultrapure LPS (200 ng/ml) for 3 h, cells were stimulated with alum (100 μg/ml), silica (50 μg/ml), monosodium urate (200 μg/ml), and alhydrogel (400 μg/ml) for 8 h and transfected poly(dA:dT) (1 μg/ml) or flagellin (1 μg/ml) for 4 h, as indicated in the figure legends. Poly(dA:dT) was transfected with X-tremeGENE at a mass-to-volume ratio of 1:2. Flagellin was transfected with Profect P1 at a mass-to-volume ratio of 1:5. For ATP or nigericin, cells were primed with LPS for 6 h followed with ATP (2 mm) or nigericin (5 μm) stimulation for 40 min. Supernatant and cell lysate were collected for ELISA and Western blot analysis.
Real-time PCR Analysis
Total RNA was extracted from in vitro cultured BMMs using TRIzol (Invitrogen). cDNA synthesis was performed with Moloney murine leukemia virus reverse transcriptase (Invitrogen) at 38 °C for 60 min. Real-time PCR was performed using SYBR Green PCR Master Mix in an ABI PRISM 7900 sequence detection system (Applied Biosystems). The -fold difference in mRNA expression between treatment groups was determined by a standard ΔΔCt method. β-Actin was analyzed as an internal control. The primer sequences of individual genes are available upon request.
Immunoblotting
Electrophoresis of proteins was performed by using the NuPAGE system (Invitrogen) according to the manufacturer's protocol. Briefly, cultured BMMs were collected and lysed with radioimmune precipitation assay buffer. Proteins were separated on a NuPAGE gel and transferred onto nitrocellulose membranes (Bio-Rad). Appropriate primary antibodies and HRP-conjugated secondary antibodies were used, and proteins were detected using the enhanced chemiluminescent (ECL) reagent (Thermo Scientific). The images were acquired with ChemiDoc MP System (Bio-Rad).
ELISA
Cytokines generated by in vitro cultured BMMs were quantified using the ELISA Set for mouse IL-1β, IL-6, or TNF-α (BD Biosciences) according to the manufacturer's protocol.
ASC Oligomerization Assay
ASC oligomerization assays were performed as previously described with minor modifications (40). BMMs were seeded in 6-well plates (3 × 106 cells per well). After the treatment with indicated stimuli, the cells were washed by cold PBS and resuspended in an ice-cold buffer (Buffer A: 20 mm HEPES-KOH, pH 7.5, 150 mm KCl, 1% Nonidet P-40, 0.1 mm PMSF, and protease inhibitor) and lysed by shearing 10 times through a 21-gauge needle. Nuclei and unlysed cells were removed by centrifugation at 250 × g for 5 min. The cell lysates were then centrifuged at 5000 × g for 10 min at 4 °C. After washing twice with PBS, the pellets were cross-linked with fresh disuccinimidyl substrate (2 mm) for 30 min at 37 °C. The cross-linked pellets were separated in 4–12% SDS-PAGE. and immunoblotting was performed.
Immunofluorescence
WT or Nrf2−/− BMMs were seeded on glass-bottom 12-well plates (P12G-1.5–14-F, MatTek Corp.). After the treatment with different stimuli, cells were fixed and permeabilized with 4% paraformaldehyde containing 0.1% Triton-X 100 for 20 min at room temperature. The cells were washed 3 times with 1× PBS and incubated with blocking buffer (5% BSA, 2% goat serum, 1% Fc block) for 30 min at room temperature. The samples were incubated with anti-ASC antibody (1:200 dilution) in the blocking buffer overnight at 4 °C followed by the incubation with Alexa Fluor® 647 goat anti-rabbit antibody (1:1000 dilution; A-21244, Invitrogen) for 2 h at room temperature. Images were acquired with a Zeiss LMS 710 confocal microscope.
LDH Release Assay
WT or Nrf2−/− BMMs were seeded on 96-well plates. After the treatment with indicated stimuli, supernatants were collected, and LDH activity was determined with the a cytotoxicity detection kit (LDH) (Roche Applied Science). Cells left untreated or treated with 1% Triton X-100 were used as negative and positive controls, respectively.
Alum-induced Peritonitis
Mice were injected intraperitoneally with 400 μg of alum diluted in sterile PBS. Sixteen hours later peritoneal lavage was performed, and peritoneal exudate cells were stained with cell surface markers. Neutrophils and inflammatory monocytes were identified as CD11b+Ly-6G+ and CD11b+Ly-6C+, respectively. Flow cytometry analysis was performed on a CYAN flow cytometer. Alternatively, mice were injected with 600 μg of alum, and peritoneal lavage was performed 4 h later. IL-1β was measured by ELISA.
Statistical Analysis
Statistical analysis was carried out with Prism 5 for Macintosh. Results were presented as the mean ± S.D., and unpaired Student's t test (one tailed) was applied to evaluate significance. p values less than 0.05 were considered statistically significant.
RESULTS
Nrf2 Protein Accumulation after LPS Stimulation
It is well accepted that Nrf2 protein level is tightly controlled by proteasomal degradation via cullin-3- and Keap1-mediated ubiquitination (16–19). To study the effect of Nrf2 in macrophages, we initially examined if Nrf2 can be induced by LPS, a widely used TLR4 ligand that causes classical macrophage activation. In BMMs, the protein level of Nrf2 increased from 4 h after LPS stimulation (Fig. 1A), whereas Nrf2 transcript did not increase (Fig. 1B). These findings suggest that LPS-induced Nrf2 protein accumulation independent of transcription. Furthermore, LPS-induced Nrf2 accumulation is largely dependent of MyD88, a key adaptor protein of TLR signaling, as MyD88 deficiency (Myd88−/−) dramatically attenuated LPS-increased Nrf2 protein level (Fig. 1C). Similarly, LPS-induced increase of NLRP3 and pro-IL-1β were also dramatically blunted in Myd88−/− BMMs.
FIGURE 1.
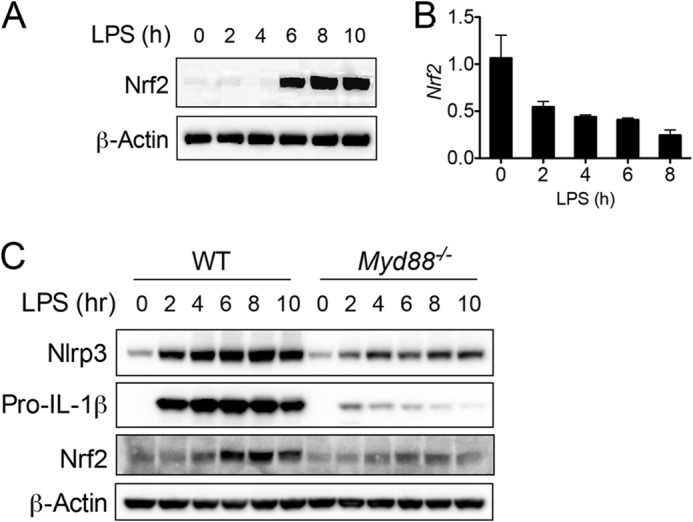
LPS induces Nrf2 protein accumulation. A and B, BMMs generated from naïve C57BL/6 mice were stimulated with LPS (400 ng/ml) for the indicated periods. Protein (A) and transcript (B) of Nrf2 was measured by immunoblotting and RT-PCR, respectively. C, BMMs generated from WT or Myd88−/− mice were stimulated with LPS (400 ng/ml) for the indicated periods. Immunoblotting was performed to measure the protein levels of indicated molecules. Values in B are expressed as the mean ± S.D., and the results are representative of three independent experiments.
Nrf2−/− Macrophages Showed Defective Activation of the NLRP3 and AIM2 Inflammasome
Previous studies suggest an inhibitory effect of Nrf2 in NF-κB-dependent cytokine production in macrophages (20, 21). Yet the significance of Nrf2 in inflammasome activation has not been fully characterized. To examine the role of Nrf2 in this capacity, we treated LPS-primed BMMs with a series of stimuli that activate the NLRP3 inflammasome. As expected, alum (Fig. 2A) and silica (Fig. 2B) induced IL-1β production from WT BMMs in a dosage (Fig. 2, A and B, left panels) and time (Fig. 2, A and B, right panels)-dependent manner. However, Nrf2−/− BMMs generated significantly less IL-1β in response to alum and silica stimulation. IL-6 production was comparable between WT and Nrf2−/− BMMs (Fig. 2C), indicating that the defective IL-1β generation was not due to a common failure in cytokine production in Nrf2−/− BMMs. Nrf2−/− BMMs showed a similar defect in IL-1β production when treated with another two crystalline reagents, alhydrogel (Fig. 2D) and monosodium urate (Fig. 2E). Furthermore, Nrf2−/− BMMs showed significantly less IL-1β production (Fig. 2F) and cell death (Fig. 2G) but similar IL-6 production (Fig. 2H) in response to ATP and nigericin, suggesting that the critical effect of Nrf2 in IL-1β production is not limited to crystalline reagents. These data indicate that Nrf2 is essential for NLRP3 inflammasome activation.
FIGURE 2.

Nrf2 is required for the NLRP3 and AIM2 inflammasome activation. A–H, LPS-primed WT or Nrf2−/− BMMs were stimulated with a series of agonists at indicated dosages, including alum (A and C), silica (B and C), alhydrogel (D), monosodium urate (MSU) (E), ATP or nigericin (F–H). Cytokines in supernatants were measured by ELISA (A–G). Cell cytotoxicity was determined by LDH release assay (H). I, LPS-primed WT or Nrf2−/− BMMs were stimulated with transfected poly(dA:dT) for 4 h (I). J, naïve WT or Nrf2−/− BMMs were stimulated with transfected poly(dA:dT) for 16 h. K–M, WT, Nrf2−/−, or Nlrc4−/− BMMs were untreated or primed with 400 ng/ml LPS for 3 h followed by the stimulation with transfected flagellin for 4 h. The treatment of cells with transfection reagent alone was included as “transfection control.” IL-1β (K) and IL-6 (L) in supernatants were measured by ELISA. Cell cytotoxicity was determined by LDH release assay (M). Values are expressed as the mean ± S.D., and the results are representative of three independent experiments. *, p < 0.05, versus controls.
We next tested if Nrf2 is important for the activation of other inflammasomes, namely AIM2 and NLRC4 inflammasome. Transfected poly(dA:dT) and flagellin are well characterized ligands that activate AIM2 and NLRC4 inflammasome, respectively. Nrf2−/− BMMs generated significantly less IL-1β in response to transfected poly(dA:dT) with (Fig. 2I) or without (Fig. 2J) LPS priming, indicating that Nrf2 is important for AIM2 inflammasome activation. In contrast, the production of IL-1β (Fig. 2K), IL-6 (Fig. 2L), and cell death (Fig. 2 m) were comparable between WT and Nrf2−/− BMMs in response to transfected flagellin, suggesting that Nrf2 is dispensable for NLRC4 inflammasome activation. As controls, we showed that IL-1β production and cell death were abolished in Nlrc4−/− BMMs.
Next we measured the cleavage of pro-IL-1β and procaspase-1 to biochemically evaluate inflammasome activation. Alum and silica caused procaspase-1 and pro-IL-1β processing to their mature forms in LPS-primed WT BMMs (Fig. 3A). However, these effects were abolished in Nrf2−/− BMMs (Fig. 3A). Similar data were acquired when the cells were stimulated with ATP, nigericin (Fig. 3B), or transfected poly(dA:dT) (Fig. 3C). These results indicate that Nrf2 is essential for NLRP3 and AIM2 inflammasome activation. In contrast, transfected flagellin promoted normal IL-1β and caspase-1 maturation in Nrf2−/− BMMs but not in Nlrc4−/− BMMs, indicating that Nrf2 is not required for NLRC4 inflammasome activation (Fig. 3D).
FIGURE 3.

Nrf2 mediates caspase-1 and IL-1β processing. A–C, resting or LPS-primed BMMs generated from WT or Nrf2−/− mice were stimulated with a series of agonists at indicated dosages, including alum or silica (A), ATP or nigericin (B), and transfected poly(dA:dT) (C). Immunoblotting for caspase-1 and IL-1β was performed in supernatants and cell lysates as indicated in the figure. E–G, WT or Nrf2−/− BMMs were stimulated with LPS (400 ng/ml) for the indicated periods. Transcripts of Nlrp3, Pycard, Casp1, and Il1b (E) and Nqo1 and Hmox1 (F) were determined by RT-PCR. Proteins of NLRP3, procaspase-1, pro-IL-1β, and ASC were measured by ELISA (G). Values in E and F are expressed as the mean ± S.D., and the results are representative of two independent experiments. *, p < 0.05, versus controls.
Nrf2 is a vital transfection factor that regulates the transcription of thousands of genes (41, 42). We asked if the defective inflammasome activation was due to the impaired gene transcription of inflammasome components in Nrf2−/− BMMs. However, Nrf2 is not required for up-regulation of genes encoding inflammasome-related molecules, including Nlrp3, Pycard, and Il1b in response to LPS stimulation (Fig. 3E). As positive controls, the induction of two classical target genes of Nrf2, Nqo1 and Hmox1, was totally abolished in Nrf2−/− BMMs (Fig. 3F). In addition, protein levels of inflammasome-related molecules were comparable between WT and Nrf2−/− BMMs in response to LPS alone (Fig. 3G) or LPS plus inflammasome stimuli (Fig. 3, A–D). These data suggest that Nrf2 is not required for expression of the acknowledged inflammasome components.
Nrf2 Is Required for ASC Speck Formation upon NLRP3 and AIM2 Inflammasome Activation
In addition to the maturation and secretion of procaspase-1 and pro-IL-1β, another hallmark of inflammasome activation is the formation of ASC speck. This large protein complex assembled by ASC is essential for caspase-1 activation (43). Next, we tested if Nrf2 controls ASC speck formation upon NLRP3 and AIM2 inflammasome activation. Endogenous ASC immunostaining showed that ATP or nigericin stimulation caused ASC speck formation in >60% of LPS-primed WT BMMs (Fig. 4A). In contrast, significantly less Nrf2−/− BMMs contained ASC speck after similar treatments (Fig. 4A), suggesting that Nrf2 is important for ASC speck formation. Similarly, Nrf2−/− BMMs showed defective ASC speck formation in response to transfected poly(dA:dT) (Fig. 4B). These data are consistent with our previous findings and further strengthens the critical role of Nrf2 in NLRP3 and AIM2 inflammasome activation.
FIGURE 4.

Nrf2 is required for ASC speck formation upon NLRP3 and AIM2 inflammasome activation. A and B, WT or Nrf2−/− BMMs were primed with 400 ng/ml LPS for 6 h followed by the stimulation with ATP (2 mm) or nigericin (10 μm) for 20 min (A) or transfected poly (dA:dT) (5 μg/ml) (B) for 4 h. Immunostaining of endogenous ASC was performed (left panels). Quantification of ASC speck was performed by counting cells in 10 individual filed (right panels). C and D, LPS-primed WT or Nrf2−/− BMMs were stimulated with ATP or nigericin (C) and alum or silica (D). Detergent-resistant ASC aggregate was isolated, and immunoblotting of ASC and Nrf2 was performed in cross-linked pellets (upper panels) and in cell lysates (lower panels). Values are expressed as the mean ± S.D., and the results are representative of two independent experiments. *, p < 0.05, versus controls.
Subsequently, we wanted to determine the role of Nrf2 in NLRP3-dependent ASC speck formation with a well established biochemical assay (40). As expected, the stimulation of LPS-primed WT BMMs with ATP, nigericin (Fig. 4C), alum, or silica (Fig. 4D) caused the formation of detergent-resistant ASC aggregates. Due to the usage of cross-linking reagent disuccinimidyl substrate, aggregated ASC formed dimers or oligomers. However, ASC aggregates and oligomerization were largely diminished in similarly treated Nrf2−/− BMMs. Interestingly, ASC appeared in ASC aggregate after inflammasome activation (Fig. 4, C and D). These findings revealed a critical role of Nrf2 in ASC speck formation and suggested a possibility that Nrf2 may mediate inflammasome activation by directly promoting ASC speck formation.
ROS Mediates Inflammasome Activation via Nrf2
As a typical Nrf2 activator, ROS modifies the critical cysteine thiols of Keap1 and Nrf2, which leads to their dissociation and Nrf2 accumulation (16–19). Interestingly, ROS has also been suggested as an important mechanism that either primes or activates the NLRP3 inflammasome (7, 11, 44), although controversy still exists (45). We thereby hypothesized that Nrf2 is an effector molecule that is involved in ROS-mediated inflammasome activation. Two inhibitors of mitochondrial respiratory function, rotenone and antimycin, have previously been shown to activate the NLRP3 inflammasome by increasing mitochondrial ROS generation (11). Indeed, we found that rotenone and antimycin caused IL-1β release (Fig. 5A) and the cleavage of pro-IL-1β and procaspase-1 (Fig. 5C), responses dependent on NLRP3 activation. To confirm that these inhibitors work primarily through NLRP3, we showed that IL-6 generation was not affected by the treatments (Fig. 5B). Importantly, these effects were largely diminished in similarly treated Nrf2−/− BMMs, indicating that Nrf2 is important for mitochondrial ROS-mediated inflammasome activation. Rotenone and antimycin did not further enhance LPS-induced Nrf2 protein accumulation, suggesting that Nrf2 protein level was saturated by LPS treatment alone.
FIGURE 5.

Nrf2 is important for ROS-mediated NLRP3 inflammasome activation. A–C, WT, Nrf2−/−, or Nlrp3−/− BMMs were untreated or primed with 400 ng/ml LPS for 3 h followed by the stimulation with rotenone (20 μm) or antimycin (40 μg/ml) for 6 h. IL-1β (A) and IL-6 (B) in supernatants were measured by ELISA. Immunoblotting for caspase-1 and IL-1β were performed in supernatants and cell lysates (C). D, WT BMMs were untreated or pretreated with NAC or DPI for 30 min followed by stimulation with LPS (400 ng/ml) for 6 h. Immunoblotting (left) and densitometric analysis (right) for Nrf2 and NLRP3 inflammasome component molecules were performed. E and F, WT or Nrf2−/− BMMs were untreated or pretreated with NAC or DPI for 30 min followed by stimulation with LPS (400 ng/ml) for 3 h then silica (100 μg/ml) for 6 h (E) or transfected poly(dA:dT) for 4 h (F). IL-1β in supernatants was measured by ELISA. G and H, WT BMMs were treated with LPS for 4 h followed by the stimulation with silica in the absence or presence of NAC or DPI for 4 h. Immunoblotting was performed (G). IL-1β in supernatants was measured by ELISA (H). Values are expressed as the mean ± S.D., and the results are representative of four independent experiments. * p < 0.05, versus controls.
ROS inhibition has been shown to block the priming or activation of the NLRP3 inflammasome (44, 46). We found that NAC and DPI, two widely used ROS inhibitors, dramatically decreased LPS-induced Nrf2 protein accumulation with minimal effect on other inflammasome-related protein such as NLRP3, procaspase-1, and pro-IL-1β (Fig. 5D). This indicates that Nrf2, but not other inflammasome components, is the major target of ROS inhibitors, a result consistent with the concept that ROS is a classical activating mechanism for Nrf2 (16–19). ROS inhibitor-mediated decreases in Nrf2 is functionally important, as NAC or DPI decreased IL-1β release induced by silica (Fig. 5E) or poly(dA:dT) (Fig. 5F) in WT, but not Nrf2−/−, BMMs. When ROS inhibitors were added 4 h after LPS priming, neither LPS-induced Nrf2 accumulation (Fig. 5G) nor IL-1β release (Fig. 5H) by silica was inhibited, suggesting that Nrf2 accumulation paralleled with inflammasome activation. These findings indicate that ROS inhibitors minimized NLRP3 inflammasome activation via Nrf2.
Attenuated Alum-induced Peritonitis in Nrf2−/− Mice
We next evaluated the in vivo effect of Nrf2-mediated inflammasome activation by utilizing an alum-induced peritonitis model in our mice. Intraperitoneal administration of alum induces IL-1 signaling-dependent inflammatory cell recruitment (47). We found that alum induced immune cell recruitment in the peritoneal cavity including Ly-6G+ neutrophils and Ly-6C+ inflammatory monocytes in WT mice (Fig. 6, A–D). However, Nrf2−/− mice showed a significantly less recruitment of immune cells, supporting a critical proinflammatory role of Nrf2 in alum-induced peritonitis. Furthermore, Nrf2−/− mice generated significantly less IL-1β in peritoneal cavity 4 h after alum intraperitoneal injection (Fig. 6E). Overall, this study suggests that Nrf2 is important for inflammasome-mediated cytokine responses both in vitro and in vivo.
FIGURE 6.

Nrf2−/− mice showed attenuated inflammation in alum-induced peritonitis. A–D, WT or Nrf2−/− mice were intraperitoneally injected with alum (400 μg/per mouse) diluted in sterile PBS. n = 6 mice per group. Sixteen hour after alum injection, peritoneal lavage was performed. Absolute number of peritoneal exudate cells (PEC) (A), CD11b+Ly-6G+ neutrophils (B), and CD11b+Ly-6C+ inflammatory monocytes (C) was evaluated by FACS analysis. Cytospin preparation followed by H&E staining was performed (D). E, WT or Nrf2−/− mice were intraperitoneally injected with alum (600 μg/per mouse) diluted in sterile PBS. n = 5 mice per group. Peritoneal lavage was performed 4 h after injection. IL-1β was measured by ELISA. Values are expressed as the mean ± S.D., and the results are representative of two independent experiments. *, p < 0.05, versus controls.
DISCUSSION
Inflammasome assembly is triggered by various danger signals, including those from both endogenous and exogenous sources. Oxidative stress, a commonly observed feature during inflammatory responses, has been indicated as an important upstream signaling mediator contributing toward inflammasome activation (7, 11, 48). Nrf2 is a pivotal transcription factor that maintains intracellular redox homeostasis through regulating the transcription of antioxidant genes (16–19). As a result, Nrf2 deficiency has been shown to cause an elevated ROS level, which is detrimental to normal cell functions and promotes cell death (16–19). Based on the well defined anti-oxidative effect of Nrf2, we initially hypothesized that Nrf2 deficiency would result in an enhance inflammasome activation via a ROS-dependent mechanism. Surprisingly, our findings suggest a positive effect of Nrf2 in the activation of the NLRP3 and AIM2 inflammasome but not the NLRC4 inflammasome. Furthermore, Nrf2 is the major target of ROS inhibition and is essential for ROS-induced inflammasome activation. From this we can conclude that Nrf2 may play a proinflammatory role in inflammasome-related diseases, particularly in the context of metabolic disorders. Interestingly, several studies reported that Nrf2 deficiency attenuates the development of atherosclerosis, obesity, and insulin resistance (27–31).
During our investigation of mechanism by which Nrf2 mediates inflammasome activation, we found that Nrf2 is not required for the transcription of genes encoding inflammasome components including Nlrp3, Pycard, Casp1, and Il1b. This finding suggested that several potential mechanisms may exist. First, we observed the appearance of Nrf2 in cytosolic ASC-enriched compartments after NLRP3 inflammasome activation, suggesting that cytosolic Nrf2 may contribute to the assembly of ASC speck. This possibility is reinforced by the fact that Nrf2 is accumulated in both cytosolic and nuclear compartments after LPS stimulation (49) and interacts with other cytosolic cell stress responses such as autophagy (17). Further in-depth biochemical analysis of the interaction between Nrf2 and other inflammasome-associated molecules may provide mechanistic insight. Second, Nrf2 may mediate inflammasome activation through transcriptional control of some unknown factor(s). This mechanism can be further tested utilizing microarray techniques. Third, a previous study reported that gene deletion of superoxide dismutase 1 (Sod1) led to elevated ROS generation, which inactivated caspase-1 through reversible oxidation of caspase-1 cysteine residues (50). However, it is unlikely that Nrf2 directly targets on caspase-1, as we show that Nrf2 is not required for NLRC4 inflammasome activation, indicating Nrf2 may be targeting signaling molecules upstream of caspase-1.
Accumulative evidence suggests that during inflammatory response, various stress signaling pathways interact, and their cross-regulations determine cell adaptive responses. Our findings reveal a close relationship between oxidative stress and inflammasome activation, both of which are controlled by Nrf2. This is of special significance regarding cell stress response against invading pathogens, including bacteria, virus, and fungi. Recent studies suggest that Nrf2−/− mice generally show an exacerbated susceptibility against pathogen infection including bacteria (24), viruses (23), and fungi (25). These observations are unlikely to be explained by the anti-oxidative effect of Nrf2, as it is largely accepted that enhanced ROS generation contributes to innate cell microbe elimination during respiratory burst. Interestingly, it has been well documented that inflammasome activation plays an essential role against invading various pathogens (7). Our findings suggest that Nrf2 is a pivotal mediator of inflammasome activation, thus highlighting the possibility that Nrf2 controls pathogen infection by activating inflammasomes. More recently, the importance of Nrf2 function has been effectively utilized clinically. Nrf2-targeted therapeutic regimen has been Food and Drug Administration-approved in the treatment of patients with relapsing-remitting multiple sclerosis. As this transcription factor has proven to be a practical target, it will be of great clinical significance to investigate the effect of Nrf2 in inflammasome activation during the context of pathogen infections.
Acknowledgments
We thank Drs. Vishva Dixit, Richard Flavell, Steven Kleeberger, and Shizou Akira for gene deletion mice. We acknowledge Kathleen Mulvaney from Dr. Michael B. Major's Laboratory and Tongde Wu from Dr. Donna Zhang's Laboratory for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grant K01DK098307 (to H. W.).
- NLR
- nucleotide binding domain (NBD)-leucine-rich repeat (LRR) or NOD-like receptor
- DPI
- diphenyleneiodonium
- Hmox1
- heme oxygenase 1
- Keap1
- kelch-like ECH-associated protein 1
- NAC
- N-acetyl-cysteine
- Nqo1
- NAD(P)H dehydrogenase, quinone 1
- Nrf2
- nuclear factor E2-related factor-2
- ROS
- reactive oxygen species
- BMM
- bone marrow-derived macrophage
- ASC
- apoptosis-associated speck-like protein containing CARD
- CARD
- caspase recruitment domain.
REFERENCES
- 1. Davis B. K., Wen H., Ting J. P. (2011) The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 29, 707–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Strowig T., Henao-Mejia J., Elinav E., Flavell R. (2012) Inflammasomes in health and disease. Nature 481, 278–286 [DOI] [PubMed] [Google Scholar]
- 3. Schroder K., Tschopp J. (2010) The inflammasomes. Cell 140, 821–832 [DOI] [PubMed] [Google Scholar]
- 4. Lamkanfi M., Dixit V. M. (2012) Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 28, 137–161 [DOI] [PubMed] [Google Scholar]
- 5. von Moltke J., Ayres J. S., Kofoed E. M., Chavarría-Smith J., Vance R. E. (2013) Recognition of bacteria by inflammasomes. Annu. Rev. Immunol. 31, 73–106 [DOI] [PubMed] [Google Scholar]
- 6. Rathinam V. A., Vanaja S. K., Fitzgerald K. A. (2012) Regulation of inflammasome signaling. Nat. Immunol. 13, 333–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wen H., Miao E. A., Ting J. P. (2013) Mechanisms of NOD-like receptor-associated inflammasome activation. Immunity 39, 432–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Menu P., Mayor A., Zhou R., Tardivel A., Ichijo H., Mori K., Tschopp J. (2012) ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis. 3, e261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee G. S., Subramanian N., Kim A. I., Aksentijevich I., Goldbach-Mansky R., Sacks D. B., Germain R. N., Kastner D. L., Chae J. J. (2012) The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 492, 123–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Murakami T., Ockinger J., Yu J., Byles V., McColl A., Hofer A. M., Horng T. (2012) Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. U.S.A. 109, 11282–11287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhou R., Yazdi A. S., Menu P., Tschopp J. (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 [DOI] [PubMed] [Google Scholar]
- 12. Wen H., Gris D., Lei Y., Jha S., Zhang L., Huang M. T., Brickey W. J., Ting J. P. (2011) Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 12, 408–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Iyer S. S., He Q., Janczy J. R., Elliott E. I., Zhong Z., Olivier A. K., Sadler J. J., Knepper-Adrian V., Han R., Qiao L., Eisenbarth S. C., Nauseef W. M., Cassel S. L., Sutterwala F. S. (2013) Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39, 311–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nakahira K., Haspel J. A., Rathinam V. A., Lee S. J., Dolinay T., Lam H. C., Englert J. A., Rabinovitch M., Cernadas M., Kim H. P., Fitzgerald K. A., Ryter S. W., Choi A. M. (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shimada K., Crother T. R., Karlin J., Dagvadorj J., Chiba N., Chen S., Ramanujan V. K., Wolf A. J., Vergnes L., Ojcius D. M., Rentsendorj A., Vargas M., Guerrero C., Wang Y., Fitzgerald K. A., Underhill D. M., Town T., Arditi M. (2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jaramillo M. C., Zhang D. D. (2013) The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 27, 2179–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ma Q. (2013) Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 53, 401–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Suzuki T., Motohashi H., Yamamoto M. (2013) Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol. Sci. 34, 340–346 [DOI] [PubMed] [Google Scholar]
- 19. Kensler T. W., Wakabayashi N., Biswal S. (2007) Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 47, 89–116 [DOI] [PubMed] [Google Scholar]
- 20. Thimmulappa R. K., Lee H., Rangasamy T., Reddy S. P., Yamamoto M., Kensler T. W., Biswal S. (2006) Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Invest. 116, 984–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kong X., Thimmulappa R., Craciun F., Harvey C., Singh A., Kombairaju P., Reddy S. P., Remick D., Biswal S. (2011) Enhancing Nrf2 pathway by disruption of Keap1 in myeloid leukocytes protects against sepsis. Am. J. Respir. Crit. Care Med. 184, 928–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rangasamy T., Guo J., Mitzner W. A., Roman J., Singh A., Fryer A. D., Yamamoto M., Kensler T. W., Tuder R. M., Georas S. N., Biswal S. (2005) Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J. Exp. Med. 202, 47–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cho H. Y., Imani F., Miller-DeGraff L., Walters D., Melendi G. A., Yamamoto M., Polack F. P., Kleeberger S. R. (2009) Antiviral activity of Nrf2 in a murine model of respiratory syncytial virus disease. Am. J. Respir. Crit. Care Med. 179, 138–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reddy N. M., Suryanarayana V., Kalvakolanu D. V., Yamamoto M., Kensler T. W., Hassoun P. M., Kleeberger S. R., Reddy S. P. (2009) Innate immunity against bacterial infection following hyperoxia exposure is impaired in NRF2-deficient mice. J. Immunol. 183, 4601–4608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Paiva C. N., Feijó D. F., Dutra F. F., Carneiro V. C., Freitas G. B., Alves L. S., Mesquita J., Fortes G. B., Figueiredo R. T., Souza H. S., Fantappié M. R., Lannes-Vieira J., Bozza M. T. (2012) Oxidative stress fuels Trypanosoma cruzi infection in mice. J. Clin. Invest. 122, 2531–2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rangasamy T., Cho C. Y., Thimmulappa R. K., Zhen L., Srisuma S. S., Kensler T. W., Yamamoto M., Petrache I., Tuder R. M., Biswal S. (2004) Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Invest. 114, 1248–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sussan T. E., Jun J., Thimmulappa R., Bedja D., Antero M., Gabrielson K. L., Polotsky V. Y., Biswal S. (2008) Disruption of Nrf2, a key inducer of antioxidant defenses, attenuates apoE-mediated atherosclerosis in mice. PLoS ONE 3, e3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barajas B., Che N., Yin F., Rowshanrad A., Orozco L. D., Gong K. W., Wang X., Castellani L. W., Reue K., Lusis A. J., Araujo J. A. (2011) NF-E2-related factor 2 promotes atherosclerosis by effects on plasma lipoproteins and cholesterol transport that overshadow antioxidant protection. Arterioscler. Thromb. Vasc. Biol. 31, 58–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Freigang S., Ampenberger F., Spohn G., Heer S., Shamshiev A. T., Kisielow J., Hersberger M., Yamamoto M., Bachmann M. F., Kopf M. (2011) Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis. Eur. J. Immunol. 41, 2040–2051 [DOI] [PubMed] [Google Scholar]
- 30. Pi J., Leung L., Xue P., Wang W., Hou Y., Liu D., Yehuda-Shnaidman E., Lee C., Lau J., Kurtz T. W., Chan J. Y. (2010) Deficiency in the nuclear factor E2-related factor-2 transcription factor results in impaired adipogenesis and protects against diet-induced obesity. J. Biol. Chem. 285, 9292–9300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chartoumpekis D. V., Ziros P. G., Psyrogiannis A. I., Papavassiliou A. G., Kyriazopoulou V. E., Sykiotis G. P., Habeos I. G. (2011) Nrf2 represses FGF21 during long-term high-fat diet-induced obesity in mice. Diabetes 60, 2465–2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Duewell P., Kono H., Rayner K. J., Sirois C. M., Vladimer G., Bauernfeind F. G., Abela G. S., Franchi L., Nuñez G., Schnurr M., Espevik T., Lien E., Fitzgerald K. A., Rock K. L., Moore K. J., Wright S. D., Hornung V., Latz E. (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Masters S. L., Dunne A., Subramanian S. L., Hull R. L., Tannahill G. M., Sharp F. A., Becker C., Franchi L., Yoshihara E., Chen Z., Mullooly N., Mielke L. A., Harris J., Coll R. C., Mills K. H., Mok K. H., Newsholme P., Nuñez G., Yodoi J., Kahn S. E., Lavelle E. C., O'Neill L. A. (2010) Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat. Immunol. 11, 897–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vandanmagsar B., Youm Y. H., Ravussin A., Galgani J. E., Stadler K., Mynatt R. L., Ravussin E., Stephens J. M., Dixit V. D. (2011) The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 17, 179–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stienstra R., van Diepen J. A., Tack C. J., Zaki M. H., van de Veerdonk F. L., Perera D., Neale G. A., Hooiveld G. J., Hijmans A., Vroegrijk I., van den Berg S., Romijn J., Rensen P. C., Joosten L. A., Netea M. G., Kanneganti T. D. (2011) Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. U.S.A. 108, 15324–15329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stienstra R., Joosten L. A., Koenen T., van Tits B., van Diepen J. A., van den Berg S. A., Rensen P. C., Voshol P. J., Fantuzzi G., Hijmans A., Kersten S., Müller M., van den Berg W. B., van Rooijen N., Wabitsch M., Kullberg B. J., van der Meer J. W., Kanneganti T., Tack C. J., Netea M. G. (2010) The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 12, 593–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sutterwala F. S., Ogura Y., Szczepanik M., Lara-Tejero M., Lichtenberger G. S., Grant E. P., Bertin J., Coyle A. J., Galán J. E., Askenase P. W., Flavell R. A. (2006) Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity 24, 317–327 [DOI] [PubMed] [Google Scholar]
- 38. Mariathasan S., Newton K., Monack D. M., Vucic D., French D. M., Lee W. P., Roose-Girma M., Erickson S., Dixit V. M. (2004) Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430, 213–218 [DOI] [PubMed] [Google Scholar]
- 39. Itoh K., Chiba T., Takahashi S., Ishii T., Igarashi K., Katoh Y., Oyake T., Hayashi N., Satoh K., Hatayama I., Yamamoto M., Nabeshima Y. (1997) An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 236, 313–322 [DOI] [PubMed] [Google Scholar]
- 40. Fernandes-Alnemri T., Yu J. W., Juliana C., Solorzano L., Kang S., Wu J., Datta P., McCormick M., Huang L., McDermott E., Eisenlohr L., Landel C. P., Alnemri E. S. (2010) The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 11, 385–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Malhotra D., Portales-Casamar E., Singh A., Srivastava S., Arenillas D., Happel C., Shyr C., Wakabayashi N., Kensler T. W., Wasserman W. W., Biswal S. (2010) Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 38, 5718–5734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hirotsu Y., Katsuoka F., Funayama R., Nagashima T., Nishida Y., Nakayama K., Engel J. D., Yamamoto M. (2012) Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 40, 10228–10239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fernandes-Alnemri T., Wu J., Yu J. W., Datta P., Miller B., Jankowski W., Rosenberg S., Zhang J., Alnemri E. S. (2007) The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 14, 1590–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bauernfeind F., Bartok E., Rieger A., Franchi L., Núñez G., Hornung V. (2011) Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J. Immunol. 187, 613–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Muñoz-Planillo R., Kuffa P., Martínez-Colón G., Smith B. L., Rajendiran T. M., Núñez G. (2013) K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dostert C., Pétrilli V., Van Bruggen R., Steele C., Mossman B. T., Tschopp J. (2008) Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guarda G., Braun M., Staehli F., Tardivel A., Mattmann C., Förster I., Farlik M., Decker T., Du Pasquier R. A., Romero P., Tschopp J. (2011) Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34, 213–223 [DOI] [PubMed] [Google Scholar]
- 48. Zhou R., Tardivel A., Thorens B., Choi I., Tschopp J. (2010) Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 11, 136–140 [DOI] [PubMed] [Google Scholar]
- 49. Piantadosi C. A., Withers C. M., Bartz R. R., MacGarvey N. C., Fu P., Sweeney T. E., Welty-Wolf K. E., Suliman H. B. (2011) Heme oxygenase-1 couples activation of mitochondrial biogenesis to anti-inflammatory cytokine expression. J. Biol. Chem. 286, 16374–16385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Meissner F., Molawi K., Zychlinsky A. (2008) Superoxide dismutase 1 regulates caspase-1 and endotoxic shock. Nat. Immunol. 9, 866–872 [DOI] [PubMed] [Google Scholar]