

Background: The roles of the adhesion molecules desmoglein (Dsg) 2 and Dsg3 for keratinocyte adhesion and signaling are poorly understood.
Results: Dsg2 compensates for Dsg3 depletion with regard to cell cohesion, but, in contrast to Dsg3, does not regulate p38 MAPK signaling.
Conclusion: Dsg2 and Dsg3 contribute differently to cell adhesion and signaling pathways.
Significance: The results demonstrate unique functions of different Dsgs expressed in the same cells.
Keywords: Cell Adhesion, Desmosome, Keratinocyte, p38 MAPK, Pemphigus
Abstract
Desmosomal cadherins are transmembrane adhesion molecules that provide cell adhesion by interacting in the intercellular space of adjacent cells. In keratinocytes, several desmoglein (Dsg1–4) and desmocollin (Dsc1–3) isoforms are coexpressed. We have shown previously that Dsg2 is less important for keratinocyte cohesion compared with Dsg3 and that the latter forms a complex with p38 MAPK. In this study, we compared the involvement of Dsg2 and Dsg3 in the p38 MAPK-dependent regulation of keratinocyte cohesion. We show that loss of cell adhesion and keratin filament retraction induced by Dsg3 depletion is ameliorated by specific p38 MAPK inhibition. Furthermore, in contrast to depletion of Dsg2, siRNA-mediated silencing of Dsg3 induced p38 MAPK activation, which is in line with immunoprecipitation experiments demonstrating the interaction of activated p38 MAPK with Dsg3 but not with Dsg2. Cell fractionation into a cytoskeleton-unbound and a cytoskeleton-anchored desmosome-containing pool revealed that Dsg3, in contrast to Dsg2, is present in relevant amounts in the unbound pool in which activated p38 MAPK is predominantly detectable. Moreover, because loss of cell adhesion by Dsg3 depletion was partially rescued by p38 MAPK inhibition, we conclude that, besides its function as an adhesion molecule, Dsg3 is strengthening cell cohesion via modulation of p38 MAPK-dependent keratin filament reorganization. Nevertheless, because subsequent targeting of Dsg3 in Dsg2-depleted cells led to drastically enhanced keratinocyte dissociation and Dsg2 was enhanced at the membrane in Dsg3 knockout cells, we conclude that Dsg2 compensates for Dsg3 loss of function.
Introduction
Desmosomal cadherins are cell adhesion molecules consisting of an extracellular, a transmembrane, and an intracellular domain (1). The protein family consists of several desmoglein (Dsg1–4) and desmocollin (Dsc1–3) isoforms. Desmosomal cadherins are especially abundant in epithelial and non-epithelial tissues that have to withstand mechanical forces, such as the epidermis, the intestine, or the myocardium. By interacting with their extracellular domain, desmosomal cadherins provide adhesive strength to adjacent cells. With their intracellular domain, they are linked to the intermediate filament cytoskeleton via plakoglobin (PG),2 plakophilin, and desmoplakin (DP) (2). All desmosomal cadherin isoforms are expressed in the human epidermis in a layer- and differentiation-dependent manner. In the human epidermis, beside building the core of desmosomes, the desmosomal cadherins are also supposed to be present in the cell membrane outside of desmosomes (3, 4), whereas the expression of the anchoring protein DP is restricted to desmosomes. The relevance of adhesion mediated by desmosomal cadherins is illustrated by the autoimmune blistering skin disease pemphigus vulgaris. Circulating autoantibodies primarily targeting Dsg3 and/or Dsg1 induce intraepidermal separation, and patients suffer from blister formation in the epidermis and oral mucosa. In pemphigus vulgaris, blistering typically affects mucous membranes only when autoantibodies against Dsg3 are formed but mucous membranes and the epidermis when autoantibodies against both Dsg3 and Dsg1 are present. This is explained by Dsg1 expressed in high amounts in the epidermis, which compensates for the loss of Dsg3 function when only this isoform is targeted (5). Although the desmoglein compensation theory is discussed controversially (6, 7), it can be considered likely that desmosomal cadherins compensate the adhesive function of each other (8). Furthermore, it is well established that autoantibody-induced signaling is necessary for pronounced blistering (9, 10). So far, an involvement in pemphigus pathogenesis is discussed primarily for protein kinase C activation (11, 12), plakoglobin (13), epidermal growth factor signaling (14–16), and, most importantly, p38 MAPK activation (17–19). Recently, we described an adhesion-dependent signaling complex consisting of Dsg3 and activated, e.g. phosphorylated, p38 MAPK (p-p38 MAPK), which established a link between p38 MAPK activation and loss of Dsg3 interaction in pemphigus vulgaris (20). Furthermore, Dsg3, via interaction with E-cadherin, has been demonstrated to be involved in Src signaling (3, 21).
Dsg2, which is the most widespread desmosomal cadherin isoform, may be involved in mediating cell signaling events via an interaction with caveolin-1 (22) and was also found to be a mediator of apoptosis (23). In intestinal epithelial cells, in which Dsg2 and Dsc2 are the only expressed desmosomal cadherin isoforms, Dsg2 is important for cell cohesion and maintaining intestinal epithelial barrier integrity (24). However, in keratinocytes, Dsg2 was shown to be important for cell cohesion under conditions of increased shear only (25). In keratinocytes, no specific role for Dsg2 in signaling cascades or overall cell cohesion has been described yet.
Therefore, in view of our recent finding of a p38 MAPK-Dsg3 complex, we investigated the contribution of Dsg2 and Dsg3 in regulating p38 MAPK activity and cell adhesion in this study. Our data provide evidence that Dsg3, in contrast to Dsg2, regulates p38 MAPK activity in human keratinocytes. Furthermore, Dsg3 contributes to cell adhesion not only by its function as an adhesion molecule but also by tuning p38 MAPK activity and keratin filament organization. In addition, our data also denote a new function for Dsg2 to compensate for Dsg3 because Dsg3 deficiency in primary murine keratinocytes resulted in pronounced membrane localization of Dsg2, and keratinocytes with simultaneous Dsg2 and Dsg3 depletion revealed a drastically increased loss of cell cohesion.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
For detection of proteins by immunostaining and/or Western blot analysis, the following primary antibodies were used: anti-α-tubulin mAb (Abcam), anti-β-actin mAb (Sigma), anti-Dsg2 mAb (clone 10G11, Progen, Heidelberg, Germany), anti-Dsg2 pAb (clone rb5, Progen), anti-Dsg2 mAb (Abcam), anti-Dsg3 pAb (clone H-145, Santa Cruz Biotechnology, Santa Cruz, CA), anti-Dsg3 mAb (clone 5G11, Invitrogen), anti-Dsg3 pAb (clone M-20, Santa Cruz Biotechnology), anti-desmoplakin mAb (Epitomics, Burlingame, CA), anti-GAPDH mAb (Santa Cruz Biotechnology), anti-PG mAb (Progen), anti-p38 MAPK pAb (Cell Signaling Technology, Danvers, MA), and anti-phospho-p38 MAPK pAb (Cell Signaling Technology). Horseradish peroxidase-linked anti-rabbit IgG antibody (Cell Signaling Technology), anti-mouse IgG and IgM antibody, and Cy3-labeled goat anti-mouse antibody (Dianova, Hamburg, Germany) were used as secondary antibodies. FITC-conjugated pan-cytokeratin (panCK) mAb was used to stain keratin filaments. AK23 (host, mouse; isotype, IgG) is a monoclonal antibody targeting Dsg3 (Biozol, Eching, Germany) and was utilized for the incubation steps in the cell culture model at a concentration of 75 μg/ml. The specific p38 MAPK inhibitor SB202190 (Merck, Darmstadt, Germany) was applied at a concentration of 30 μmol/liter for 24 h either alone or 1 h before AK23 incubation started.
Genotyping
One-day-old littermates of heterozygous B6;129X1-Dsg3tm1Stan/J mice (26) (The Jackson Laboratory, Bar Harbor, ME) were used for the isolation of primary murine keratinocytes. Homozygous Dsg3+/+ and Dsg3−/− mice were used for cell preparation only. To genotype neonatal mice, animals were decapitated, and 2-mm tail pieces were heated with 25 mmol/liter NaOH and 0.2 mmol/liter EDTA for 1 h at 98 °C. After addition of 40 mmol/liter Tris-HCl (pH 5.5) and centrifugation at 8000 rpm for 3 min, 2-μl aliquots per DNA were taken for PCR analysis. PCR was carried out as described elsewhere (27).
Cell Culture
The spontaneously immortalized human keratinocyte cell line HaCaT (ATCC) was grown in DMEM (Invitrogen, 1.8 mmol/liter Ca2+) supplemented with 10% fetal bovine serum (Biochrom, Berlin, Germany), 50 units/ml penicillin (AppliChem, Darmstadt, Germany), and 50 μg/ml streptomycin (AppliChem) and maintained in a humidified atmosphere containing 5% CO2 at 37 °C. Normal human epidermal keratinocytes were purchased from PromoCell (Heidelberg, Germany) and cultured in keratinocyte growth medium 2 with supplement mix and 0.06 mmol/liter Ca2+ (PromoCell) according to the protocol of the manufacturer. Normal human epidermal keratinocytes cells were switched to 1.8 mmol/liter Ca2+ (for 24 h) before scraping for immunoprecipitation. Primary murine epidermal keratinocytes (MEKs) were isolated from the epidermises of neonatal mice. The skins of 1-day-old mice were incubated for 16 h in a dispase solution (>2.4 units/ml Dispase II in Hanks' buffered saline solution (Sigma)) supplemented with gentamycin and amphotericin B (CELLnTEC, Bern, Switzerland). After carefully separating the dermis and the epidermis, the latter was incubated for 20 min on Accutase (CELLnTEC) at room temperature to dissociate cells. Keratinocytes were resuspended in keratinocyte growth medium 2 and maintained in a humidified atmosphere containing 5% CO2 at 37 °C. Cells were switched to 1.8 mmol/liter Ca2+ 24 h before they were subjected to immunofluorescence or Western blot analysis.
RNA Isolation and Complementary DNA Preparation
Total RNA was isolated from murine epidermal keratinocytes by using an RNeasy Plus mini kit (Qiagen, Venlo, Netherlands). In short, cells were cultured in 10-cm2 dishes until they reached 90% confluence and then switched to a high calcium concentration (1.8 mmol/liter Ca2+) for 24 h. Lysis of the cultured keratinocytes and RNA extraction were performed according to the instructions of the manufacturer. The RNA was quantified spectrophotometrically with an Infinite M200 PRO Tecan reader (Tecan, Maennedorf, Switzerland). First-strand cDNA was synthesized from 3 μg of total RNA using oligo(dT) 15 primers (Promega, Madison, WI) and a SuperScript II reverse transcriptase kit (Invitrogen) as described in the protocol of the manufacturer.
Quantitative Real-time (RT) PCR
The efficiency of all primer pairs was validated by PCR, and the size of primer products was proven by ethidium bromide staining on agarose gels. 4 μl of complementary cDNA (equivalent to 60 ng of total RNA) were used in a 20-μl PCR mix containing EvaGreen Supermix (Bio-Rad), RNase-free water, and a final concentration of 350 nm gene-specific primers: Dsg2, 5′-CGCACCAGGAAAGTACCAG-3′ (forward) and 5′-CCACAGTGGCATATCAACAGC-3′ (reverse); β-actin, 5′-TTCGTTGCCGGTCCACA-3′ (forward) and 5′-ACCAGCGCAGCGATATCG-3′ (reverse). Quantitative PCR experiments were performed using the CFX96 real-time system (Bio-Rad). After an initial denaturation step at 95 °C for 2 min, the amplification was carried out in 40 cycles involving denaturation for 5 s at 95 °C and annealing for 3 s at 56 °C. The specificity of the PCR products was confirmed by a subsequent melting curve analysis from 60–95 °C with an increment of 0.5 °C for 5 s. All samples were analyzed in duplicate. Dsg2 expression was normalized to the expression of a reference gene such as β-actin involving efficiency (E) and quantification cycle (Ct). Because the reaction efficiencies of both primer pairs were close to 100%, E was set to 2. The relative RNA expression was calculated using the following formula: 2(Ct reference − Ct gene of interest), where Ct corresponds to the number of cycles needed to generate a fluorescent signal above a predefined threshold. The average Ct value for the reference gene (β-actin) was subtracted from the average Ct value of the selected gene of interest (Dsg2). Paired Student's t test was applied to compare the analyzed groups.
siRNA-mediated Gene Silencing
siRNA-mediated gene silencing was performed as described previously (25). ON-Target plus SMARTpool siRNA was purchased from Thermo Scientific/Dharmacon (Lafayette, CO) (human Dsg2 (catalog no. L-011645-00-0005), human Dsg3 (L-011646-00-0005), non-targeting control siRNA (D-001810-10-05)), and the transfection reagent TurboFectTM was purchased from Fermentas (Thermo Scientific, Waltham, MA). Cells were seeded in 24-well plates and transfected at a confluence stage of 70–80% according to the TurboFectTM transfection protocol. The medium was replaced by FCS containing DMEM 24 h after transfection, and incubation steps were started after another 24 h.
Immunostaining
To visualize proteins by immunostaining, cells were seeded on glass coverslips. After transfection or incubation steps for the indicated times, cells were fixed for 10 min with 2% formalin (freshly prepared from paraformaldehyde) in PBS at room temperature and then permeabilized with 0.1% Triton X-100 in PBS for 5 min. To prevent unspecific binding, a solution of 3% bovine serum albumin and 1% normal goat serum was applied for 40 min before incubation with the respective primary antibodies overnight at 4 °C. After three PBS washing steps, appropriate secondary antibodies were applied for 1 h at room temperature. FITC-conjugated pan-cytokeratin antibody was incubated another 24 h on the same coverslips, and then coverslips were mounted with n-propyl gallate as an antifading compound. A Leica SP5 confocal microscope with a 63× numerical aperture 1.4 PL APO objective (both Leica Microsystems, Wetzlar, Germany) was used to image the immunostained cells.
Western Blot Analysis
Cell lysates for Western blot analysis were prepared either 3 days after siRNA transfection or immediately after performing a Triton assay or immunoprecipitation. After siRNA transfection, cells were scraped into SDS lysis buffer containing 25 mmol/liter HEPES, 2 mmol/liter EDTA, 25 mmol/liter NaF, 1% sodium dodecyl sulfate (pH 7.4), and a protease inhibitor mixture (Roche) and sonicated immediately. Cell lysates for Western blot analysis after the Triton assay or immunoprecipitation were handled in the same way except for the use of the appropriate lysis buffer. For all samples, a BCA protein assay kit (Pierce/Thermo Scientific) was used to determine the protein concentration according to the protocol of the manufacturer. For each experimental setup, samples were mixed with Laemmli buffer (28), and for each condition, equal protein amount were loaded onto polyacrylamide gels (self-made). After transfer to a nitrocellulose membrane and blocking with 5% milk solution, primary antibodies were incubated at 4 °C overnight. Horseradish peroxidase-conjugated secondary antibodies and an ECL reaction system (self-made solutions) were used to visualize the proteins.
Immunoprecipitation
To concentrate Dsg2 or Dsg3 in cell lysates and to investigate whether these proteins form a complex with p-p38 MAPK, immunoprecipitation was performed. T75 flasks of confluent cells were washed once with ice-cold PBS and incubated with 1 ml of radioimmune precipitation assay buffer (0.05 mol/liter Tris-HCl, 0.15 mol/liter NaCl, 0.1% SDS, 1% Nonidet P-40, 0.0001 mol/liter EDTA) for 30 min on ice under gentle shaking. After centrifugation at 13,000 rpm for 5 min on 4 °C, the protein concentration of the cell lysate supernatant was measured with a BCA protein assay kit (Pierce/Thermo Scientific). A protein amount of 600–1000 μg was filled up to 500 μl with radioimmune precipitation assay buffer, and, after preclearing with protein A/G beads (Santa Cruz Biotechnology), antibodies (1 μg of a-Dsg2 mAb and 1 μg of a-Dsg3 pAb) were incubated for 3 h at 4 °C on a rotating incubator. 40 μl of protein A/G beads were then applied to this antibody/lysate mixture, incubated overnight at 4 °C, and afterward washed with radioimmune precipitation assay wash buffer (0.05 mol/liter Tris-HCl, 0.15 mol/liter NaCl, 0.1% SDS, and 0.1% Nonidet P-40). Following centrifugation and washing, beads were mixed with Laemmli buffer (28), boiled for 10 min at 95 °C, and subjected to Western blot analysis.
Triton X-100 Protein Fractionation
Protein fractionation was carried out as described previously (25). Incubation of HaCaT cells with a buffering system containing 0.5% Triton X-100, 50 mmol/liter MES, 25 mmol/liter EGTA, and 5 mmol/liter MgCl2 for 15 min on ice under gentle shaking divided the whole protein pool into a cytoskeleton-anchored (Triton-insoluble) and a cytoskeleton-unanchored (Triton-soluble) fraction. To distinguish between both fractions, the cell lysates were centrifuged at 13,000 rpm for 5 min and separated into supernatant (Triton-soluble) and pellet (Triton-insoluble). The pellets were resuspended in SDS lysis buffer, and the protein concentration of the pellet solution and the supernatant was determined as described above.
Dispase-based Dissociation Assay
For the dissociation assay, HaCaT cells were seeded in 24-well plates, transfected 24 h later, or incubated with AK23 or SB202190 3 days after seeding. For all experiments, a dispase-based dissociation assay was performed 4 days after seeding. After removing the medium and washing with PBS, 150 μl of the enzyme dispase (>2.4 units/ml Dispase II in Hanks' buffered saline solution (Sigma)) were incubated for 20 min at 37 °C to detach the whole cell monolayer from the well bottom. After addition of 200 μl of Hanḱs buffered saline solution, cell monolayers were exposed to mechanical stress by pipetting them five times up and down with a 1-ml electrical pipette. In each well, the resulting fragments were counted with a binocular stereo microscope (Leica). For the dispase-based dissociation assays with MEK cells, SB202190 was applied for 1 h starting 23 h after the Ca2+ switch.
Statistics
For the quantification of the band density of Western blot experiments, ImageJ software was used. ImageJ was also used for the quantification of fluorescence intensity of the indicated proteins on the cell membrane and for measuring the extent of cytokeratin retraction of immunofluorescence experiments. The distance between the aggregation of intermediate filaments of opposing cells was measured essentially as described previously (20). To rule out variations resulting from different cell densities, a quotient was calculated by dividing the length of the retraction of keratin filaments between adjacent cells with the distance between nuclei of the same cells. The standard error of the mean is illustrated by the error bars in all column graphs. Statistical significance was either calculated by Student's t test or by analysis of variance followed by Bonferroni's correction for multiple-group samples. Significance was assumed at p < 0.05.
RESULTS
Loss of Cell Cohesion in Dsg3-depleted Keratinocytes Is in Part Dependent on p38 MAPK Signaling
We have shown previously that Dsg2 is of minor importance for keratinocyte cohesion when compared with Dsg3 under conditions of low shear but contributed significantly to cell cohesion under higher mechanic stress (25). Moreover, we detected that Dsg3 forms a complex with p38 MAPK (20). In this study, we investigated the relevance of both desmogleins for intracellular signaling involved in cell cohesion, specifically p38 MAPK. First, Dsg3 was targeted either by a monoclonal antibody derived from a pemphigus mouse model (AK23) that directly interferes with Dsg3 binding (29–31) or by siRNA. Cell cohesion was quantified by dispase-based dissociation assays. Both targeting Dsg3 with AK23 (fragment number 24.0 ± 2.5) or by siRNA (12.5 ± 2.7) resulted in a loss of cell cohesion compared with cells transfected with non-targeting siRNA (2.0 ± 0.3). Strikingly, 1 h of preincubation with the p38 MAPK inhibitor SB202190 (3.4 ± 1.2) or 24-h incubation of Dsg3-depleted cells with SB202190 significantly reduced the number of fragments (5.6 ± 1.5). SB202190 treatment for 24 h alone had no effect (2.9 ± 0.8) (Fig. 1A).
FIGURE 1.

Targeting of Dsg3 leads to p38 MAPK-dependent loss of cell cohesion. A, targeting of Dsg3 function by either incubation with AK23 or siRNA-mediated depletion of Dsg3 protein levels in HaCaT cells resulted in increased cell monolayer fragmentation in dispase-based dissociation assays. Both effects were blocked by 24-h incubation with the specific p38 MAPK inhibitor SB202190 (n ≥ 6). *, p < 0.05. B, representative Western blot analysis for the detection of successful Dsg3 knockdown in HaCaT cells. Lysates were prepared in parallel with the experiments presented in A. α-Tubulin was used as a loading control (n ≥ 6). C, dispase-based dissociation assays were performed in primary MEKs 24 h after Ca2+ switch from 0.06 mmol/liter to 1.8 mmol/liter Ca2+. The loss of cell cohesion detectable in Dsg3−/− cells was blocked significantly by 1 h of incubation with SB202190 (n ≥ 8). *, p < 0.05 versus Dsg3−/− control. D, representative PCR analysis of Dsg3+/−, Dsg3+/+, and Dsg3−/− mice. E, representative Western blot analysis performed with whole cell lysates of primary keratinocytes from Dsg3+/+ and Dsg3−/− mice 24 h after Ca2+ switch from 0.06 mmol/liter to 1.8 mmol/liter Ca2+. α-Tubulin was used as a loading control (n = 3).
To exclude possible effects caused by differences in knockdown efficiency, primary keratinocytes were isolated from the epidermis of neonatal mice carrying a targeted mutation of Dsg3 to gain cells with a complete depletion of Dsg3 (Dsg3−/−) and appropriate control cells with endogenous Dsg3 expression (Dsg3+/+). Compared with cells with endogenous Dsg3 expression (fragment number 2.2 ± 0.5), Dsg3-depleted cells were not able to withstand mechanically induced stress (47.1 ± 4.8). Similar to HaCaT cells after Dsg3 silencing, fragment numbers were significantly reduced by 1 h of incubation with SB202190 (26.6 ± 4.6). Inhibition of p38 MAPK phosphorylation by SB202190 incubation for 1 h had no effect on Dsg3+/+ cells (3.4 ± 0.7) (Fig. 1C). The fact that SB202190 ameliorated the loss of cell adhesion induced by Dsg3 depletion in two different model systems demonstrates that Dsg3 controls cell cohesion not only via its function as adhesion molecule but also by orchestrating p38 MAPK signaling.
Dsg2 Compensates for Loss of Dsg3 Adhesive Function
To further characterize the role of Dsg2 in keratinocytes, we targeted Dsg3 function in Dsg2-depleted cells, again by application of AK23 or by siRNA (Fig. 2A). Knockdown efficiency was controlled by Western blot analysis in parallel with the dissociation assays (Fig. 2B). Simultaneous siRNA-mediated depletion of Dsg2 and Dsg3 resulted in drastically enhanced loss of cell cohesion (fragment number 120.4 ± 23.7, Fig. 2A) compared with single depletion of either Dsg2 (1.3 ± 0.2, Fig. 2A) or Dsg3 (12.5 ± 2.7, already shown in Fig. 1A) and was reduced significantly by 24-h incubation with SB202190 (48.2 ± 19.5, Fig. 2A). Similar results were obtained by subsequent incubation of Dsg2-depleted HaCaT cells with AK23 (364.6 ± 30.6). This effect was also blunted by 1 h of preincubation with SB202190 (209.8 ± 20.1) (Fig. 2A). This dramatic increase in fragment numbers indicates that Dsg2 compensates for the adhesive function of Dsg3 in HaCaT cells. Dsg2-depleted HaCaT cells (44.36 ± 5.77% of n.t. siRNA) showed reduced Dsg3 protein levels (75.26 ± 7.50% of n.t. siRNA). In contrast, following Dsg3 knockdown (40.57 ± 8.32%), Dsg2 levels were not altered (100.17 ± 2.56%) which is in line with previous data (25) (Fig. 2, B–D). Dsg2 protein levels were also not changed significantly in Dsg3−/− MEK cells compared with Dsg3+/+ cells (85.22 ± 25.53%, Fig. 3, A and B). Nevertheless, the loss of Dsg3 in these primary keratinocytes resulted in significantly decreased PG protein levels (43.91 ± 13.53%). Interestingly, Dsg2 as well as p-p38 MAPK localized to cell borders in Dsg3−/− MEK cells but not in Dsg3+/+ controls (Fig. 3, C and D). Although Dsg2 protein levels were not changed in Dsg3−/− mice, mRNA expression of Dsg2 was increased slightly (155.70 ± 10.75% of Dsg2 mRNA expression in Dsg3+/+ MEK cells) (Fig. 3E). Similar to the results obtained in MEK cells, Dsg2 staining appeared to be more pronounced along junctions (209.24 ± 6.81% versus n.t. siRNA) in HaCaT cells that were depleted for Dsg3 via siRNA (38.73 ± 2.81% versus n.t. siRNA) (Fig. 4, A and B).
FIGURE 2.

Dsg2 compensates for Dsg3 loss of function. A, fragment numbers were increased drastically after double knockdown of Dsg2 and Dsg3 and after incubation of Dsg2-depleted cells with AK23. Both effects were reduced by 24-h incubation with the specific p38 MAPK inhibitor SB202190 (n ≥ 6). *, p < 0.05. B, representative Western blot analysis of lysates prepared in parallel with the dispase-based dissociation assays shown in Figs. 1A and 2A to prove knockdown efficiency. C, band density of at least five representative experiments per siRNA-silencing condition. Values for Dsg2 protein levels per indicated knockdown condition were normalized to α-tubulin and calculated as percent of the obtained value for n.t. siRNA condition (n ≥ 5). *, p < 0.05 versus n.t. siRNA. D, values for Dsg3 protein levels per indicated knockdown condition normalized to α-tubulin and calculated as percent of the obtained value for n.t. siRNA condition (n ≥ 5). *, p < 0.05 versus n.t. siRNA.
FIGURE 3.

Dsg3 depletion recruits Dsg2 and p-p38 MAPK to the plasma membrane, accompanied by alterations in PG protein and Dsg2 mRNA expression. A, representative Western blot analysis with detection of Dsg2 and PG in total cell lysates of MEK cells 24 h after Ca2+ switch. α-Tubulin was used as a loading control. Dsg3−/− keratinocytes showed decreased PG protein levels compared with cells isolated from Dsg3+/+ mice (n ≥ 6). B, the band density of at least six different mice per genotype was analyzed. Values were normalized to α-tubulin and calculated as percent of protein levels detected in keratinocytes of Dsg3+/+ mice (n ≥ 6). *, p < 0.05 versus Dsg3+/+. C, immunofluorescence staining of Dsg3+/+ and Dsg3−/− cells (MEK) after 24-h Ca2+ switch confirmed depletion of Dsg3 in Dsg3−/− cells and revealed a more membrane-associated localization of Dsg2 and p-p38 MAPK in Dsg3−/− cells compared with Dsg3+/+ cells (n ≥ 3). Scale bar = 25 μm. D, the fluorescence intensity of Dsg2 on the cell membrane was analyzed in Dsg3+/+ and Dsg3−/− cells, and values obtained in Dsg3−/− cells were calculated as percent of values obtained in Dsg3+/+ cells. Sixty different cells were analyzed in total of three independent keratinocyte preparations per group. *, p < 0.05 versus Dsg3+/+. E, Dsg2 mRNA expression evaluated by quantitative RT-PCR. Dsg2 mRNA expression was normalized to the mRNA expression of β-actin. The gene expression values for Dsg3−/− MEK cells were calculated as percent of values obtained for Dsg3+/+ MEK cells (n = 4). *, p < 0.05 versus Dsg3+/+.
FIGURE 4.
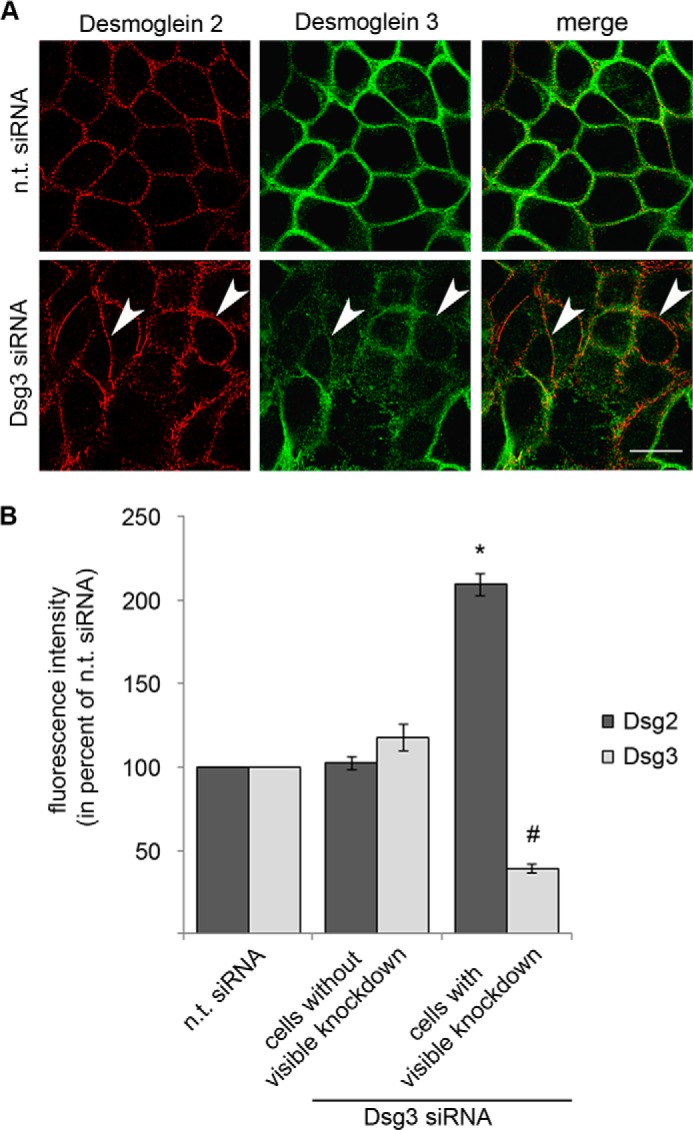
Dsg2 cell membrane staining is increased following Dsg3 depletion. A, immunostaining of HaCaT cells after either n. t. siRNA or Dsg3 siRNA transfection. Dsg2 is shown in red, and Dsg3 is shown in green. A more pronounced and linearized membrane staining of Dsg2 was detectable in cells with Dsg3 depletion (arrowheads) (n = 3). Scale bar = 20 μm. B, fluorescence intensity was analyzed for Dsg2 and Dsg3 after n.t. siRNA or Dsg3 siRNA transfection. After transfection with Dsg3 siRNA, cells with and without visible knockdown were distinguished. Three different membrane spots per cell were measured for Dsg2 and Dsg3 in parallel. In total, 45 cells of three independent experiments were analyzed per condition (15 for each experiment). *, p < 0.05 versus n.t. siRNA; #, p < 0.05 versus n.t. siRNA.
Dsg2, in Contrast to Dsg3, Is Not Involved in p38 MAPK Signaling in Human Keratinocytes
In view of the protective effect of p38 MAPK inhibition on cell cohesion in Dsg3-depleted cells, we evaluated the protein levels of p-p38 MAPK under conditions of reduced Dsg2 or Dsg3 content. Loss of cell cohesion correlated with the activity of p38 MAPK because enhanced phosphorylation of p38 MAPK was observed by Western blot analysis after siRNA-mediated Dsg3 silencing (146.9 ± 10.9%). In contrast, depletion of Dsg2 by siRNA had no effect on p38 MAPK phosphorylation compared with cells transfected with non-targeting siRNA (116.2 ± 5.1%) (Fig. 5, A and B). Because we have previously demonstrated a complex of Dsg3 and active p38 MAPK (20), we tested whether Dsg2 is also capable to bind p-p38 MAPK. However, in contrast to Dsg3, complex formation of Dsg2 and p-p38 MAPK was not detectable (Fig. 5C). Similar results were obtained in primary human keratinocytes (Fig. 5D). Taken together, these data indicate that Dsg3, but not Dsg2, is involved in the regulation of p38 MAPK signaling.
FIGURE 5.

p38 MAPK is activated following Dsg3 depletion and forms a complex with Dsg3. A, HaCaT cells were transfected with siRNA targeting either Dsg2 or Dsg3, and successful knockdown was proven by reduced protein levels in a Western blot analysis against non-targeting controls. β-Actin was used as a loading control. In contrast to Dsg2, gene silencing of Dsg3 by siRNA led to an increase in p-p38 MAPK protein levels. Total p38 MAPK was used as a loading control (n ≥ 3). B, band density of p-p38 MAPK protein levels after indicated knockdown conditions. p38 MAPK protein levels were used as loading controls, and all values were calculated as percent of values under n.t. siRNA conditions (n ≥ 3). *, p < 0.05. C, following immunoprecipitation (IP) in whole HaCaT cell lysates with either a-Dsg2 monoclonal antibody or a-Dsg3 polyclonal antibody, phosphorylated p38 MAPK and p38 MAPK were detectable after Dsg3 pulldown only (n = 3). D, following immunoprecipitation in whole normal human epidermal keratinocyte (NHEK) cell lysates with either a-Dsg2 monoclonal antibody or a-Dsg3 polyclonal antibody, phosphorylated p38 MAPK was detectable after Dsg3 pulldown only (n = 3).
Dsg3, via p38 MAPK, Regulates the Spatial Distribution of the Keratin Filament Network
Our data indicate that Dsg3 strengthens cell adhesion by mechanisms independent of its adhesive properties. One of the hallmarks of pemphigus is the so-called keratin filament retraction (32). Because we have shown previously that the AK23-mediated collapse of the keratin filament network was mediated via p38 MAPK (20), we tested the effects of Dsg2 and Dsg3 depletion on keratin filament distribution by immunostaining for cytokeratins. In cells treated with Dsg2 siRNA, keratin filaments were spanning the entire cytosol and reached the region underneath the cell membrane, regardless of whether an effective Dsg2 depletion was detectable (Fig. 6, A, a–c, arrows, and B) or not (arrowheads). However, following depletion of Dsg3, keratin filaments appeared to be retracted from cell borders and clustered around the nucleus in cells without Dsg3 staining (Fig. 6, A, d–f, arrows, and B), whereas, in cells with a normal distribution of Dsg3, the keratin filament network appeared unaffected (arrowheads). Interestingly, incubation with SB202190 largely prevented keratin filament retraction in Dsg3-depleted cells (Fig. 6, A, g–i, and B). Incubation of cells transfected with Dsg3 siRNA with SB202190 did not affect the efficiency of Dsg3 depletion (Fig. 6, C and D). These data suggest that keratin filament retraction is mediated by activation of p38 MAPK subsequent to Dsg3 depletion and indicate that keratin filament retraction does not result from depletion of the adhesion molecules itself but, rather, is caused by p38 MAPK activation, which is regulated by Dsg3 but not by Dsg2.
FIGURE 6.
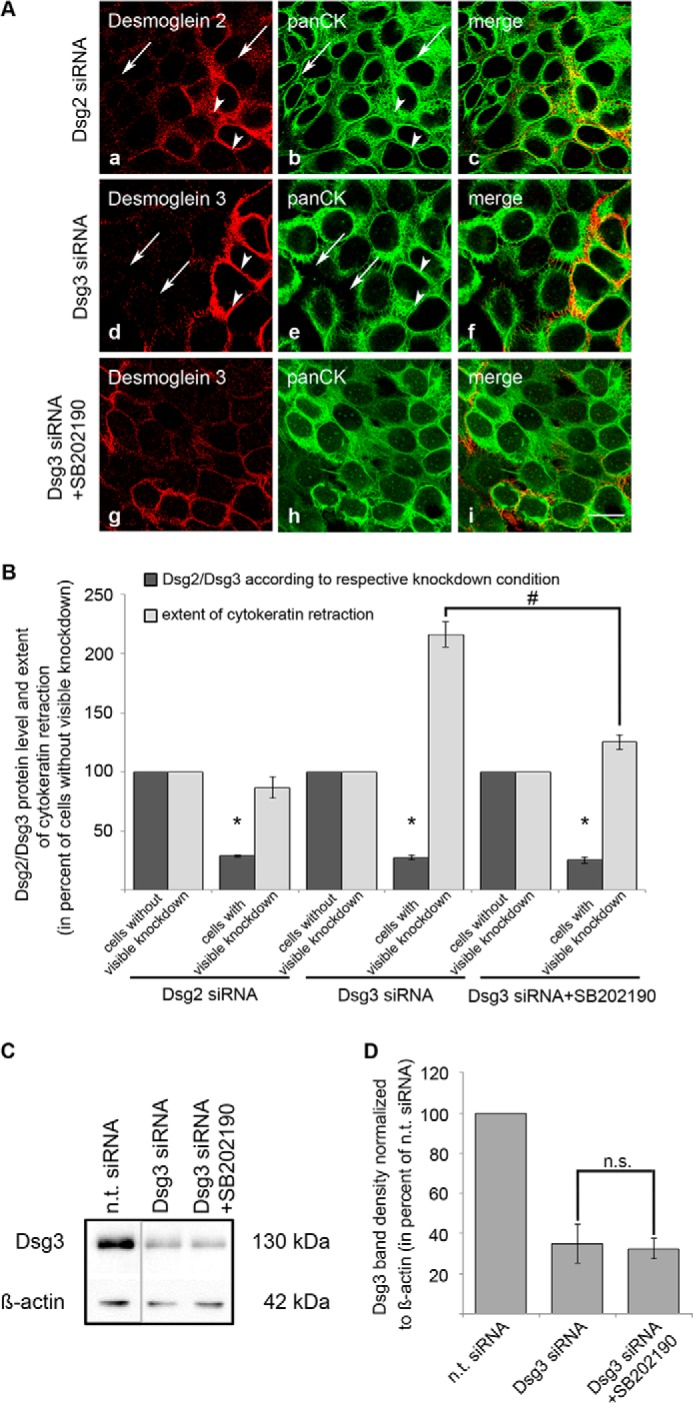
Keratin filament retraction induced by Dsg3 silencing was largely prevented by SB202190. A, a–c, siRNA-mediated silencing of Dsg2 (red) did not affect the organization of the keratin filament network (green) in HaCaT cells. Arrows indicate cells with Dsg2 depletion, and arrowheads show cells with unaffected endogenous Dsg2 expression (n = 3). d–f, in cells with abolished Dsg3 expression after siRNA-mediated silencing, keratin filament retraction was detectable (green, arrows) compared with adjacent cells with unaffected Dsg3 expression (red, arrowheads) (n = 3). g–i, this effect was blocked by 24-h incubation with SB202190 (n = 3). Scale bar = 20 μm. B, to evaluate the extent of cytokeratin retraction, cells were first classified in cells with or without visible knockdown by measuring the fluorescence intensity of either Dsg2 or Dsg3 of three membrane spots per cell. In the same cells, the distance between the bulk mass of cytokeratin filaments of adjacent cells was measured and divided by the distance between cell nuclei. This was also calculated on three different areas per cell. Under the condition of Dsg2 depletion, 25 cells were analyzed of two independent experiments. After transfection with Dsg3 siRNA, 45 cells of three independent experiments were evaluated. *, p < 0.05 versus cells without visible knockdown under the same condition; #, p < 0.05. C, representative Western blot analysis after the indicated transfections showing that incubation of Dsg3-depleted HaCaT cells with SB202190 did not affect Dsg3 protein levels. β-Actin was used as a loading control (n = 3). D, band density for Dsg3 was normalized to β-actin, and values were calculated as percent of values obtained after n.t. siRNA transfection (n = 3). n.s., not significant.
Phosphorylated p38 MAPK Is Attached to both Cytoskeleton-bound and Non-cytoskeleton-associated Dsg3
Dsg3 is not only present in desmosomes but is also found outside of desmosomes in the cell membrane. The latter may be relevant for signaling into the cell (4). Thus, we performed Triton extraction to separately investigate the cytoskeleton-anchored protein pool (insoluble) and the cytoskeleton-unanchored pool (soluble). The insoluble pool was clearly identified as the desmosome-containing fraction by the presence of DP (Fig. 7A). Dsg2, in contrast to Dsg3, was hardly detectable in the Triton-soluble pool (4.4 versus 18.6% of total amount of the respective protein, Fig. 7B). This indicates that Dsg2, in contrast to Dsg3, is restricted predominantly to desmosomes. In contrast, the majorities of both p38 MAPK and p-p38 MAPK were found in the soluble pool (86.8 and 97.1%, respectively). Although the soluble pool also contains all cytoplasmic proteins, these data suggest that, predominantly, the non-cytoskeleton-bound pool of Dsg3 is responsible for modulating p38 MAPK activity. To further elucidate this possibility, we combined Triton extraction with immunoprecipitation experiments (Fig. 7C). Interestingly, after separate pulldown of Dsg3 from the soluble and the insoluble pools, Dsg3-associated p38 MAPK and p-p38 MAPK was detectable in both pools. Because only 5% of p-p38 MAPK was found in the insoluble pool, these data suggest that most of the cytoskeletal p38 MAPK was attached to Dsg3.
FIGURE 7.

Dsg3 forms a complex with p-p38 MAPK in both the Triton X-100-soluble and Triton X-100-insoluble pool. Western blot analysis following Triton X-100-mediated cell fractionation in HaCaT cells revealed that Dsg3 was present in the soluble (S) and insoluble (I) protein pools, whereas Dsg2 was restricted largely to the cytoskeletal fraction. DP served as an indicator to identify the insoluble fraction as the desmosome-containing pool. p38 MAPK and p-p38 MAPK were primarily detectable in the soluble pool of proteins. A, Western blot analysis of the indicated proteins after Triton X-100-mediated cell fractionation. B, calculation of band density for each protein in the soluble and insoluble fraction of four independent experiments. The bars colored in dark gray represent the percentage of total protein amount detectable in the Triton X-100-insoluble fraction, and the bars colored in light gray represent the protein amount in the Triton X-100-soluble fraction (n = 4). C, after cell fractionation of HaCaT cells by incubation with Triton X-100, a complex formation between Dsg3 and p-p38 MAPK was detectable in both the soluble and the insoluble fractions. DP was used to indicate the insoluble pool, and GAPDH served to denote the soluble fraction (n = 3). IP, immunoprecipitation.
DISCUSSION
The results of this study reveal that Dsg3 contributes to cell cohesion in keratinocytes by both its adhesive function and by regulating keratin filament organization via p38 MAPK. Additionally, our data indicate that Dsg2 serves to compensate for the loss of Dsg3 binding.
Dsg2 Compensates for the Loss of Dsg3 Function
In contrast to Dsg3, reduced protein levels of Dsg2 did not result in increased loss of cell adhesion when exposed to low shear, although both proteins are expressed in similar amounts in HaCaT cells (25). However, our recent data demonstrate that, under conditions of Dsg2 depletion and simultaneous targeting of Dsg3 function by AK23 or siRNA-knockdown, fragment numbers were increased dramatically compared with AK23 treatment or Dsg3 depletion alone. Moreover, Dsg2 was present at the membrane in Dsg3−/− keratinocytes only. This was accompanied by an increased expression of Dsg2 on mRNA level in Dsg3−/− cells but no detectable changes on Dsg2 protein level. This might be explained with an increased Dsg2 turnover at cell junctions after Dsg2 was being transported to the cell membrane to compensate for Dsg3 function in primary keratinocytes isolated from Dsg3 knockout mice. In contrast, Baron et al. (33) found an increased expression of Dsg2 in Dsg3−/− MEK cells. However, Baron et al. (33) used an antibody that detects both Dsg1 and Dsg2. According to our results, enhanced Dsg2 membrane localization may contribute to the compensation for loss of Dsg3 function. This is in line with a study in which superficially expressed Dsg2 was compensating for the loss of Dsg1 in mouse epidermis (34). Interestingly, in our study, p38 MAPK inhibition largely prevented the effect of AK23 but only partially reduced cell dissociation under simultaneous targeting of Dsg2 and Dsg3. This strengthens our data demonstrating that Dsg2 is not involved in the regulation of p38 MAPK activity. Nevertheless, it is possible that Dsg2 contributes to signaling events independently of the p38 MAPK/keratin filament pathway. This scenario is conceivable because Dsg2 is discussed to be associated with tumorigenesis in epithelial tissues (35, 36) and has been implicated in the regulation of apoptosis in intestinal cells (23). Furthermore, loss of Dsc2 has been reported recently to contribute to malignant transformation of intestinal epithelial cells via activation of Akt/β-catenin signaling (37). Thus, similar to classical cadherins, desmosomal cadherins should be recognized not only as adhesion molecules but, rather, as adhesion receptors with a variety of functions independent of their adhesive properties.
Dsg3 Stabilizes Cell Adhesion by Controlling p38 MAPK Signaling
Our data indicate fundamental differences for the function of Dsg3 and Dsg2 in keratinocytes. In line with previous data using AK23 to inhibit binding of Dsg3 (20), Dsg3 depletion induced loss of cell adhesion, p38 MAPK activation, and cytokeratin retraction. In contrast, Dsg2 depletion yielded none of these results and seemed relevant under conditions of simultaneous loss of Dsg3 function only. The fact that cell dissociation by Dsg3 depletion was prevented partially by p38 MAPK inhibition indicates a dual role for Dsg3. It serves as an adhesion molecule mediating trans-interaction to desmogleins on adjacent cells (30, 31, 38). In addition, Dsg3 controls p38 MAPK signaling, which is also relevant for cell adhesion. Accordingly, we found p38 MAPK to be associated with Dsg3 but not with Dsg2. The trigger for p38 MAPK activation seems to be the loss of binding of Dsg3 molecules. After addition of AK23 or specific peptides (both inhibiting Dsg3 interaction), p38 MAPK is activated rapidly in a complex with Dsg3 (20). However, under long term depletion of Dsg3 by siRNA, p38 MAPK may also be activated when not associated with Dsg3. Thus, trans-interacting Dsg3 may sequester a pool of p38 MAPK at the cell membrane and, by suppressing its activation, may stabilize cell adhesion. In this scenario, PG may play an important role as modulator of p38 MAPK signaling (39). In line with this, primary keratinocytes isolated from Dsg3 knockout mice showed reduced PG protein levels, which was reported to result in p38 MAPK activation and cytokeratin retraction (39). It has to be noted that p38 MAPK activation is apparently not an unspecific response to general loss of cell cohesion because cell dissociation by desmoplakin depletion does not lead to activation of p38 MAPK (39). Additionally, our data do not rule out the possibility that p38 MAPK may modulate cell adhesion independently of Dsg3 regulation. p38 MAPK regulates the keratin filament cytoskeleton (20, 40) and, thereby, modulates cell adhesion. In this study, the keratin filament collapse induced by Dsg3 depletion was prevented by p38 MAPK inhibition. It is likely that the partial decrease of cell dissociation observed under p38 MAPK inhibition is primarily a result of the restored keratin filament distribution. This result is intriguing because it demonstrates that cytokeratin filament retraction is not the consequence of desmoglein depletion itself but, rather, is a process orchestrated by p38 MAPK, which, in turn, is regulated by Dsg3 but not by Dsg2. Desmosomal cadherins are not restricted to desmosomes but are also found unanchored to intermediate filaments in the cell membrane (4). In HaCaT cells, Dsg2 is apparently restricted mainly to desmosomes, whereas relevant amounts of Dsg3 are found in the Triton-soluble pool. The roles for these extradesmosomal Dsg3 molecules remain unclear. It is tempting to speculate that, primarily, the extradesmosomal Dsg3 molecules serve as signaling initiators (41), which is supported by the notion that the Dsg3-p38 MAPK complex is found in the Triton-soluble pool. However, our immunoprecipitation experiments also revealed that the complex is not detectable exclusively in the soluble pool but also within desmosomes. Moreover, AK23, which induces p38 MAPK activation (20), has been shown to also bind to Dsg3 within desmosomes (42). Thus, the contribution of desmosomal versus non-desmosomal Dsg3 to signaling events needs to be clarified further.
Taken together, a scenario is conceivable in which loss of function of desmosomal and/or non-desmosomal Dsg3, either by pathogenic autoantibodies or by siRNA-mediated depletion, results in phosphorylation of p38 MAPK. Subsequently, loss of cell adhesion is augmented by p38 MAPK-mediated keratin filament retraction from desmosomes (Fig. 8).
FIGURE 8.

Mechanisms of Dsg2- and Dsg3-mediated cell-cell adhesion in keratinocytes. Targeting of Dsg3 binding either by pemphigus antibodies or siRNA leads to p38 MAPK activation and following retraction of the keratin filament network, which both negatively affect cell-cell cohesion in keratinocytes. In contrast, Dsg2 may serve primarily as a compensation partner for the loss of Dsg3 function. P designates phosphorylated p38 MAPK.
Taken together, our data demonstrate unique roles for specific desmosomal cadherins. Dsg3 is relevant for cell adhesion, at least in part, by controlling p38 MAPK activity. In contrast, Dsg2 apparently exhibits compensating properties for loss of Dsg3 function in keratinocytes.
Acknowledgments
We thank Martina Hitzenbichler and Cathleen Plietz for technical assistance.
Footnotes
- PG
- plakoglobin
- DP
- desmoplakin
- p-p38 MAPK
- phosphorylated p38 MAPK
- mAb
- monoclonal antibody
- pAb
- polyclonal antibody
- MEK
- murine epidermal keratinocyte
- n.t.
- non-targeting.
REFERENCES
- 1. Delva E., Tucker D. K., Kowalczyk A. P. (2009) The desmosome. Cold Spring Harb. Perspect. Biol. 1, a002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Waschke J. (2008) The desmosome and pemphigus. Histochem. Cell Biol. 130, 21–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tsang S. M., Brown L., Lin K., Liu L., Piper K., O'Toole E. A., Grose R., Hart I. R., Garrod D. R., Fortune F., Wan H. (2012) Non-junctional human desmoglein 3 acts as an upstream regulator of Src in E-cadherin adhesion, a pathway possibly involved in the pathogenesis of pemphigus vulgaris. J. Pathol. 227, 81–93 [DOI] [PubMed] [Google Scholar]
- 4. Kitajima Y. (2013) New insights into desmosome regulation and pemphigus blistering as a desmosome-remodeling disease. Kaohsiung J. Med. Sci. 29, 1–13 [DOI] [PubMed] [Google Scholar]
- 5. Stanley J. R., Amagai M. (2006) Pemphigus, bullous impetigo, and the staphylococcal scalded-skin syndrome. N. Engl. J. Med. 355, 1800–1810 [DOI] [PubMed] [Google Scholar]
- 6. Spindler V., Drenckhahn D., Zillikens D., Waschke J. (2007) Pemphigus IgG causes skin splitting in the presence of both desmoglein 1 and desmoglein 3. Am. J. Pathol. 171, 906–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grando S. A., Bystryn J. C., Chernyavsky A. I., Frusić-Zlotkin M., Gniadecki R., Lotti R., Milner Y., Pittelkow M. R., Pincelli C. (2009) Apoptolysis: a novel mechanism of skin blistering in pemphigus vulgaris linking the apoptotic pathways to basal cell shrinkage and suprabasal acantholysis. Exp. Dermatol. 18, 764–770 [DOI] [PubMed] [Google Scholar]
- 8. Wu H., Wang Z. H., Yan A., Lyle S., Fakharzadeh S., Wahl J. K., Wheelock M. J., Ishikawa H., Uitto J., Amagai M., Stanley J. R. (2000) Protection against pemphigus foliaceus by desmoglein 3 in neonates. N. Engl. J. Med. 343, 31–35 [DOI] [PubMed] [Google Scholar]
- 9. Getsios S., Waschke J., Borradori L., Hertl M., Müller E. J. (2010) From cell signaling to novel therapeutic concepts: international pemphigus meeting on advances in pemphigus research and therapy. J. Invest. Dermatol. 130, 1764–1768 [DOI] [PubMed] [Google Scholar]
- 10. Spindler V., Waschke J. (2011) Role of Rho GTPases in desmosomal adhesion and pemphigus pathogenesis. Ann. Anat. 193, 177–180 [DOI] [PubMed] [Google Scholar]
- 11. Spindler V., Vielmuth F., Schmidt E., Rubenstein D. S., Waschke J. (2010) Protective endogenous cyclic adenosine 5′-monophosphate signaling triggered by pemphigus autoantibodies. J. Immunol. 185, 6831–6838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Osada K., Seishima M., Kitajima Y. (1997) Pemphigus IgG activates and translocates protein kinase C from the cytosol to the particulate/cytoskeleton fractions in human keratinocytes. J. Invest. Dermatol. 108, 482–487 [DOI] [PubMed] [Google Scholar]
- 13. Caldelari R., de Bruin A., Baumann D., Suter M. M., Bierkamp C., Balmer V., Müller E. (2001) A central role for the armadillo protein plakoglobin in the autoimmune disease pemphigus vulgaris. J. Cell Biol. 153, 823–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bektas M., Jolly P. S., Berkowitz P., Amagai M., Rubenstein D. S. (2013) A pathophysiologic role for epidermal growth factor receptor in pemphigus acantholysis. J. Biol. Chem. 288, 9447–9456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heupel W.-M., Engerer P., Schmidt E., Waschke J. (2009) Pemphigus vulgaris IgG cause loss of desmoglein-mediated adhesion and keratinocyte dissociation independent of epidermal growth factor receptor. Am. J. Pathol. 174, 475–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Williamson L., Raess N. A., Caldelari R., Zakher A., de Bruin A., Posthaus H., Bolli R., Hunziker T., Suter M. M., Müller E. J. (2006) Pemphigus vulgaris identifies plakoglobin as key suppressor of c-Myc in the skin. EMBO J. 25, 3298–3309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jolly P. S., Berkowitz P., Bektas M., Lee H. E., Chua M., Diaz L. A., Rubenstein D. S. (2010) p38MAPK signaling and desmoglein-3 internalization are linked events in pemphigus acantholysis. J. Biol. Chem. 285, 8936–8941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Berkowitz P., Hu P., Warren S., Liu Z., Diaz L. A., Rubenstein D. S. (2006) p38MAPK inhibition prevents disease in pemphigus vulgaris mice. Proc. Natl. Acad. Sci. U.S.A. 103, 12855–12860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Berkowitz P., Hu P., Liu Z., Diaz L. A., Enghild J. J., Chua M. P., Rubenstein D. S. (2005) Desmosome signaling: inhibition of p38MAPK prevents pemphigus vulgaris IgG-induced cytoskeleton reorganization. J. Biol. Chem. 280, 23778–23784 [DOI] [PubMed] [Google Scholar]
- 20. Spindler V., Rötzer V., Dehner C., Kempf B., Gliem M., Radeva M., Hartlieb E., Harms G. S., Schmidt E., Waschke J. (2013) Peptide-mediated desmoglein 3 crosslinking prevents pemphigus vulgaris autoantibody-induced skin blistering. J. Clin. Invest. 123, 800–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hotchin N. A., Tsang S. M., Liu L., Teh M.-T., Wheeler A., Grose R., Hart I. R., Garrod D. R., Fortune F., Wan H. (2010) Desmoglein 3, via an interaction with E-cadherin, is associated with activation of Src. PLoS ONE 5, e14211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brennan D., Peltonen S., Dowling A., Medhat W., Green K. J., Wahl J. K., 3rd, Del Galdo F., Mahoney M. G. (2012) A role for caveolin-1 in desmoglein binding and desmosome dynamics. Oncogene 31, 1636–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nava P., Laukoetter M. G., Hopkins A. M., Laur O., Gerner-Smidt K., Green K. J., Parkos C. A., Nusrat A. (2007) Desmoglein-2: a novel regulator of apoptosis in the intestinal epithelium. Mol. Biol. Cell 18, 4565–4578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schlegel N., Meir M., Heupel W. M., Holthöfer B., Leube R. E., Waschke J. (2010) Desmoglein 2-mediated adhesion is required for intestinal epithelial barrier integrity. Am. J. Physiol. Gastrointest. Liver Physiol. 298, G774–783 [DOI] [PubMed] [Google Scholar]
- 25. Hartlieb E., Kempf B., Partilla M., Vigh B., Spindler V., Waschke J. (2013) Desmoglein 2 is less important than desmoglein 3 for keratinocyte cohesion. PLoS ONE 8, e53739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Koch P. J., Mahoney M. G., Ishikawa H., Pulkkinen L., Uitto J., Shultz L., Murphy G. F., Whitaker-Menezes D., Stanley J. R. (1997) Targeted disruption of the pemphigus vulgaris antigen (desmoglein 3) gene in mice causes loss of keratinocyte cell adhesion with a phenotype similar to pemphigus vulgaris. J. Cell Biol. 137, 1091–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mahoney M. G., Wang Z., Rothenberger K., Koch P. J., Amagai M., Stanley J. R. (1999) Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J. Clin. Invest. 103, 461–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 29. Tsunoda K., Ota T., Aoki M., Yamada T., Nagai T., Nakagawa T., Koyasu S., Nishikawa T., Amagai M. (2003) Induction of pemphigus phenotype by a mouse monoclonal antibody against the amino-terminal adhesive interface of desmoglein 3. J. Immunol. 170, 2170–2178 [DOI] [PubMed] [Google Scholar]
- 30. Heupel W. M., Zillikens D., Drenckhahn D., Waschke J. (2008) Pemphigus vulgaris IgG directly inhibit desmoglein 3-mediated transinteraction. J. Immunol. 181, 1825–1834 [DOI] [PubMed] [Google Scholar]
- 31. Heupel W. M., Müller T., Efthymiadis A., Schmidt E., Drenckhahn D., Waschke J. (2009) Peptides Targeting the desmoglein 3 adhesive interface prevent autoantibody-induced acantholysis in pemphigus. J. Biol. Chem. 284, 8589–8595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lever W. F. (1953) Pemphigus. Medicine 32, 1–123 [DOI] [PubMed] [Google Scholar]
- 33. Baron S., Hoang A., Vogel H., Attardi L. D. (2012) Unimpaired skin carcinogenesis in Desmoglein 3 knockout mice. PLoS ONE 7, e50024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brennan D., Hu Y., Medhat W., Dowling A., Mahoney M. G. (2010) Superficial dsg2 expression limits epidermal blister formation mediated by pemphigus foliaceus antibodies and exfoliative toxins. Dermatol. Res. Pract. 2010, 410278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brennan D., Mahoney M. G. (2009) Increased expression of Dsg2 in malignant skin carcinomas: a tissue-microarray based study. Cell Adh. Migr. 3, 148–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Khan K., Hardy R., Haq A., Ogunbiyi O., Morton D., Chidgey M. (2006) Desmocollin switching in colorectal cancer. Br. J. Cancer 95, 1367–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kolegraff K., Nava P., Helms M. N., Parkos C. A., Nusrat A. (2011) Loss of desmocollin-2 confers a tumorigenic phenotype to colonic epithelial cells through activation of Akt/β-catenin signaling. Mol. Biol. Cell 22, 1121–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Spindler V., Heupel W. M., Efthymiadis A., Schmidt E., Eming R., Rankl C., Hinterdorfer P., Müller T., Drenckhahn D., Waschke J. (2009) Desmocollin 3-mediated binding is crucial for keratinocyte cohesion and is impaired in pemphigus. J. Biol. Chem. 284, 30556–30564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Spindler V., Dehner C., Hubner S., Waschke J. (2014) Plakoglobin but not desmoplakin regulates keratinocyte cohesion via modulation of p38MAPK signaling. J. Invest. Dermatol. 10.1038/jid.2014.21 [DOI] [PubMed] [Google Scholar]
- 40. Wöll S., Windoffer R., Leube R. E. (2007) p38 MAPK-dependent shaping of the keratin cytoskeleton in cultured cells. J. Cell Biol. 177, 795–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Müller E. J., Williamson L., Kolly C., Suter M. M. (2008) Outside-in signaling through integrins and cadherins: a central mechanism to control epidermal growth and differentiation? J. Invest. Dermatol. 128, 501–516 [DOI] [PubMed] [Google Scholar]
- 42. Tsunoda K., Ota T., Saito M., Hata T., Shimizu A., Ishiko A., Yamada T., Nakagawa T., Kowalczyk A. P., Amagai M. (2011) Pathogenic relevance of IgG and IgM antibodies against desmoglein 3 in blister formation in pemphigus vulgaris. Am. J. Pathol. 179, 795–806 [DOI] [PMC free article] [PubMed] [Google Scholar]