

Background: Glucocorticoid receptor (GR) is essential for early phase of adipogenesis.
Results: Depletion of coregulator CCAR1 compromised adipogenesis in cell culture and GR-mediated chromatin remodeling of peroxisome proliferator-activated receptor (PPARγ) gene enhancer elements.
Conclusion: GR and CCAR1 are required for transcriptional activation of PPARγ and adipocyte differentiation.
Significance: Mechanistic analysis identifies specific critical roles for GR and coregulator CCAR1 in the early stages of adipogenesis.
Keywords: Adipogenesis, Chromatin Modification, Glucocorticoid Receptor, Transcription Enhancer, Transcription Regulation, Cell Cycle and Apoptosis Regulator 1 (CCAR1), Peroxisome Proliferator-activated Receptor
Abstract
Glucocorticoids contribute to adipocyte differentiation by cooperating with transcription factors, such as CCAAT/enhancer-binding protein β (C/EBPβ), to stimulate transcription of the gene encoding peroxisome proliferator-activated receptor (PPARγ), a master regulator of adipogenesis. However, the mechanism of PPARγ gene regulation by glucocorticoids, the glucocorticoid receptor (GR), and its coregulators is poorly understood. Here we show that two GR binding regions (GBRs) in the mouse PPARγ gene were responsive to glucocorticoid, and treatment of 3T3-L1 preadipocytes with glucocorticoid alone induced GR occupancy and chromatin remodeling at PPARγ GBRs, which also contain binding sites for C/EBP and PPARγ proteins. GR recruited cell cycle and apoptosis regulator 1 (CCAR1), a transcription coregulator, to the PPARγ gene GBRs. Notably, CCAR1 was required for GR occupancy and chromatin remodeling at one of the PPARγ gene GBRs. Moreover, depletion of CCAR1 markedly suppressed differentiation of mouse mesenchymal stem cells and 3T3-L1 preadipocytes to mature adipocytes and decreased induction of PPARγ, C/EBPα, and C/EBPδ. Although CCAR1 was required for stimulation of several GR-regulated adipogenic genes in 3T3-L1 preadipocytes by glucocorticoid, it was not required for GR-activated transcription of certain anti-inflammatory genes in human A549 lung epithelial cells. Overall, our results highlighted the novel and specific roles of GR and CCAR1 in adipogenesis.
Introduction
Obesity is a major risk factor for many diseases, such as type 2 diabetes, hypertension, and cardiovascular disease (1). Hence, it is recognized as a prevalent health hazard worldwide. This excess of white adipose tissue caused by increases in the size and number of white adipocytes is the major cause of obesity. The number of white adipocytes present in an organism is determined largely by adipocyte differentiation (1). Therefore, elucidation of mechanisms of adipocyte differentiation is essential for understanding the pathogenesis of obesity and obesity-related diseases and thereby providing important information for developing new strategies to prevent and treat obesity.
Adipocytes, the major fat-containing components of adipose tissue, are developed from mesenchymal stem cells (MSC)3 in adipose tissue. This process involves an initial commitment phase where MSC are committed to the preadipocyte lineage followed by a differentiation phase (adipogenesis) where preadipocytes differentiate into mature fat-laden adipocytes. The major discoveries of key adipogenic signaling pathways have relied heavily on investigations in preadipocyte cell lines, such as mouse 3T3-L1 cells, which undergo a highly conserved and efficient program of adipogenesis in culture and upon transplantation in vivo (2). Because subsequent experiments in mice have convincingly validated the physiological significance of these major signaling pathways, the in vitro differentiation model represents a powerful and valid tool for deciphering the complex regulatory network of adipocyte differentiation.
The adipogenic conversion of most mouse and human preadipocyte cell lines requires stimulation with a hormonal mixture that consists of the synthetic glucocorticoid analogue dexamethasone (dex), insulin, and the cAMP phosphodiesterase inhibitor isobutylmethylxanthine (IBMX). Temporary exposure of preadipocytes to the adipogenic stimuli induces a complex network of pro-adipogenic transcription factors that act at different stages of differentiation and cooperatively promote adipogenesis. Significant players in this transcriptional cascade include CCAAT/enhancer binding protein (C/EBP) family members (i.e. C/EBPα, C/EBPβ, and C/EBPδ) and the nuclear receptor peroxisome proliferator-activated receptor γ (PPARγ), which is considered as the master positive regulator of preadipocyte differentiation to adipocytes and is both indispensable and under some conditions sufficient for adipogenesis (2). The expression levels of C/EBPβ and C/EBPδ are low in preadipocytes but are greatly enhanced after treatment of the cells with adipogenic mixture. During the first hour of adipogenesis, both of these C/EBPs associate with regulatory elements of a variety of adipogenic genes, including the two major late-acting transcription factors, C/EBPα and PPARγ, and subsequently activate gene expression. Raised levels of C/EBPα and PPARγ then induce each other's expression in a positive feedback loop promoting and maintaining the differentiated state of the mature adipocyte (3).
Dex is a key component of the adipogenic mixture and is involved in transcriptional regulation of adipogenesis via binding to and activating the glucocorticoid receptor (GR), a hormone-dependent transcription factor (4). Several lines of evidence have demonstrated the important role of GR in adipogenesis. First, cells with decreased GR levels were unable to efficiently store lipids and express adipocyte-selective proteins C/EBPα and PPARγ in response to adipogenic stimuli. Second, GR is involved in induction of key pro-adipogenic transcription factor C/EBPδ (4) as well as several other adipogenic genes such as RGS2 (5) and EPAS1 (6). Finally, during the early stages of adipogenesis, transient enrichment of GR, C/EBPβ, mediator subunit 1 (Med1), p300, and histone acetylation at regulatory sites near adipogenic genes has been previously reported (7); target genes include the gene encoding the master regulator of adipogenesis, PPARγ. Thus, GR cooperates with C/EBPβ to initiate transcriptional activation of key adipogenic genes that are required to specify adipogenic cell fate. However, both the precise molecular contribution by GR and the identities of the transcriptional coregulators that support and modulate GR activity during the adipocyte differentiation process remain largely unexplored and are the subject of the study presented here.
In addition to the role of glucocorticoids and GR in regulating adipogenesis, glucocorticoids are well known for their anti-inflammatory and immunosuppressive properties and are, therefore, widely and successfully used in the treatment of numerous inflammatory conditions, including autoimmune diseases, inflammatory bowel disease, and lymphoma. Nevertheless, glucocorticoids are also infamous for adverse metabolic side effects associated with the chronic administration required to treat these diseases effectively. For approximately two decades, great efforts have been made to identify and develop for clinical use selective GR ligands that dissociate beneficial anti-inflammatory effects from adverse metabolic effects of glucocorticoids (8–11), but unfortunately with little success. Another possible approach to this problem may be offered by coregulators, which have been shown to function in a gene-specific manner with GR and other transcription factors and, therefore, may represent potential targets for combination therapy to accompany the use of classical anti-inflammatory steroids such as dex. For example, inhibition of a coregulator required for regulation of genes involved in the metabolic actions of glucocorticoids but not required for regulation of genes involved in the anti-inflammatory response to glucocorticoids could limit some side effects associated with glucocorticoid treatment of inflammatory diseases.
Toward that end we performed a limited screen of coregulators by assessing the effect of depleting each coregulator from 3T3-L1 cells on the induction of several adipogenic genes by dex. In parallel, we tested the effect of coregulator depletion on dex-induced expression of several anti-inflammatory genes in A549 human lung adenocarcinoma cells. From this screen cell cycle and apoptosis regulator 1 (CCAR1), previously identified as a coactivator for GR (12), was preferentially required for dex-induced expression of adipogenic genes versus anti-inflammatory genes. Subsequently, we characterized the role of CCAR1 in the differentiation of 3T3-L1 cells to mature adipocytes and the mechanisms by which GR and CCAR1 contribute to the activation of the key adipogenic transcription factor, PPARγ. We found that CCAR1 determines adipogenic competency, acting in part through positive regulation of GR transcriptional activity that drives early adipogenesis. Thus, targeting glucocorticoid coregulators, such as CCAR1, could be an effective approach to separate beneficial anti-inflammatory effects of glucocorticoids from certain unfavorable effects of glucocorticoids, such as obesity involving aberrantly activated adipogenesis.
EXPERIMENTAL PROCEDURES
Materials
Antibodies against the following proteins were purchased and used for this study: GR from Santa Cruz Biotechnology; β-actin and α-tubulin from Sigma; CCAR1 and IgG from Bethyl Laboratory. Secondary antibodies (horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG) were purchased from Promega. Insulin, dex, and IBMX were purchased from Sigma. Transfection reagent Dharmafect Duo was purchased from Thermo Fisher Scientific-Dharmacon, and Oligofectamine was purchased from Invitrogen.
Lentivirus-mediated Coregulator Depletion
For lentivirus production, the packaging vector pCMV-ΔR8.91, the envelop plasmid pMD.G1, and the transfer vector pHRCMVpuroSin8 targeting nonspecific sequence as control were described previously (13). For depletion of mouse CCAR1, two pLKO.1 vectors expressing shRNA against different regions of CCAR1 mRNA were purchased from Sigma. Lentiviral particle production and virus transduction were performed according to a previously described protocol (13).
Cell Cultures and RNA Interference Screen
A549 human lung carcinoma cells were maintained in DMEM with 10% fetal bovine serum (FBS). 3T3-L1 mouse preadipocyte cells were maintained in DMEM supplemented with 10% bovine calf serum. For coregulator depletion in 3T3-L1 cells, siRNA was transfected into the detached 3T3-L1 cells in suspension by using DharmaFECT duo as mentioned previously (14) or Hiperfect (Qiagen). The siRNA-cell mixture was plated and incubated at 37 °C with 5% CO2 for 2 days before the media was exchanged for phenol-red free DMEM supplemented with 5% charcoal-stripped FBS. The next day, cells were treated with 100 nm dex or ethanol as control for 6 h. For coregulator depletion in A549 cells, siRNA was transfected into attached cells by using Oligofectamine or Hiperfect (Qiagen) according to the manufacturer's protocol. Two days after transfection, the medium was changed to phenol red-free DMEM containing 5% charcoal-stripped FBS. The sequences of the sense siRNA used were as follows: nonspecific control siRNA (15); si-mBAF57 (Santa Cruz, sc-45941); si-mCARM1, 5′-GCCAUGAAGAUGUGUGUGU-3′; si-mCCAR1, 5′-GCUACAUGAUACAUUUGGATT-3′; si-mCoCoA, 5′-CCUUGCGGGAAUAGAAUUATT-3′; si-mDBC1, 5′-AUGACUAUGACUCGAAGAATT-3′; si-mFli-I, 5′-CACATTCAGTCTTCAGAAGAA-3′; si-mMed14 (Santa Cruz, sc-142586); si-mTIF1α (Santa Cruz, sc-38549); si-mTip60, 5′-CGGAGUAUGACUGCAAAGGTT-3′; si-mZNF282 (Santa Cruz, sc-108045); si-hBAF57, 5′-AACCGCGTACCTTGCTTACAT-3′; si-hCARM1 (15); si-hCCAR1 (12); si-hCoCoA (16); si-hDBC1 (17); si-hFli-I, 5′-AACAACCTGACCACGCTTCAT-3′; si-hMed14 (Santa Cruz, sc-38579); si-hTip60 (18); si-hTIF1α (19); si-hZNF282, 5′-AATTTGGGAACCACATGGAGA-3′.
Differentiation into Adipocytes
For induction of differentiation with adipogenic mixture, 3T3-L1 cells were grown for an additional 2 days after confluence, and the 2-day post-confluent cells (designated as day 0) were induced to differentiate with DMEM containing 10% FBS, 0.5 mm IBMX, 1 μm dex, and 1 μg/ml insulin. After 2 days, medium was changed with fresh DMEM containing 10% FBS and again every other day thereafter until processing for analysis. Mouse MSC derived from the bone marrow of C57BL/6 mice were obtained from Cyagen Biosciences. MSC were cultured in OriCell mMSC growth medium (Cyagen Biosciences) and differentiated into adipocytes according to manufacturer's instructions. Briefly, 3 days after MSC reached confluence, MSC were induced to differentiate with adipogenic medium containing FBS, IBMX, dex, insulin, and rosiglitazone (a PPARγ agonist). After cells had differentiated, acquisition of adipocyte phenotype was determined by Oil Red O staining as described previously (4).
Chromatin Immunoprecipitation (ChIP)
Two days after 3T3-L1 cells reached confluence, culture medium was removed, and cells were grown in DMEM supplemented with 5% charcoal-stripped FBS overnight. The next day, cells were treated with or without 100 nm dex for the indicated times. ChIP assays were carried out as described previously (20). For immunoprecipitation, 8 μg of CCAR1 antibodies, 4 μg of antibodies against GR, or 4 μg of normal rabbit IgG were used. Purified immunoprecipitated DNA and input DNA were used as the template for amplification by quantitative PCR with the following primer pairs: 5′-AGCTTTGCTGGCTAGAGGTG-3′ (forward) and 5′-TTTCGCAGAACTGAGGTTGA-3′ (reverse) (−183 kb relative to the PPARγ2 transcription start site (TSS)) (7) and 5′-TTCTTCCCAGTAGGAACTGCAT-3′ (forward) and 5′-GATCACTCAGTTGGCATTTCTC (reverse) (−10 kb relative to PPARγ2 TSS) (7). After amplification, a melting curve analysis was performed to confirm the homogeneity of PCR products from each reaction. The immunoprecipitated DNA and input signals were quantified using the relative standard curve method, and results were expressed as the percentage of input by normalizing the immunoprecipitated signal to its corresponding input signal.
Formaldehyde-assisted Isolation of Regulatory Elements (FAIRE) Assays
3T3-L1 cells were grown with culture medium for an additional 2 days after confluence and were incubated with phenol-red free DMEM containing 5% charcoal-stripped FBS overnight. Cells were then treated with or without 100 nm dex for 90 min, and subsequently cells were cross-linked with 1% formaldehyde and prepared for FAIRE analysis. FAIRE was performed essentially as previously described (21). In brief, nuclei were isolated from harvested cells and submitted to sonication to shear chromatin. Samples were subjected to phenol/chloroform/isoamyl alcohol extraction to recover free DNA not bound by nucleosomes in the aqueous phase. After extraction cross-linking was reversed, and the FAIRE DNAs were purified. Input chromatin samples were obtained before the phenol/chloroform/isoamyl alcohol extraction, and DNA was purified as described above. Representation of specific GR binding sites in the FAIRE DNA samples (which represent open chromatin regions) was determined relative to input DNA by quantitative PCR using the same PCR primers specified above for ChIP.
Quantitative Reverse Transcriptase-PCR
RNA was extracted from cells using the TRIzol method (Invitrogen), resuspended in diethylpyrocarbonate-treated H2O, and adjusted to 100 ng/μl. cDNA was synthesized by reverse-transcribing 0.9 μg of total RNA using iScript cDNA synthesis kit (Bio-Rad). The cDNA was mixed with appropriate primers and LightCycler 480 SYBR Green I Master (Roche Applied Science), and the mixture was then analyzed with the LightCycler 480 System (Roche Applied Science). The primers used are listed in Table 1 or as follows: mC/EBPα, mC/EBPβ, mC/EBPδ, and mGlut4 (4); hβ-actin (20); hGilz (12); hDUSP1 (22), hCARM1 (23), hCCAR1 (12), hCoCoA (16), hDBC1 (17), hTip60 (18), and hZNF282 (24). Results shown are mean and range of variation for duplicate PCR reactions from a single cDNA preparation and are representative of a minimum of two independent experiments. Relative mRNA expression levels were determined by normalizing against GAPDH for mouse mRNA and β-actin for human mRNA.
TABLE 1.
Primer sequences for RT-PCR
m-, mouse; h-, human.
| Gene | Forward primer sequence (5′→3′) | Reverse primer sequence (5′→3′) |
|---|---|---|
| mBAF57 | GGAATCGTAATGCCAGAGGA | ACACCAGGGTTTGTGGGATA |
| mCARM1 | ATCGCCCTCTACAGCCATGA | CTGTCTGCCCACACGACTG |
| mCCAR1 | ATTGCCCACAAGCCTTAGCC | TGTAGCTTTGTAACCACTCCTGT |
| mCoCoA | CAAGGTGGAATGTCACTACACTT | CGTGTGATAATCTCGAACGCAG |
| mDBC1 | TTTAAGCGGCAGAGGATCAAC | GGAGGAGTAAGTAGACCAGGG |
| mFli-I | GGACGATGTGTTGACAGTGG | CTGCCGAGGTAGGAAGAGTG |
| mMed14 | TTATTTGTGGATACCGCTGA | CAATCGCATATGGAATAGCA |
| mTIF1α | GTTGTCTATGATGTGTCTCTCAGC | GGTGGTCCTTCGCCATTC |
| mTip60 | AGACACCTACCAAGAACGGAC | GTGTGATCTGGACCGGGATTG |
| mZNF282 | AGCAATGTCGACGAAGGTCA | TGGAGAACCTGGAGAACCTG |
| mEpas1 | ATCACGGGATTTCTCCTTCC | GGTTAAGGAACCCAGGTGCT |
| mRgs2 | CTTTTCTTGCCAGTTTTGGG | GAGGAGAAGCGGGAGAAAAT |
| mSphk1 | TGCAGTTGATGAGCAGGTCT | ACCCCTGTGTAGCCTCCCT |
| mLpin1 | TGAAGACTCGCTGTGAATGG | CGCCAAAGAATAACCTGGAA |
| mPPARγ2 | CGCTGATGCACTGCCTATGA | AATGGCATCTCTGTGTCAACCA |
| mGAPDH | ATCCATGACAACTTTGGCATT | ATGACCTTGCCCACAGCCTT |
| hBAF57 | GGGGTTTTGGAATCGTGATA | CACCAGGGTTTGTGGGATAC |
| hFli-I | ACAAGTGTTCCACCTTCTGC | CTCAGCGGCAACGACTTC |
| hMed14 | AAGCCATCCTGTTTGTGGAC | GCATATGGGATGGCAAAACT |
| hTIF1α | CAAACCCAATGGACCAGTTC | TGGCTTTATTGCTTGTCGTG |
| hZFP36 | GAGAACTTGGAGAACTTGCTGCGC | GTAGTTCTCCTTAACAAGGTTGTT |
Plasmids, Transfection, and Luciferase Reporter Assay
GR binding regions (GBRs) 1 and 2 from mouse PPARγ gene were amplified by Expand Long Template PCR System (Roche Applied Science) from mouse genomic DNA using the following primers: GBR1_5′, GCTGCAGGTACCGCAGAGGACCAGGTAGGTC; GBR1_3′, CGCTCTCTCGAGCTCTGGCCTTAATGTCCTGT; GBR2_5′, GCTGCAGGTACCCCAACGCATGCAAGTCTTAC; GBR2_3′, CGCTCTCTCGAGCAGTGGGTATTCCTAACTGC. These PCR fragments were cloned into the pGL4.10-E4TATA vector with KpnI/XhoI sites. pGL4.10-E4TATA reporter plasmid was generated by insertion of a 50-bp minimal E4 TATA promoter sequence (25) into the BglII to HindIII sites of vector pGL4.10 to drive luciferase expression (26). Lipofectamine 2000 (Invitrogen) was used to transfect 3T3-L1 preadipocytes according to the technical manual. Twenty-four hours post-transfection cells were treated with either 1 μm dex or control ETOH for 16–20 h. Cells were then harvested, and their luciferase activities were measured with the Dual Luciferase Reporter Assay kit (Promega) according to procedures in the technical manual.
RESULTS
Identification of GR Coregulators Involved in Adipogenic Versus Anti-inflammatory Gene Transcription
GR belongs to the steroid hormone receptor subclass of nuclear receptors and controls physiological processes through activation or repression of distinct sets of causative target genes driving different physiological pathways. Previous studies reported that coregulators function in a gene-specific manner. We then tried to define coregulator utilization patterns by performing a limited RNA interference screen. Ten different coregulators were individually down-regulated by transfection of cells with small interfering RNA (siRNA) to investigate their involvement in glucocorticoid-induced expression of five adipogenic genes in 3T3-L1 preadipocytes and three anti-inflammatory genes in A549 lung carcinoma cells (Fig. 1). The mRNA levels of coregulators were substantially reduced by their respective siRNAs when compared with nonspecific siRNA (Fig. 1A). In agreement with previous studies, gene-specific coregulator requirements were observed (Fig. 1B). Moreover, coregulators CCAR1, CARM1, DBC1, and Tip60 are required for optimum dex-induced expression of the selected adipogenic genes but not for dex-induced expression of the selected anti-inflammatory genes (Fig. 1B). Hence, we propose that single coregulators or specific combinations of coregulators may dictate specific glucocorticoid-controlled biological functions by selectively regulating important causative genes in distinct physiological pathways.
FIGURE 1.
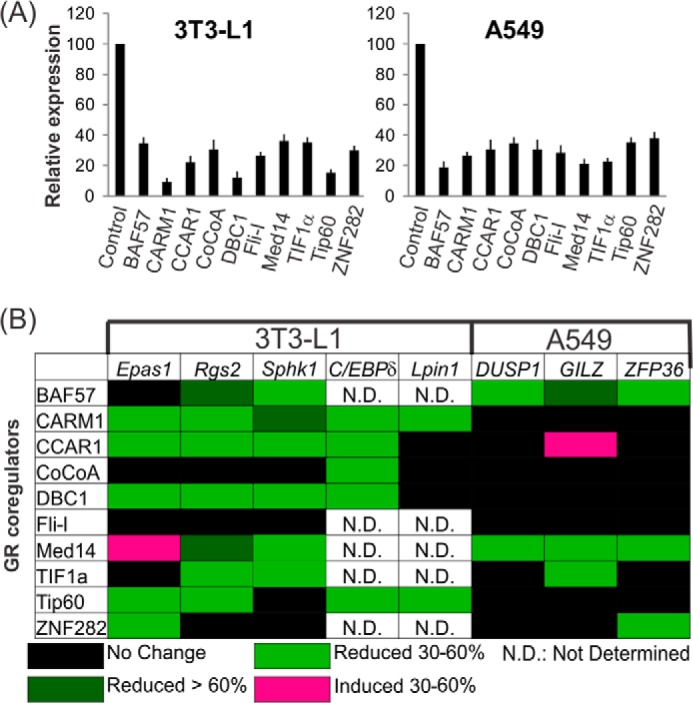
Effect of depletion of endogenous coregulators on the expression of GR-regulated adipogenic genes or anti-inflammatory genes. A, human A549 or mouse 3T3-L1 cells were transfected with control siNS or siRNA specific for each coregulator listed and grown in culture media for 2 days. The culture media were replaced with hormone-free media overnight, and the cells were treated with 100 nm dex or control ETOH for 6 h before harvesting. Total RNA was analyzed by quantitative real-time-PCR to determine the relative mRNA level for each coregulator in the cells transfected with siNS or siRNA against the coregulator. The coregulator mRNA expression was normalized to the level of human β-actin mRNA in A549 cells or mouse GAPDH mRNA in 3T3-L1 cells. For each experiment, the control nonspecific (NS) signal was set to 100, and the relative mRNA expression of the specific coregulator was calculated by dividing the si-coregulator signal by the siNS signal followed by multiplying by 100. Data are the means ± S.E. B, total RNA was analyzed for each target gene mRNA by quantitative real-time-PCR and normalized to the level of human β-actin mRNA in A549 cells or mouse GAPDH mRNA in 3T3-L1 cells. Colors represent degree of change in dex-induced expression level after depletion of the indicated coregulator. The color code is shown on the bottom. Results shown are representative of at least two independent experiments.
CCAR1 Is Required for Adipogenesis
Our siRNA screen indicated that CCAR1 is important for the expression of adipogenic genes. This result prompted us to examine the requirement of CCAR1 in adipocyte differentiation using short hairpin RNA (shRNA)-mediated knockdown. Adipose-derived MSC from C57BL/6 mice were infected with lentivirus encoding shRNA against CCAR1 (shCCAR1) or against a nonspecific target (shNS) as control. Efficient depletion of CCAR1 protein was achieved by two different shRNAs directed against different regions of the CCAR1 mRNA (Fig. 2A). CCAR1-depleted MSC and the control culture expressing shNS were induced to differentiate with adipogenic stimuli containing dex, insulin, IBMX, and rosiglitazone (a PPARγ agonist); the differentiation efficiency was determined 3 weeks later by Oil Red O staining. The control culture underwent efficient differentiation to lipid droplet-containing adipocytes (Fig. 2B). In contrast, differentiation efficiency of CCAR1-depleted MSC was drastically reduced. Thus, CCAR1 depletion suppressed adipocyte differentiation, suggesting that CCAR1 is essential for adipogenesis.
FIGURE 2.
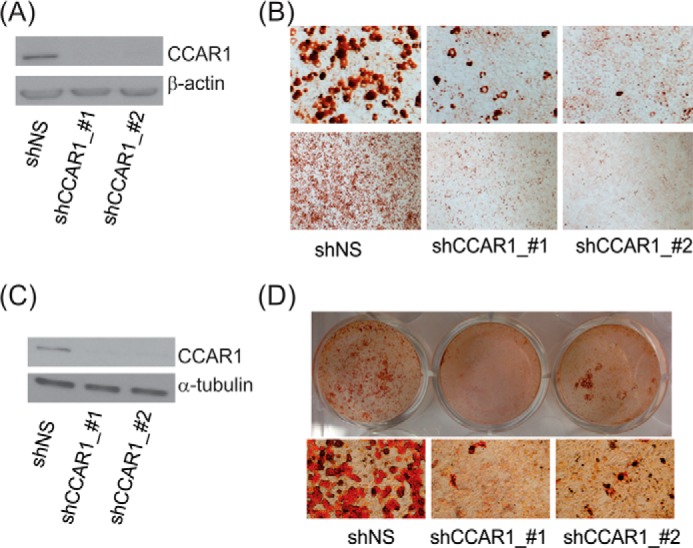
Depletion of CCAR1 blocks adipogenesis. A and B, MSC were infected with lentiviral vectors expressing shRNA against CCAR1 or nonspecific (NS) sequence and subsequently differentiated with complete differentiation media. A, immunoblot verifying depletion of CCAR1 protein level at the start of the differentiation process. B, Oil Red O staining of cells expressing the indicated shRNA and differentiated in completed differentiation media for 3 weeks. Results are representative of a minimum of two independent experiments. C and D, 3T3-L1 cells were transduced with lentivirus expressing shRNAs targeting either CCAR1 or a nonspecific sequence as control. Infected cells were selected with puromycin and subsequently were replated at confluent cell density. After 2 days the cells were induced to differentiate. C, cells were harvested at the beginning of differentiation, and cell lysates were separated by SDS-PAGE and analyzed by immunoblot with antibodies against CCAR1 or β-actin protein. D, cells were stained with Oil Red O to visualize the accumulation of lipid droplets at day 8 of differentiation. Results are representative of a minimum of two independent experiments.
Many discoveries of key adipogenic events, signaling pathways, and transcriptional control mechanisms have been based on studies in cell lines derived from mouse embryonic fibroblasts, particularly the 3T3-L1 cell line. Importantly, given that most of the findings from cell lines have been convincingly validated in vivo, the in vitro 3T3-L1 differentiation model represents a valuable tool for identification of novel molecules and signaling networks regulating adipogenesis. When we infected 3T3-L1 cells with lentivirus encoding shNS or two different species of shCCAR1, CCAR1 protein levels were specifically reduced in cells expressing CCAR1 shRNA, but not in cells expressing shNS (Fig. 2C). As we observed for the MSC, shCCAR1 greatly decreased the efficiency of adipogenesis, as assessed by Oil Red O dye staining after 8 days of treatment with adipogenic stimuli containing dex, insulin, and IBMX (Fig. 2D). These results demonstrate the critical involvement of CCAR1 in adipocyte differentiation.
CCAR1 Functions in Adipogenesis at an Early Stage of 3T3-L1 Adipocyte Differentiation
We further analyzed the expression of adipocyte-specific markers at various time points after adding adipogenic stimuli to 3T3-L1 cells expressing shCCAR1 or control shNS. In particular, we examined the expression of key adipogenic transcription factors that are induced during adipogenesis, including PPARγ. Two different mRNAs are produced from two different TSSs of the PPARγ gene; because the PPARγ2 TSS is the major isoform induced during adipogenesis (27), we specifically examined transcripts produced from this TSS. CCAR1 protein levels remained depleted by shCCAR1 throughout the adipogenesis process (Fig. 3A). As a consequence of CCAR1 knockdown, induction of mRNA expression for terminal differentiation markers (e.g. PPARγ2 and C/EBPα) by the adipogenic mixture was greatly reduced (Fig. 3B). Expression of Glut4 (glucose transporter 4), the insulin-regulated glucose transporter that is found in mature adipose tissues, was also inhibited by down-regulation of CCAR1 (Fig. 3B). For early adipocyte-specific markers CCAR1 depletion moderately reduced the induction of C/EBPδ but had little or no effect on induction of C/EBPβ (Fig. 3B). Thus, CCAR1 functions early in adipogenesis to facilitate induction of C/EBPδ, PPARγ2, and C/EBPα.
FIGURE 3.
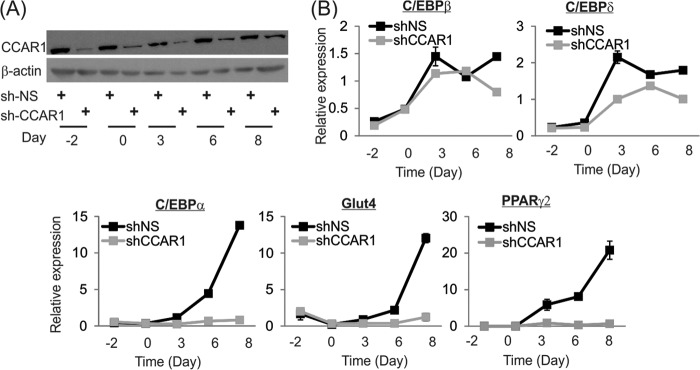
Time course of adipogenic gene expression in 3T3-L1 cells after CCAR1 depletion. A, immunoblot analysis of the CCAR1 protein expression from 3T3-L1 cells infected with lentivirus expressing control (NS) or CCAR1 shRNAs and subsequently treated with adipogenic mixture. Day −2, sub-confluent growing cells before differentiation induction; day 0, confluent cells at time of induction with differentiation mixture; days 3–8, time after the addition of adipogenic mixture. B, quantitative real-time PCR analysis of total RNA during differentiation of 3T3-L1 preadipocytes. Cells were treated as in A. Results shown are normalized to the level of GAPDH mRNA, are the mean and range of variation for duplicate PCR reactions from a single experiment, and are representative of three independent experiments.
Both Major Regulatory Elements of the PPARγ Gene Confer Glucocorticoid Response
During the process of adipocyte differentiation, PPARγ expression is strongly induced, and subsequently PPARγ is directly involved in the activation of most adipocyte genes. Hence, PPARγ is recognized as the master regulator of adipogenesis (28). Within hours of initiation of adipogenesis, GR is activated by dex, and expression of early adipogenic transcription factors C/EBPβ and C/EBPδ is induced; all three transcription factors occupy key PPARγ gene regulatory elements and remodel local chromatin structure. Chromatin remodeling facilitates enhanced transcription factor binding and recruitment of coregulators and basal transcription machinery, including RNA polymerase II, ultimately resulting in gene transcription. Thus, the importance of GR and the C/EBP proteins for induction of PPARγ expression by adipogenic mixture has been addressed. However, the specific molecular roles of GR and its associated coregulators in PPARγ gene activation are not known.
Two important regulatory elements for controlling PPARγ gene expression have been reported (7). The GBRs within the regulatory elements of PPARγ gene are located at −183 kb (GBR1) and −10 kb (GBR2) upstream from the TSS of the PPARγ2 promoter (Fig. 4A). There are also confirmed binding sites for C/EBP proteins and PPARγ near the GBRs.
FIGURE 4.
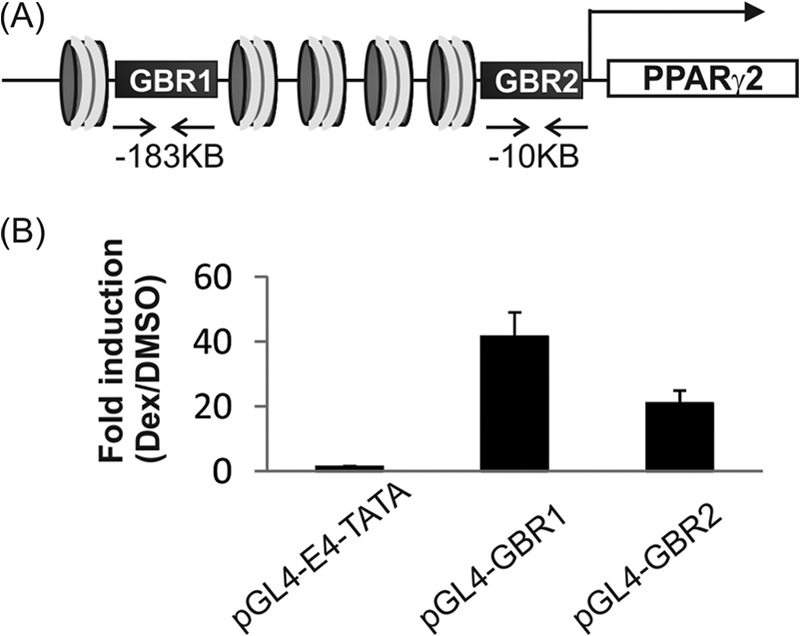
Two major regulatory elements of the PPARγ gene confer glucocorticoid regulation. A, genomic locations of two PPARγ regulatory elements. The PPARγ gene contains two major regulatory sites containing closely associated binding sites for GR, C/EBP proteins, and PPARγ itself. The GBRs are located at −183 kb (GBR1) and −10 kb (GBR2) upstream from the PPARγ2 gene TSS. B, GBR1 (chr6: 115238568-115238963) or GBR2 (chr6: 115412360-115412835) was inserted into reporter plasmid pGL4.10-E4TATA encoding firefly luciferase reporter. The reporter plasmids were transfected into 3T3-L1 cells, and cells were then treated with dex or vehicle for 16–20 h before harvesting. Cells lysates were prepared and subjected to luciferase assay. Data are the means ± S.E. Results shown are representative of three independent experiments.
To investigate the functional role of GR in PPARγ gene activation, we first tested whether the two GBRs are functionally responsive to glucocorticoids. We inserted GBR1 (chr6: 115238568-115238963) or GBR2 (chr6: 115412360-115412835) into reporter plasmid pGL4.10-E4TATA encoding firefly luciferase. The reporter plasmids were transfected into 3T3-L1 cells, and the cells were then treated with dex alone for 16–20 h before harvesting. Luciferase expression from both reporters was dex-responsive (Fig. 4B), indicating that both GBRs confer glucocorticoid-directed transcriptional responses.
Next, to focus on the mechanism by which GR regulates PPARγ gene activation, 3T3-L1 preadipocytes were treated with dex alone instead of the full adipogenic mixture. Using FAIREs assays to assess chromatin accessibility, we observed that treatment of 3T3-L1 cells with dex alone for only 90 min was sufficient to induce significant chromatin opening as well as significant GR occupancy at both PPARγ GBRs (Fig. 5). This demonstrates that GR directs the opening of chromatin conformation at both of the key PPARγ enhancer elements.
FIGURE 5.
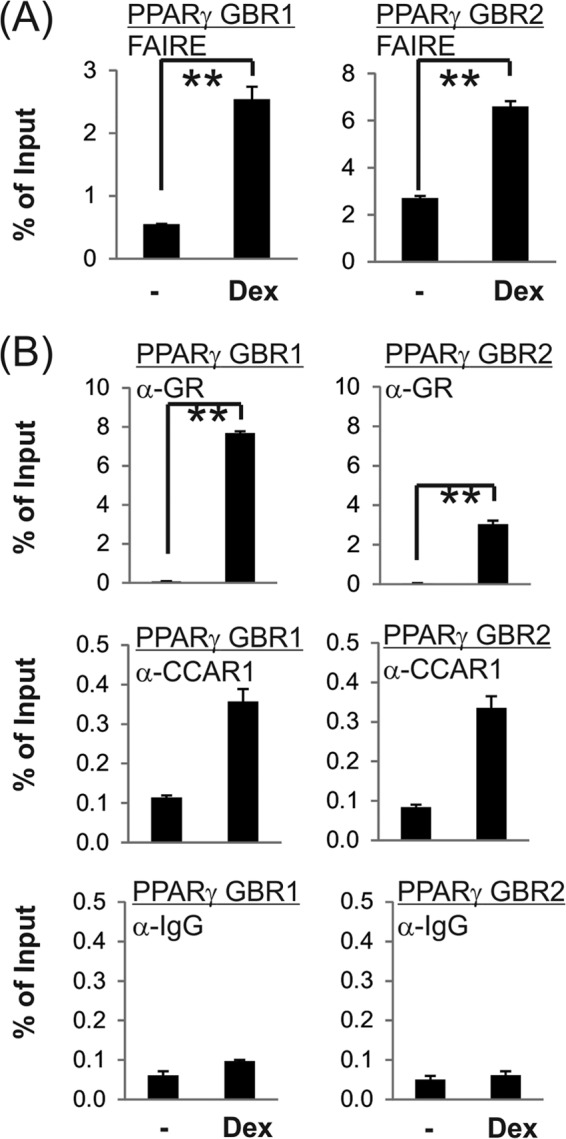
Dex-induced chromatin remodeling and transcription complex assembly at GBRs of the PPARγ gene. 3T3-L1 cells confluent for 2 days were incubated with charcoal-stripped medium overnight and then treated with 100 nm dex for 90 min. Cells were harvested for FAIRE (A) or ChIP (B) assays performed as described under “Experimental Procedures.” Quantitative PCR analyses were performed using primers specific for the PPARγ gene enhancers. Results shown are percentage of input, are the mean and range of variation for duplicate PCR reactions from a single experiment, and are representative of four (for ChIP) or five (for FAIRE) independent biological replicates. p value was determined from all of the biological replicates by Student's paired t test. **, p ≤ 0.01.
CCAR1 Is Important for Dex-induced GR Binding and Chromatin Remodeling at a Key Regulatory Element Controlling PPARγ Gene Expression
Previously, CCAR1 has been shown to interact with GR and positively regulate GR-controlled gene expression (12). Our results show CCAR1 is required for efficient induction of several adipogenic genes by dex (Fig. 1). Furthermore, CCAR1 depletion significantly inhibited the induction of PPARγ2 gene expression (Fig. 3B) as well as adipogenesis (Fig. 2) by the adipogenic mixture containing dex. Therefore, we tested whether CCAR1 acts through GR signaling to regulate the expression of PPARγ2 during adipocyte differentiation. In four independent ChIP experiments CCAR1 was recruited to PPARγ enhancer elements in a dex-dependent manner (Fig. 5B); however, the results were not statistically significant by Student's paired t test, possibly due to the relatively weak signal enrichment obtained in ChIP experiments using antibodies against CCAR1 and many other coregulators. Because these results were produced by a 90-min treatment with dex alone, it suggests that CCAR1 is recruited to the PPARγ enhancer elements by GR and is thus in position to act directly on the PPARγ gene to facilitate transcriptional activation.
To investigate the mechanism of CCAR1 action on the PPARγ gene during the early hours of adipocyte differentiation, we assessed the effect of reduced CCAR1 levels on transcription complex assembly on the PPARγ gene. Reduction of CCAR1 levels by shRNA had no measurable effect on the cellular levels of GR (Fig. 6A). However, CCAR1 depletion significantly reduced the ligand-dependent occupancy of GR on PPARγ GBR1 (Fig. 6B); for GBR2 a similar trend was observed but was not significant. The result indicates that CCAR1 is required for optimal association of GR with the GBR1 enhancer of PPARγ gene. We further assessed whether CCAR1 is involved in chromatin remodeling at the PPARγ gene enhancer elements by FAIRE analysis. CCAR1 depletion significantly inhibited the hormone-induced enhancement of chromatin accessibility at GBR1 but had only a modest (non-significant) effect on chromatin remodeling at GBR2 (Fig. 6C). Thus, activation of GR alone is sufficient to initiate chromatin remodeling of the PPARγ gene, and CCAR1 is required for optimal ligand-induced chromatin remodeling and optimal binding of GR to the PPARγ gene.
FIGURE 6.

CCAR1 is essential for dex-induced chromatin remodeling and GR binding to the PPARγ gene. 3T3-L1 cells expressing shNS or shCCAR1 and confluent for 2 days were incubated with or without 100 nm Dex for 90 min. Protein levels were monitored by immunoblot using the indicated antibodies (A). ChIP (B) and FAIRE (C) assays were conducted as described under “Experimental Procedures.” Quantitative PCR analyses were performed using primers specific for the PPARγ enhancers. Results shown are from a single experiment, which is representative of five (ChIP) or three (FAIRE) independent biological replicates. The mean and range of variation of duplicate PCR reactions from a single experiment are shown. The p value was determined from all of the biological replicates by Student's paired t test. *, p ≤ 0.05.
DISCUSSION
Glucocorticoids play an important role in inducing adipocyte cell fate and differentiation both in vivo and ex vivo (29–31). This regulatory process is critically important in normal physiology and numerous disease states; patients exposed chronically to excess glucocorticoids frequently experience problems such as increased adiposity, insulin resistance, hyperglycemia, hyperlipidemia, muscle wasting, osteoporosis, or a variety of other pathological conditions. The fact that coregulators function in a gene-specific manner to support transcriptional regulation by GR suggests that coregulators may regulate specific physiological pathways among the many that are targeted by GR and glucocorticoids. Currently, the coregulators that mediate or modulate gene regulation by GR during adipocyte differentiation remain to be determined. In addition, although GR is known to play a critical role in adipogenesis, other transcription factors (e.g. three C/EBP proteins) are also required. As a result, the precise molecular role of GR in this process is not well understood. Thus, a better understanding of GR and coregulator function and gene-specificity during the adipogenic process will ultimately provide critical information that may advance development of novel remedies for obesity or obesity-associated diseases, such as diabetes and hypertension.
During early adipogenesis, GR is activated by glucocorticoids, and expression of multiple other transcription factors, including C/EBPβ and C/EBPδ, is activated in response to the adipogenic signals. These transcription factors subsequently associate with enhancer elements of key adipogenic genes (32) including the PPARγ gene, which encodes the master regulator of adipogenesis. By recruiting coregulators the early adipogenic transcription factors trigger dynamic epigenetic modifications and modulation of the chromatin landscape at the two major PPARγ enhancer elements located at −10 and −183 kb from the PPARγ2 TSS (28). In preadipocytes, the repressive histone H3 lysine 9 dimethylation (H3K9me2) marks are found throughout the PPARγ locus, leading to transcriptional repression (33). Within hours of adding the adipogenic mixture to preadipocytes, the two upstream PPARγ enhancer elements are characterized with open chromatin configuration and are enriched for active transcription marks, histone H3 lysine 9 and 27 acetylation, replacing the previously deposited H3K9me2 (33). Moreover, these regions are occupied by GR, C/EBPβ, C/EBPδ, and other transcription factors as well as coregulators p300 (a histone acetyltransferase) and Med1 (7, 32).
The importance of coregulators for the efficient expression of gene sets controlling adipogenesis has started to be explored in adipose tissue biology. For instance, SRC-2−/− or SRC-3−/− mice exhibit reduced weight compared with control mice, and it was reported that this effect is at least partly due to a defect in adipocyte differentiation and fat accumulation in white adipose tissue (34, 35). At the molecular level, it has been shown that SRC-2 acts synergistically with transcription factor PPARγ (34), whereas SRC-3 functions with C/EBP transcription factors (35). However, the coregulators that mediate the function of GR have not been reported yet.
The fact that GR and C/EBP proteins co-occupy the PPARγ gene enhancer elements after treatment with the adipogenic mixture makes it difficult to assess the specific role of GR in this process. To specifically study GR function in PPARγ gene activation, 3T3-L1 preadipocytes were treated with dex alone (without the other adipogenic stimuli). Treatment with dex for only 90 min was sufficient to induce chromatin opening in both enhancer regions of PPARγ as well as GR occupancy (Fig. 5). At this early time point and in the absence of the other components of the adipogenic mixture, these results indicate that activated GR directs the early chromatin remodeling at the PPARγ enhancer elements. Furthermore, our finding that CCAR1 is recruited to the same GBRs in a dex-dependent manner (Fig. 5) and is also required for optimal chromatin remodeling at one of these sites (Fig. 6) also implicates GR in the recruitment of CCAR1 and in the chromatin remodeling process. Because occupancy of C/EBPβ and GR on the PPARγ gene enhancer elements was previously shown to be co-dependent at a later time point in the adipogenic process (7, 32), we cannot rule out the possibility that a low level of C/EBPβ occupancy at these sites is required to facilitate GR binding and CCAR1 recruitment. Nevertheless, the activation of GR by dex is clearly the triggering event for initiation of chromatin remodeling on the two PPARγ gene enhancer elements.
In this study we also identified CCAR1 as a novel coregulator required for adipogenic differentiation of 3T3-L1 preadipocytes and MSC (Fig. 2) and for expression of key adipogenic genes (PPARγ2, C/EBPα, and C/EBPδ) induced by the adipogenic mixture in 3T3-L1 cells (Fig. 3B). Furthermore, we showed that CCAR1, which is known to interact with and support the activity of GR in other cell lines (12), positively regulates expression of a subset of GR-mediated adipogenic genes in a dex-dependent manner (Fig. 1B). ChIP data demonstrated the dex-induced occupancy of CCAR1 at the two key enhancer elements of the PPARγ gene (Fig. 5B), indicating that CCAR1 can exert its positive regulator effect directly on this gene (although we cannot rule out the possibility that CCAR1 also acts through an indirect mechanism). Interestingly, CCAR1 is necessary for full dex-induced binding of GR as well as chromatin remodeling at PPARγ enhancer regions (Fig. 6). Although CCAR1 itself is not known to possess chromatin remodeling activity, CCAR1 could be involved in facilitating GR interaction with or recruitment of an ATP-dependent or other type of chromatin remodeling complex. Thus, CCAR1 acts at a very early stage of the gene activation process to influence the occupancy of GR and its ability to direct chromatin remodeling and subsequent assembly of an active transcription complex on the PPARγ2 promoter. This contrasts from our earlier finding for other GR target genes in a different cell line that CCAR1 is required for recruitment of the mediator complex and not for efficient GR occupancy of the gene (12). These contrasting results on different GR target genes that require CCAR1 indicate that CCAR1 is a very versatile coregulator protein that acts in a gene-specific manner and uses different mechanisms of action on different target genes. Collectively, our results indicate that CCAR1 is a key pro-adipogenic coregulator that positively regulates adipogenic gene expression through the GR signaling pathway and provides essential functions during the early stage of adipogenesis.
Acknowledgments
We thank Daniel Gerke, Kelly Chang, and Farah Alammari for technical assistance. We thank Professor Jeong Hoon Kim (Sungkyunkwan University, Korea) for information on various siRNA sequences and Western blots in A549 cells.
This work was supported, in whole or in part, by National Institutes of Health Grants DK043093 (to M. R. S.), DK083591 (to J.-C. W.), and P30 CA014089 (a Cancer Center Support Grant to the University of Southern California).
- MSC
- mesenchymal stem cells
- GR
- glucocorticoid receptor
- PPARγ
- peroxisome proliferator-activated receptor
- C/EBP
- CCAAT/enhancer-binding protein
- GBR
- glucocorticoid receptor binding region
- CCAR1
- cell cycle and apoptosis regulator 1
- dex
- dexamethasone
- IBMX
- isobutylmethylxanthine
- Med1
- mediator subunit 1
- FAIRE
- formaldehyde-assisted isolation of regulatory elements
- siRNA
- small interfering RNA
- shRNA
- short hairpin RNA
- TSS
- transcription start site.
REFERENCES
- 1. Cristancho A. G., Lazar M. A. (2011) Forming functional fat: a growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell Biol. 12, 722–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Siersbæk R., Mandrup S. (2011) Transcriptional networks controlling adipocyte differentiation. Cold Spring Harb. Symp Quant. Biol. 76, 247–255 [DOI] [PubMed] [Google Scholar]
- 3. Siersbæk R., Nielsen R., Mandrup S. (2012) Transcriptional networks and chromatin remodeling controlling adipogenesis. Trends Endocrinol. Metab 23, 56–64 [DOI] [PubMed] [Google Scholar]
- 4. Pantoja C., Huff J. T., Yamamoto K. R. (2008) Glucocorticoid signaling defines a novel commitment state during adipogenesis in vitro. Mol. Biol. Cell 19, 4032–4041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nishizuka M., Honda K., Tsuchiya T., Nishihara T., Imagawa M. (2001) RGS2 promotes adipocyte differentiation in the presence of ligand for peroxisome proliferator-activated receptor γ. J. Biol. Chem. 276, 29625–29627 [DOI] [PubMed] [Google Scholar]
- 6. Shimba S., Wada T., Hara S., Tezuka M. (2004) EPAS1 promotes adipose differentiation in 3T3-L1 cells. J. Biol. Chem. 279, 40946–40953 [DOI] [PubMed] [Google Scholar]
- 7. Steger D. J., Grant G. R., Schupp M., Tomaru T., Lefterova M. I., Schug J., Manduchi E., Stoeckert C. J., Jr., Lazar M. A. (2010) Propagation of adipogenic signals through an epigenomic transition state. Genes Dev. 24, 1035–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shah N., Scanlan T. S. (2004) Design and evaluation of novel nonsteroidal dissociating glucocorticoid receptor ligands. Bioorg Med. Chem. Lett 14, 5199–5203 [DOI] [PubMed] [Google Scholar]
- 9. Roohk D. J., Varady K. A., Turner S. M., Emson C. L., Gelling R. W., Shankaran M., Lindwall G., Shipp L. E., Scanlan T. S., Wang J. C., Hellerstein M. K. (2010) Differential in vivo effects on target pathways of a novel arylpyrazole glucocorticoid receptor modulator compared with prednisolone. J. Pharmacol. Exp. Ther. 333, 281–289 [DOI] [PubMed] [Google Scholar]
- 10. Ali A., Thompson C. F., Balkovec J. M., Graham D. W., Hammond M. L., Quraishi N., Tata J. R., Einstein M., Ge L., Harris G., Kelly T. M., Mazur P., Pandit S., Santoro J., Sitlani A., Wang C., Williamson J., Miller D. K., Thompson C. M., Zaller D. M., Forrest M. J., Carballo-Jane E., Luell S. (2004) Novel N-arylpyrazolo[3,2-c]-based ligands for the glucocorticoid receptor: receptor binding and in vivo activity. J. Med. Chem. 47, 2441–2452 [DOI] [PubMed] [Google Scholar]
- 11. Bungard C. J., Hartman G. D., Manikowski J. J., Perkins J. J., Bai C., Brandish P. E., Euler D. H., Hershey J. C., Schmidt A., Fang Y., Norcross R. T., Rushmore T. H., Thompson C. D., Meissner R. S. (2011) Discovery of selective glucocorticoid receptor modulator MK-5932. Bioorg Med. Chem. 19, 7374–7386 [DOI] [PubMed] [Google Scholar]
- 12. Kim J. H., Yang C. K., Heo K., Roeder R. G., An W., Stallcup M. R. (2008) CCAR1, a key regulator of mediator complex recruitment to nuclear receptor transcription complexes. Mol. Cell 31, 510–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ou C. Y., LaBonte M. J., Manegold P. C., So A. Y., Ianculescu I., Gerke D. S., Yamamoto K. R., Ladner R. D., Kahn M., Kim J. H., Stallcup M. R. (2011) A coactivator role of CARM1 in the dysregulation of β-catenin activity in colorectal cancer cell growth and gene expression. Mol. Cancer Res. 9, 660–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kilroy G., Burk D. H., Floyd Z. E. (2009) High efficiency lipid-based siRNA transfection of adipocytes in suspension. PLoS ONE 4, e6940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bittencourt D., Wu D. Y., Jeong K. W., Gerke D. S., Herviou L., Ianculescu I., Chodankar R., Siegmund K. D., Stallcup M. R. (2012) G9a functions as a molecular scaffold for assembly of transcriptional coactivators on a subset of glucocorticoid receptor target genes. Proc. Natl. Acad. Sci. U.S.A. 109, 19673–19678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim J. H., Li H., Stallcup M. R. (2003) CoCoA, a nuclear receptor coactivator which acts through an N-terminal activation domain of p160 coactivators. Mol. Cell 12, 1537–1549 [DOI] [PubMed] [Google Scholar]
- 17. Yu E. J., Kim S. H., Heo K., Ou C. Y., Stallcup M. R., Kim J. H. (2011) Reciprocal roles of DBC1 and SIRT1 in regulating estrogen receptor α activity and co-activator synergy. Nucleic Acids Res. 39, 6932–6943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jeong K. W., Kim K., Situ A. J., Ulmer T. S., An W., Stallcup M. R. (2011) Recognition of enhancer element-specific histone methylation by TIP60 in transcriptional activation. Nat. Struct. Mol. Biol. 18, 1358–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Teyssier C., Ou C. Y., Khetchoumian K., Losson R., Stallcup M. R. (2006) Transcriptional intermediary factor 1α mediates physical interaction and functional synergy between the coactivator-associated arginine methyltransferase 1 and glucocorticoid receptor-interacting protein 1 nuclear receptor coactivators. Mol. Endocrinol. 20, 1276–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ou C. Y., Kim J. H., Yang C. K., Stallcup M. R. (2009) Requirement of cell cycle and apoptosis regulator 1 for target gene activation by Wnt and β-catenin and for anchorage-independent growth of human colon carcinoma cells. J. Biol. Chem. 284, 20629–20637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Seo W. Y., Jeong B. C., Yu E. J., Kim H. J., Kim S. H., Lim J. E., Kwon G. Y., Lee H. M., Kim J. H. (2013) CCAR1 promotes chromatin loading of androgen receptor (AR) transcription complex by stabilizing the association between AR and GATA2. Nucleic Acids Res. 41, 8526–8536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shipp L. E., Lee J. V., Yu C. Y., Pufall M., Zhang P., Scott D. K., Wang J. C. (2010) Transcriptional regulation of human dual specificity protein phosphatase 1 (DUSP1) gene by glucocorticoids. PLoS ONE 5, e13754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim Y. R., Lee B. K., Park R. Y., Nguyen N. T., Bae J. A., Kwon D. D., Jung C. (2010) Differential CARM1 expression in prostate and colorectal cancers. BMC Cancer 10, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yu E. J., Kim S. H., Kim M. J., Seo W. Y., Song K. A., Kang M. S., Yang C. K., Stallcup M. R., Kim J. H. (2013) SUMOylation of ZFP282 potentiates its positive effect on estrogen signaling in breast tumorigenesis. Oncogene 32, 4160–4168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lin Y. S., Carey M. F., Ptashne M., Green M. R. (1988) GAL4 derivatives function alone and synergistically with mammalian activators in vitro. Cell 54, 659–664 [DOI] [PubMed] [Google Scholar]
- 26. Bolton E. C., So A. Y., Chaivorapol C., Haqq C. M., Li H., Yamamoto K. R. (2007) Cell- and gene-specific regulation of primary target genes by the androgen receptor. Genes Dev. 21, 2005–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Saladin R., Fajas L., Dana S., Halvorsen Y. D., Auwerx J., Briggs M. (1999) Differential regulation of peroxisome proliferator activated receptor γ1 (PPARγ1) and PPARγ2 messenger RNA expression in the early stages of adipogenesis. Cell Growth Differ. 10, 43–48 [PubMed] [Google Scholar]
- 28. Tontonoz P., Spiegelman B. M. (2008) Fat and beyond: the diverse biology of PPARγ. Annu. Rev. Biochem. 77, 289–312 [DOI] [PubMed] [Google Scholar]
- 29. Masuzaki H., Paterson J., Shinyama H., Morton N. M., Mullins J. J., Seckl J. R., Flier J. S. (2001) A transgenic model of visceral obesity and the metabolic syndrome. Science 294, 2166–2170 [DOI] [PubMed] [Google Scholar]
- 30. MacDougald O. A., Mandrup S. (2002) Adipogenesis: forces that tip the scales. Trends Endocrinol. Metab. 13, 5–11 [DOI] [PubMed] [Google Scholar]
- 31. Roberge C., Carpentier A. C., Langlois M. F., Baillargeon J. P., Ardilouze J. L., Maheux P., Gallo-Payet N. (2007) Adrenocortical dysregulation as a major player in insulin resistance and onset of obesity. Am. J. Physiol. Endocrinol. Metab 293, E1465–E1478 [DOI] [PubMed] [Google Scholar]
- 32. Siersbæk R., Nielsen R., John S., Sung M. H., Baek S., Loft A., Hager G. L., Mandrup S. (2011) Extensive chromatin remodelling and establishment of transcription factor “hotspots” during early adipogenesis. EMBO J. 30, 1459–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang L., Xu S., Lee J. E., Baldridge A., Grullon S., Peng W., Ge K. (2013) Histone H3K9 methyltransferase G9a represses PPARγ expression and adipogenesis. EMBO J. 32, 45–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Picard F., Géhin M., Annicotte J., Rocchi S., Champy M. F., O'Malley B. W., Chambon P., Auwerx J. (2002) SRC-1 and TIF2 control energy balance between white and brown adipose tissues. Cell 111, 931–941 [DOI] [PubMed] [Google Scholar]
- 35. Louet J. F., Coste A., Amazit L., Tannour-Louet M., Wu R. C., Tsai S. Y., Tsai M. J., Auwerx J., O'Malley B. W. (2006) Oncogenic steroid receptor coactivator-3 is a key regulator of the white adipogenic program. Proc. Natl. Acad. Sci. U.S.A. 103, 17868–17873 [DOI] [PMC free article] [PubMed] [Google Scholar]