

Background: Tomosyn's WD40 domain affects its ability to inhibit exocytosis.
Results: Unstructured loops in the WD40 domain are involved in tomosyn's diffusion and organization on the plasma membrane.
Conclusion: These key loops mediate tomosyn's binding to the SNARE protein SNAP25.
Significance: Novel findings regarding tomosyn's membranal distribution and interactions shed new light on regulation of exocytosis by the SNARE complex and tomosyn.
Keywords: Exocytosis, Membrane Fusion, Microscopy, Neurobiology, SNARE Proteins, FRAP, Tomosyn
Abstract
Neuronal exocytosis depends on efficient formation of soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complexes and is regulated by tomosyn, a SNARE-binding protein. To gain new information about tomosyn's activity, we characterized its mobility and organization on the plasma membrane (PM) in relation to other SNARE proteins and inhibition of exocytosis. By using direct stochastic optical reconstruction microscopy (dSTORM), we found tomosyn to be organized in small clusters adjacent to syntaxin clusters. In addition, we show that tomosyn is present in both syntaxin-tomosyn complexes and syntaxin-SNAP25-tomosyn complexes. Tomosyn mutants that lack residues 537–578 or 897–917 from its β-propeller core diffused faster on the PM and exhibited reduced binding to SNAP25, suggesting that these mutants shift the equilibrium between tomosyn-syntaxin-SNAP25 complexes on the PM to tomosyn-syntaxin complexes. As these deletion mutants impose less inhibition on exocytosis, we suggest that tomosyn inhibition is mediated via tomosyn-syntaxin-SNAP25 complexes and not tomosyn-syntaxin complexes. These findings characterize, for the first time, tomosyn's dynamics at the PM and its relation to its inhibition of exocytosis.
Introduction
Upon arrival at the plasma membrane (PM)2 and before fusion, vesicles undergo a priming reaction that leaves them fusion-competent. One of the processes occurring during priming is the assembly of the three soluble NSF attachment protein receptor (SNARE) proteins into a stable and highly conserved complex, termed SNARE complex (1). The SNARE complex is composed of the vesicular SNARE synaptobrevin and the PM SNARE proteins syntaxin and SNAP25 (2). Once assembled, the SNARE complex catalyzes vesicle fusion during exocytosis in neurons and neuroendocrine cells by overcoming the energy barrier required for this process (3). Several SNARE-interacting proteins are thought to regulate priming, one of them being tomosyn, which is known as a negative regulator of exocytosis and, in particular, priming (4, 5).
Tomosyn was initially discovered as a binding partner of syntaxin, creating a complex with syntaxin, SNAP25, and synaptotagmin (4). A few years later, Hatsuzawa et al. (6) showed that tomosyn forms a dead-end tomosyn-SNARE complex with syntaxin and SNAP25. Tomosyn contains a synaptobrevin-like SNARE motif but lacks a transmembrane anchor, which might preclude the SNARE molecules from forming fusogenic SNARE complexes, thereby inhibiting priming and fusion. It was also claimed that the N-terminal WD40 repeat domain of tomosyn catalyzes oligomerization of the SNARE complex, leading to inhibition of neurotransmitter release (7). Furthermore, it was shown that tomosyn can inhibit exocytosis independently of its SNARE interaction with syntaxin and that the integrity of the WD40 domain is crucial for tomosyn's inhibitory function (8). Hence, there is conflicting evidence for tomosyn's mode of action.
In 2007, the crystal structure of one of tomosyn's homologs in yeast (Sro7p) was published, revealing a backbone composed of two 7-bladed WD40 β-propellers leading to a “tail” domain bound to the β-propeller loops (9). We used this structure as a template for homology modeling to build a putative structural model for tomosyn (10). In this model, tomosyn also possesses two WD40 β-propellers followed by a tail domain. However, tomosyn's sequence is longer than that of Sro7p, creating regions in the model with no template. Three major insertions of this type were termed loop 1 (537–578 aa), loop 2 (675–752 aa), and loop 3 (897–917 aa) of rat tomosyn isoform m-1. PC12 cells expressing tomosyn lacking loop 1 or loop 3 (Δ-loop 1 and Δ-loop 3, respectively) lost their ability to inhibit secretion of human growth hormone compared with cells expressing the wild type tomosyn (10). Interestingly, the interaction of the tomosyn Δ-loop mutants with syntaxin was not altered as measured by GST-pulldown experiments, and they exhibited similar translocalization to the PM upon coexpression with syntaxin to that of the wild type tomosyn (10). Therefore, the reason for the loss of function of Δ-loop 1 and Δ-loop 3 mutants remained unknown.
Until recently, the organization and dynamics of presynaptic proteins involved in vesicle priming and fusion had not been extensively studied. Studies using super-resolution imaging showed that syntaxin and SNAP25 form separate clusters with very little overlap, although they require each other for fusion (11–13). The clusters of syntaxin and SNAP25 are at equilibrium with free SNARE molecules, which are much more mobile (13, 14). These free molecules occasionally form syntaxin-SNAP25 dimers termed “acceptor complex,” which can interact with synaptobrevin during the fusion process (Ref. 12 but see Ref. 15 for additional view). Syntaxin's diffusive motion was also the interest of Ribrault et al. (16), which demonstrated, using FRAP in neurons, that syntaxin was rapidly exchanged between synaptic and extrasynaptic regions and characterized dynamic interactions with its partners. Knowles et al. (17) measured the diffusion coefficients of syntaxin and SNAP25 in PC12 cells and demonstrated two populations of syntaxin, with rapid and slow diffusion rates, that might represent the distribution of molecules outside and inside the clusters, respectively. Here, we characterize tomosyn's mobility and organization on the PM, further investigate its interactions, with and without the loops, with syntaxin and SNAP25, and suggest a possible structure-function mechanism.
MATERIALS AND METHODS
Expression Constructs
Rat syntaxin-1A and m-tomosyn-1 were fused to the C terminus of the fluorescent proteins EGFP and monomeric red fluorescent protein. Preparation of the deletion mutants was as described previously (10). C-terminal deletion of tomosyn aa 1–1067 (tomosyn-ΔCT, D1068X) was constructed using the PCR-based QuikChange site-directed mutagenesis kit (Stratagene).
Monomeric mutants (A206K) of pECFP-C1 and pEcYFP-C1 (citrine) vectors harboring the LoxP sequence at the C-terminal end of the fluorophore were used as recipients for subcloning using the Cre recombinase-mediated Creator system (Clontech). Nucleotide sequences encoding rat syntaxin-1A, m-tomosyn-1, and SNAP25 were fused to the C terminus of each fluoroprotein sequence. A flexible 14–17-aa linker was inserted between the fluoroprotein and the gene of interest to reduce restrictions on fluoroprotein orientation. Site-directed mutagenesis was carried out using the QuikChange site-directed mutagenesis kit to generate a mutant of SNAP25.
Cell Culture, Transfection, and Immunocytochemistry
PC12 cells were cultured at 37 °C in 5% CO2 in culture dishes that contained glass coverslips pretreated with poly-d-lysine (100 μg/ml). The culture medium was Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% horse serum, 5% fetal bovine serum, and 1% penicillin/streptomycin (Invitrogen). Cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol, except that one-half of the suggested Lipofectamine concentration was used. Cells were used for experiments 24–48 h post-transfection.
For PLA staining, cells were fixed for 20 min at room temperature in 4% paraformaldehyde in phosphate-buffered saline (PBS), permeabilized for 10 min in 0.2% Triton X-100, and blocked for 30 min in normal goat serum (200 μg/ml). Then cells were probed with affinity-purified primary antibodies for 1 h at room temperature and labeled with secondary antibodies according to the company's protocol (Olink Bioscience, Uppsala, Sweden).
To image the distribution of tomosyn in the PM using dSTORM, PC12 cells were grown overnight on poly-l-lysine-coated glass coverslips. Coverslips were sonicated to remove the upper parts of the cells, leaving the native PM sheets attached. These were immediately fixed, washed, and immunostained as described previously (18).
Antibodies
Primary antibody against tomosyn (homemade affinity-purified rabbit anti-tomosyn polyclonal, a kind gift from Dr. Ulf Matti and Prof. Jens Rettig, Saarland University, Germany) was diluted 1:200 for immunocytochemistry and 1:1000 for Western blotting after pulldown. Preparation was described previously (6). Primary antibody against syntaxin was diluted 1:100 for PLA (catalog no. 110111, Synaptic Systems, Goettingen, Germany) and for dSTORM staining (mouse monoclonal antibody HPC-1). Primary antibody against SNAP25 was diluted to 1:400 (catalog no. 111011, Synaptic Systems). For dSTORM staining and for single color imaging, Cy5-coupled goat anti-mouse was used. For dual-color imaging, both Cy5-coupled goat anti-mouse and Alexa488-coupled goat anti-rabbit were used as the secondary antibodies (Dianova).
Cell Culture and Transfection for GST Pulldown
Human embryonic kidney (HEK) 293A cells were cultured at 37 °C and 5% CO2 in DMEM supplemented with 10% fetal calf serum, 2 mm l-glutamine, and 100 units/ml penicillin/streptomycin. Transfections were performed using Jet-PEI (PolyPlus Transfection, New York) according to the manufacturer's instructions using a 1:2 (DNA/reagent) ratio.
FRAP
FRAP studies were conducted in external solution (140 mm NaCl, 3 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm HEPES, and 2 mg/ml glucose) on PC12 cells transfected with EGFP-tomosyn together with nonfluorescently tagged syntaxin. FRAP measurements were performed at 37 °C. An argon-ion laser beam (Innova 70C, Coherent) was focused through a fluorescence microscope (Axio Imager.D1, Carl Zeiss MicroImaging) to a Gaussian spot of 0.77 ± 0.03 μm (Plan Apochromat 63×/1.4 NA oil-immersion objective). Focus was determined at the membrane of the cell where higher fluorescence was measured relative to the cytoplasm. After a brief measurement at the monitoring intensity (488 nm, 1 microwatt), a 5-microwatt pulse (5–10 ms) bleached 60–75% of the fluorescence in the spot, and recovery was followed by the monitoring beam for 30 s. The resulting curve was fitted (see supporting information in Ref. 19) by entering as fixed inputs the cytosolic diffusion value (Dc, 3 μm2/s; measured for tomosyn-ΔCt, which lacks the SNARE motif) and the Rm value (measured for each condition/mutant as the ratio between fluorescence intensity on the PM to the sum of average fluorescence on the PM and in the cytosol). Unlike Ref. 19, each cell was fitted separately, and the values were averaged to obtain standard deviations instead of averaging the curves to obtain an averaged fitting curve.
The derivation of the analytical expressions and the MatLab program used for the fitting and the derivation of the best fit parameters (Rf, Dm, Koff, and Kon) are presented in the supporting information in Ref. 19.
FRET
FRET imaging was measured using the sensitized emission imaging methodology. The imaging setup was described previously (10).
Image correction and analysis were performed off line using the method of FRET stoichiometry (20) and implemented using custom-written MatLab scripts. Calibration parameters specific to the optical system and fluorophores that are required for FRET analysis were determined as described previously (21). The apparent FRET efficiency of the acceptor in complex with the donor (EA), apparent FRET efficiency of the donor in complex with the acceptor (ED), and molar ratio of total acceptor/donor (ratio) were determined by pixel-by-pixel analysis of the images (21).
GST Pulldown
Syntaxin-1A was expressed in Escherichia coli as a GST fusion protein. GST and GST-SNAP25 were expressed in a similar manner, and all three proteins were purified by glutathione-Sepharose (Sigma). Purified GST, GST-syntaxin-1A, or GST-SNAP25 (150 pmol) were incubated with glutathione-Sepharose at 4 °C, and excess proteins were removed by three washes with PBS. EGFP-tagged tomosyn mutants were expressed in HEK293 cells by transfection using JetPei (PolyPlus Transfection, New York) according to the manufacturer's instructions using a 1:2 (DNA/reagent) ratio. Lysates prepared from the cells 48 h after transfection were incubated with the coated beads for 2 h at 4 °C in lysis buffer (50 mm Tris, pH 7.6, 10 mm MgCl2, 0.5 mg/ml BSA, 0.5 mm DTT, 0.1 m NaCl) with protease inhibitors. The beads were washed four times with PBS, and bound proteins were analyzed by SDS-PAGE and immunoblotting with anti-tomosyn antibody (diluted 1:1000). GST-syntaxin-1A, GST-SNAP25, and GST plasmids were a kind gift from Prof. Ilana Lotan (Tel Aviv University).
Confocal Microscopy
Images were obtained with a Leica TCS SP5 II confocal microscope (Leica Microsystems) under the control of LAS-AF software. Confocal images were acquired using 561 DPSS laser and 436/488/514 argon laser at 400 Hz and 1024 × 1024 pixel with 26 × 26 nm pixel size.
Confocal imaging of the PLA experiment was carried out with a Zeiss (Oberkochen, Germany) LSM 510 META confocal microscope. Images were acquired using 633 HeNe laser and 488 argon laser and 512 × 512 pixel with 60 × 60 nm pixel size. PLA signals were collected by acquiring a Z-stack and creating an image of its projection. Signals were quantified by FIJI software (ImageJ) using “FindPeaks” plugin.
dSTORM Imaging and Analysis
For single color imaging, shortly before imaging, samples were incubated in a “switching buffer” consisting of PBS, pH 7.4, containing oxygen scavenger (oxygen was removed by adding 0.5 mg ml−1 glucose oxidase (Sigma), 40 μg ml−1 catalase (Roche Applied Science), 10% w/v glucose, and 100 mm β-mercaptoethylamine) (11). For dual-color imaging, shortly before imaging, samples were incubated in a “switching buffer” consisting of 100 mm β-mercaptoethylamine in PBS, pH 8.3.
dSTORM imaging and analysis were performed as detailed previously (11, 22, 23). Briefly, we used an inverted microscope (Olympus IX-71) equipped with an oil-immersion objective (×60, NA 1.45; Olympus). A 641-nm diode laser (Cube 640-100C, Coherent, Santa Clara, CA) was used for excitation of Cy5, and a 488-nm laser (Sapphire 488LP; Coherent) was used for excitation of Alexa488. A polychromatic mirror (HC 410/504/582/669; Semrock, Rochester, NY) was used to separate laser and fluorescence light; the latter was imaged on an electron-multiplying CCD (EMCCD; Ixon DU897, Andor) camera. Additional bandpass filters were used in the detection path of the setup (ET700/75 and HQ535/50, Chroma). Additional lenses were used to achieve a final camera pixel size of 105 nm. 20,000–30,000 frames were recorded with frame rates of 100–140 Hz at irradiation intensities of 1–5 kilowatts/cm2. Applying a laser power of 1–5 kilowatts cm−2, we detected 500–5000 photons/molecule and frame, corresponding to a localization precision of ∼10 nm (24).
In the current imaging setup, a nosepiece stage (e.g. IX2-NPS, Olympus) was used to minimize lateral and especially axial drift. To check for lateral drift, fluorescent beads that do not photobleach during the experiment were immobilized at low density, and their positions were determined as a function of time. By generating histograms of the x and y position of single beads and by determining the standard deviation of the distributions in x and y, lateral drift can be monitored (24). During the imaging experiments used for the reconstruction of the dSTORM maps, the lateral drift was negligible.
For the meta-analysis of the fluorophore blinking events, in case the same molecule was “on” for several (n >1) sequential frames, all blinking events were united to a single blinking event. In case blinking from the same fluorophore occurred several times during the movie (nonsequential), all events were taken into account as part of the dSTORM reconstructed map.
For the localization map, we used a photon threshold of 1000 photons applied for the raw data localization text file, i.e. only localizations above 1000 photon counts were used for image reconstruction and further analysis. After these steps, no further threshold or image processing was applied for the reconstructed dSTORM maps. Hence, all the remaining localizations in the map were taken for the Mander's colocalization test and for the clustering analysis. Mander's coefficient calculation was performed on images reconstructed with a pixel size of 10 nm.
Colocalization analysis with Mander's coefficient was carried out using JACoP plugin version 2.0 (25), FIJI software. As a control, a randomized image of the green channel was created, and colocalization was calculated. The control showed 50% decrease in the colocalization with the corresponding syntaxin image compared with the original picture.
RESULTS
Tomosyn Localizes to the PM in the Presence of Syntaxin and Has a Similar Diffusion Coefficient on the Membrane
To visualize tomosyn-SNARE interactions in living cells, N-terminal fluoroprotein-tagged constructs were generated for tomosyn and syntaxin. Confocal microscopy of PC12 cells transfected with wild type tomosyn demonstrated a primarily cytosolic distribution. However, when tomosyn was coexpressed with syntaxin, it demonstrated clear colocalization with syntaxin in the PM region (Fig. 1A), as reported previously (26). To ensure that tomosyn indeed localizes at the PM in the presence of syntaxin and not in submembranal regions, we compared its staining with the membranal staining of the FM dye FM5-95 and observed full overlap between tomosyn and the FM dye (data not shown). The C-terminal SNARE motif of tomosyn is critical for its interaction with other SNARE proteins, and it is through these interactions that tomosyn is believed to localize to the PM (8). Indeed, a truncation mutant of tomosyn in which the R-SNARE was deleted (tomosyn-ΔCT, aa 1–1067) failed to colocalize to the PM in the presence of syntaxin and showed cytoplasmic distribution, confirming that tomosyn's R-SNARE motif is critical for colocalization with syntaxin to the PM (Fig. 1A).
FIGURE 1.

Characterization of tomosyn's dynamics on the PM. A, expression and subcellular localization of fluoroprotein-tagged tomosyn and syntaxin-1A proteins in PC12 cells as visualized by confocal fluorescence microscopy. Note that tomosyn alone presents cytosolic localization of tomosyn, whereas coexpression with syntaxin results in membranal localization of tomosyn. Tomosyn-ΔCT, lacking the SNARE motif, remains cytosolic when coexpressed with syntaxin. Scale bar, 2 μm. B, recovery curve of tomosyn's fluorescence after photobleaching while coexpressed with syntaxin. The specific curve shown yielded R2 = 0.876, Koff = 0.183/s, Kon = 0.22/s, Dm = 0.015 μm2/s, and Rf (mobile fraction) = 0.82. C, summary table of FRAP experiments for tomosyn coexpressed with syntaxin. Dc used for the analysis 3.07 ± 0.16 μm2/s for tomosyn-ΔCT. Values indicate mean ± S.E. D, homology model of m-tomosyn-1 based on Sro7p demonstrating preservation of the β-propellers and addition of three undefined loops (loop 1, loop 2, and loop 3) (10). E–G, tomosyn deletion mutants have altered kinetics on the PM. Deletion mutants in loops 1 and 3 (d1 and d3, respectively) diffuse faster on the PM (Dm; p < 0.001) and display lower exchange rates with the PM (Koff WT versus d3, p < 0.05; Kon WT versus d1, p < 0.05; Kon WT versus d3, p < 0.001). *, p < 0.05; ***, p < 0.001.
On the PM, tomosyn interacts directly with syntaxin (26). However, it is not clear whether this interaction involves SNAP25. This interaction and interactions with other proteins can influence tomosyn's mobility on the PM and its ability to inhibit exocytosis. In addition, we have recently reported that deletion of two amino acid stretches in the WD40 domain of tomosyn (loop 1, aa 537–578, and loop 3, aa 897–917) alters its ability to inhibit exocytosis (10). We assume that such mutations alter tomosyn's interaction with syntaxin or syntaxin-SNAP25 and hence influence its dynamics on the PM. However, these dynamics were never examined. Thus, we decided to examine tomosyn's biophysical properties on the PM under different experimental conditions. We used FRAP measurements together with a published fitting algorithm that provides simultaneous quantification of the membrane (un)binding rates and the diffusion coefficient on the PM (19). Initially, PC12 cells were cotransfected with syntaxin and tomosyn, resulting in PM localization of tomosyn. FRAP experiments were performed by laser bleaching an average 70% of the tomosyn in a small area of the PM and monitoring a typical fluorescence recovery curve demonstrating the recovery rate of the mobile fraction of tomosyn and the fraction of immobile tomosyn that does not recover. Fluorescence recovery of tomosyn after photobleaching can stem from either lateral diffusion of tomosyn on the PM or diffusion of cytosolic tomosyn, as tomosyn can be found in equilibrium between these two pools. The resulting curve was then fitted to a function expressing the normalized fluorescence (see Equation 9 in Ref. 19). As fixed parameters, we calculated two values. The first is Dc, which reflects the cytoplasmic diffusion coefficient of tomosyn. This parameter was measured using expression of tomosyn-ΔCT, which lacks the SNARE motif. The second fixed parameter was the Rm value (the relative number of molecules of tomosyn on the PM and in the cytoplasm), which was measured for each condition/mutant. All other parameters (Dm, diffusion coefficient on the PM; Kon, rate constant of PM binding; Koff, rate constant of PM dissociation) were determined by the fitting procedure.
The Rm value of wild type tomosyn coexpressed with syntaxin was calculated as the ratio between the fluorescence on the membrane and the fluorescence in the cytosol and was found to be 0.55 ± 0.006 (n = 29). The cytoplasmic diffusion coefficient (Dc) of tomosyn was measured by FRAP (calculated as D = ω2/4τD, where ω is the Gaussian radius of the laser beam) in the cytoplasm of cells transfected with tomosyn-ΔCT (lacking the SNARE motif). This mutant does not interact with syntaxin or with SNAP25 (8), and its expression pattern is solely cytoplasmic. Accordingly, this value was found to be faster than the Dc for wild type tomosyn, which is inevitably slower as a result of its interactions with proteins on the PM. The Dc value obtained for this mutant was 3.07 ± 0.16 μm2/s (Fig. 1C; n = 32), and this value was used as a reference for all tomosyn experiments. Using two fixed values, Rm = 0.55 and Dc = 3.07 μm2/s, we analyzed 29 cells coexpressing tomosyn and syntaxin. Fig. 1B displays the FRAP recovery curve for a wild type EGFP-tomosyn-expressing cell by the full-fitting approach. The best fit values were Koff = 0.139 ± 0.026/s, Kon = 0.180 ± 0.032/s, and Dm = 0.024 ± 0.004 μm2/s. For comparison, we performed the same analysis on cells expressing only monomeric red fluorescent protein-tagged syntaxin; the measured Dm value for syntaxin in these cells was 0.03025 ± 0.002 μm2/s, similar to that of tomosyn in the presence of syntaxin (Fig. 1C, p > 0.05) and also similar to the value calculated for syntaxin previously (17, 27). This finding, together with the Förster (fluorescence) resonance energy transfer (FRET) between syntaxin and tomosyn (26), suggests that at the PM tomosyn moves with a dynamics similar to that of syntaxin. As for the obtained binding constants, they reflect a rather fast association/dissociation rate of tomosyn with PM syntaxin, with time constants of 5.5 and 7.5 s, respectively. Based on the published values for other proteins (19), these values support the notion that tomosyn acts as a nonintegral membrane protein and that it undergoes exchange between the cytoplasm and the PM, unlike a membrane-associated protein. Nonetheless, its Dm value resembles syntaxin's diffusion on the PM, in line with the knowledge of their interaction. It also suggests that with overexpression of tomosyn and syntaxin, about 55% of the tomosyn population is bound to PM syntaxin (Rm). These data represent an accurate determination of the biophysical parameters of tomosyn dynamics at the PM under cotransfection with syntaxin. The data provide, for the first time, information about the kinetics of tomosyn's interaction with syntaxin on the PM and can be used as a data set for comparison of tomosyn-PM interactions following various manipulations.
Tomosyn Mutants Display Altered Binding and Diffusion Kinetics
We previously published a putative structure of tomosyn based on homology modeling with the yeast homolog Sro7p (10), which is composed of two 7-bladed WD40 β-propellers. In the model (Fig. 1D), the primary m-tomosyn-1 sequence is significantly longer than that of Sro7p and exhibits three loops that deviate from the Sro7p template as follows: loop 1, aa 537–578; loop 2, aa 675–752; loop 3, aa 897–917, all emanating from the second β-propeller backbone. Upon expression in PC12 cells, deletion of loop 1 (Δ-loop 1) or loop 3 (Δ-loop 3) resulted in complete elimination of tomosyn inhibition of K+-induced human growth hormone secretion. In comparison, deletion of loop 2 (Δ-loop 2) had no significant effect on the ability of tomosyn to inhibit secretion (10).
We hypothesized that the lack of tomosyn inhibition in the tomosyn Δ-loop 1 and Δ-loop 3 mutants might result from changes in their interaction with other proteins and that this would affect their dynamic exchange and mobility on the PM. Accordingly, we tested whether the Δ-loop mutants demonstrate different binding kinetics or altered diffusion coefficients on the PM upon coexpression with syntaxin. PC12 cells were cotransfected with syntaxin and EGFP-Δ-loop mutants. No significant differences were found between the distribution of the tomosyn Δ-loop mutants and wild type tomosyn. The calculated Rm values were 0.505± 0.004, 0.523 ±0.007, and 0.513 ± 0.005 for Δ-loop 1, Δ-loop 2, and Δ-loop 3 mutants, respectively, suggesting that all mutants translocate similarly to the PM upon syntaxin expression. We then performed FRAP experiments with the mutants and calculated the diffusion coefficient and binding and unbinding kinetics using the full-fitting approach. Interestingly, the results indicated significant differences in the binding and diffusion kinetics between wild type tomosyn and Δ-loop 1 or Δ-loop 3. As shown in Fig. 1E, Dm values of Δ-loop 1 and Δ-loop 3 were significantly larger than that calculated for wild type tomosyn (0.051 ± 0.005 μm2/s for Δ-loop 1 and 0.048 ± 0.005 μm2/s for Δ-loop 3 versus 0.024 ± 0.004 μm2/s and 0.024 ± 0.003 μm2/s for the wild type and Δ-loop 2, respectively). Hence Δ-loop 1 and Δ-loop 3 mutants exhibited faster lateral diffusion on the PM than wild type tomosyn. Note that the change in diffusion was not caused by the change in mass because Δ-loop 2, which contains the largest deletion, diffuses like the wild type tomosyn, and in addition such a small deletion should not cause more than a 5% change in the diffusion coefficient according to the Stokes-Einstein equation. Koff and Kon values were also statistically different in these mutants as they displayed faster binding constants than the wild type protein (Koff = 0.084 ± 0.020/s and Kon 0.086 ± 0.020/s for Δ-loop 1; Koff = 0.051 ± 0.010/s and Kon 0.053 ± 0.011/s for Δ-loop 3, see Fig. 1, F and G). Notably, none of the kinetic parameters for the Δ-loop 2 mutant were statistically different from those of the wild type tomosyn. As mentioned earlier, this mutant retains tomosyn's ability to inhibit secretion, unlike Δ-loop 1 and Δ-loop 3 which lack this ability (10). These results suggest that Δ-loop 1 and Δ-loop 3 mutants may have altered interactions with syntaxin or with other proteins in this complex that affect their binding kinetics to the PM. These altered interactions may also be the cause for their loss of ability to inhibit secretion, as discussed below.
Endogenous Distribution of Tomosyn on Plasma Membrane Sheets of PC12 Cells
To complete the characterization of tomosyn on the PM, we wanted to determine the endogenous distribution of tomosyn and focus only on the distribution of its membranal component by imaging native PM sheets of PC12 cells. We employed super-resolution imaging by the dSTORM technique, which was used successfully in a previous study to characterize the distribution of syntaxin and SNAP25 at the PM of PC12 cells (11). The underlying mechanism of this method includes stochastic reversible photo-switching of synthetic organic fluorophores, followed by accurate localization of single molecules and reconstruction of a super-resolution image from a large number of single molecule coordinates (28). dSTORM exhibits exquisite single molecule sensitivity and achieves a lateral resolution of 10–20 nm (24). Using further analytic approaches that we developed, we were able to distinguish single molecules from clusters of proteins and determine the size, shape, and density of protein clusters (11). Accordingly, single- and dual-color dSTORM imaging was performed on native inside-out plasma membrane sheets generated by gentle disruption of PC12 cells as described before (18) and stained for either tomosyn or tomosyn and syntaxin (see “Materials and Methods”). The results of the analysis indicated that tomosyn distributed into nano-sized clusters and a nonclustered population. The characteristics of the tomosyn clusters, as presented in Fig. 2A, were found to be quite similar to those of syntaxin clusters; for example, the mean cluster diameter of tomosyn clusters was found to be around 97 nm, similarly to the size found for syntaxin clusters (93.4 nm (11)) but differed from the mean cluster diameter of SNAP25 clusters (129.6 nm (11)). Taking into consideration the similarity between the syntaxin and tomosyn clusters in size and shape and the interaction of syntaxin with tomosyn that enables its presence on the membrane, we tested the relative distribution of the two proteins and measured the extent of colocalization between them by dual-color dSTORM imaging (Fig. 2B). The dual-color dSTORM maps of tomosyn and syntaxin were analyzed using the Mander's coefficient, which is defined as the proportion of the red signal coincident with a signal in the green channel over its total intensity. Randomized images of the green channel were used as control (JACoP plugin, see “Materials and Methods”). The colocalization of syntaxin with tomosyn was found to be 18%, and the colocalization of tomosyn with syntaxin was found to be 66%. The results suggest that the majority of tomosyn colocalizes with syntaxin molecules. However, a large percentage of syntaxin (∼82%) does not colocalize with tomosyn (Fig. 2A, bottom). The results demonstrating high colocalization of tomosyn with syntaxin nicely coincide with the dependence of tomosyn's presence at the PM on its interaction with syntaxin. Conversely, syntaxin, as expected, does not necessarily localize with tomosyn and can interact or overlap with other proteins besides tomosyn, such as Munc18 and SNAP25, or can also contain solely syntaxin molecules (11).
FIGURE 2.

Tomosyn distribution at the plasma membrane. A, table includes mean cluster diameter, percentage of the single-molecule population, and the ratio of diameters representing cluster shape (circular or elliptical). Values indicate mean ± S.E. B, dSTORM map of the membranal staining of tomosyn (green) and syntaxin (red) with single molecule resolution was reconstructed from syntaxin 97,076 localizations and tomosyn, 7595 localizations. Insets are magnified sections of the regions in the membrane marked by rectangles, showing close proximity between tomosyn and syntaxin. Scale bar, 500 nm. Scale bar in the inset, 100 nm.
FRET Signals between Tomosyn and Syntaxin and between Tomosyn and SNAP25
We next wanted to examine whether tomosyn on the PM interacts primarily with syntaxin, the syntaxin-SNAP25 complex, or only with SNAP25. To thoroughly explore the interactions between tomosyn, syntaxin, and SNAP25 in living cells, sensitized emission FRET experiments were performed on PC12 cells coexpressing citrine-tomosyn and either CFP-syntaxin or CFP-SNAP25 (Fig. 3A). Note that upon coexpression with syntaxin, tomosyn translocates to the PM; however, with SNAP25 it is only partially localizes to the membrane (Fig. 3A). Sensitized emission FRET efficiency from shade- and background-corrected images was quantified by the FRET stoichiometry method (20). According to this method, the maximum FRET efficiency between CFP and citrine is 37% as measured inside cells (20). In PC12 cells coexpressing citrine-tomosyn and CFP-syntaxin, an average apparent FRET efficiency (ED) of 7.6 ± 0.3% (n = 55) was measured when the expression of acceptor and donor fluoroproteins ranged between molar ratios of 0.9 and 1.1 (Fig. 3B, left column). The 0.9–1.1 ratio pixels represent 15.5 ± 1.8% of all double-labeled pixels measured from the cells. This apparent FRET efficiency is severalfold greater than the background FRET level between citrine-tomosyn-ΔCT and CFP-syntaxin in these cells (1.6 ± 0.06%), although the value is lower than that previously reported in HEK293 cells and in bovine adrenal chromaffin cells (26). This is likely the result of the different intracellular milieu in PC12 cells. PC12 cells cotransfected with citrine-tomosyn and CFP-SNAP25 also demonstrated FRET, with an average apparent ED of 12.8 ± 0.8% (n = 55) with a molar ratio for expression ranging from 0.9 to 1.1 (Fig. 3C, left). The 0.9–1.1 ratio pixels represent 10.6 ± 2% of all double-labeled pixels measured from the cells. Background levels for this FRET pair (citrine-tomosyn-ΔCT and CFP-SNAP25) were 2.6 ± 0.17% for all ratios. It should be noted that this is the first report of FRET occurring between a fluorophore-labeled tomosyn-SNAP25 pair. These results suggest that tomosyn can interact with either syntaxin, as reported previously, or SNAP25. However, it is possible that the interaction with SNAP25 occurs via the tomosyn-syntaxin-SNAP25 complex, and we therefore explored this possibility.
FIGURE 3.

Visualization of tomosyn-SNARE complexes in living cells using sensitized emission FRET. A, FRET experiments were performed on PC12 cells coexpressing citrine-tomosyn and either CFP-syntaxin or CFP-SNAP25. Scale bar, 2 μm. B, FRET signal between tomosyn and syntaxin decreases after SNAP25 cleavage by BoNT-E, but is not completely eliminated (p < 0.001), indicating the presence of tomosyn-syntaxin dimers. Upper panel demonstrates cleavage of SNAP-25-CFP by BoNT-E. C, FRET signal between tomosyn and SNAP25 decreases after syntaxin cleavage by BoNT-C (p < 0.001). Upper panel demonstrates cleavage of CFP-syntaxin by BoNT-C. To test for possible cleavage of SNAP25 by BoNT-C, FRET was measured using a SNAP25 cleavage-mimicking mutant, SNAP25(1–198). Cotransfection with the light chain of BoNT-C resulted in a clear reduction of FRET between tomosyn and SNAP25(1–198) (p < 0.001), similar to the reduction seen with SNAP25(WT). D, FRET signal between tomosyn and syntaxin (left) decreases upon coexpression with Munc18 (p < 0.0001) but is not completely eliminated (p < 0.05 when compared with background). FRET signal between tomosyn and SNAP-25 does not change significantly upon coexpression with Munc18 (p > 0.05). ***, p < 0.001.
Tomosyn is known to interact with SNAP25 via formation of a ternary SNARE core complex with syntaxin (6), but whether tomosyn also interacts directly with SNAP25 in vivo via a binary interaction remains unclear. Initially, blot overlay assays demonstrated that glutathione S-transferase (GST)-SNAP25 does not bind tomosyn from a lysate of COS7 cells overexpressing tomosyn (4). However, tomosyn's yeast homolog Sro7p directly binds the SNAP25 yeast homolog, Sec9 (29). Furthermore, GST-SNAP23 was found to pull down tomosyn from adipocyte lysates (30). A later publication (8) demonstrated that GST-SNAP25 binds to EGFP-tomosyn from a PC12 lysate overexpressing EGFP-tomosyn; however, it is unclear whether syntaxin was also present in this complex.
To test whether the FRET signal between citrine-tomosyn and CFP-SNAP25 resulted from a binary interaction (i.e. tomosyn-SNAP25 complex) or a ternary interaction (i.e. tomosyn-syntaxin-SNAP25 complex), we initially determined whether the tomosyn-SNAP25 FRET signal requires the presence of syntaxin. We took advantage of the fact that botulinum neurotoxin C (BoNT-C) cleaves syntaxin in its juxtamembrane region, precluding it from forming SNARE complexes at the PM (31). Transfection of the catalytic light chain of BoNT-C into PC12 cells results in effective cleavage of syntaxin and strong knockdown of regulated exocytosis (32). Fig. 3C (upper panel) demonstrates cleavage of CFP-syntaxin by BoNT-C as the fluorescent signal translocates from the membrane into the cytosol. We hypothesized that if the tomosyn-SNAP25 FRET signal results from a ternary SNARE interaction and is dependent on syntaxin, then coexpression of BoNT-C in these cells would result in a decrease in tomosyn-SNAP25 FRET. Conversely, if tomosyn-SNAP25 FRET results from a binary interaction and is independent of syntaxin, then BoNT-C should have little or no effect on the tomosyn-SNAP25 FRET signal. Cotransfection of the light chain of BoNT-C with citrine-tomosyn and CFP-SNAP25 in PC12 cells resulted in a significant reduction in the apparent FRET efficiency between m-tomosyn and SNAP25 (BoNT-C, 4.5 ± 0.4%; control, 12.8 ± 0.8%, Fig. 3C, ED over molar ratio range 0.9–1.1). The signal was reduced by 64%, whereas the reduction to background level was 79%. These data demonstrate that the tomosyn-SNAP25 FRET signal reports primarily on the ternary tomosyn-SNARE complex (tomosyn-syntaxin-SNAP25) and that tomosyn-SNAP25 complexes are not abundant.
Excessive BoNT-C has also been shown to cleave SNAP25 at its C terminus (33, 34); thus, the effect of overexpressed BoNT-C on the tomosyn-SNAP25 FRET signal could potentially be attributed to cleavage of SNAP25 rather than to cleavage of syntaxin. To test this possibility, we measured FRET between tomosyn and a truncation mutant of SNAP25 (aa 1–198) that mimics the BoNT-C-cleaved form of SNAP25. At baseline, the apparent FRET efficiency between citrine-tomosyn and CFP-SNAP25(1–198) was similar to that with CFP-SNAP25(WT) (ED over molar ratio range 0.9–1.1: SNAP25(1–198), 13.4 ± 0.7%; SNAP25(WT), 12.8 ± 0.8%; Fig. 3C, right). Moreover, cotransfection with the light chain of BoNT-C resulted in a clear reduction of FRET between tomosyn and SNAP25(1–198), similar to the reduction seen with SNAP25(WT) (ED: SNAP25(1–198), 6.5 ± 0.7%; SNAP25(WT), 4.5 ± 0.4%). These data demonstrate that the reduction in FRET signal between tomosyn and SNAP25 in the presence of BoNT-C is due to the cleavage of syntaxin and not of SNAP25. Moreover, the data demonstrate that the tomosyn-SNAP25 FRET signal reports primarily on the tomosyn ternary SNARE complex.
The tomosyn-syntaxin FRET pair had been previously reported to produce a FRET signal, but it was unclear whether this FRET signal corresponded to a specific tomosyn-syntaxin complex or a tomosyn-SNARE complex (i.e. binary or ternary) (26). To investigate whether the FRET signal between citrine-tomosyn and CFP-syntaxin requires the presence of SNAP25, we performed experiments using the light chain of BoNT-E. This neurotoxin protease cleaves a 26-amino acid C-terminal fragment from SNAP25 and inactivates the protein for exocytic SNARE complex formation (35–37). BoNT-E light chain was previously shown to be able to achieve complete conversion of overexpressed EGFP-SNAP25 to the EGFP-SNAP25(1–180) fragment (38). As expected, Fig. 3B (upper panel) demonstrates cleavage of SNAP25-CFP by BoNT-E as the fluorescent signal translocates from the membrane into the cytosol. The rationale behind these experiments was analogous to that for the BoNT-C experiments with tomosyn-SNAP25 FRET; if the tomosyn-syntaxin FRET signal requires the presence of SNAP25 (i.e. ternary complex), then the light chain of BoNT-E that cleaves SNAP25 should decrease the FRET signal. Conversely, if the tomosyn-syntaxin FRET signal is independent of SNAP25 (i.e. binary complex), then the light chain of BoNT-E should not affect the FRET signal. The results of this experiment are shown in Fig. 3B. Coexpression of the light chain of BoNT-E in PC12 cells resulted in a decrease in the FRET signal between tomosyn and syntaxin but did not completely abolish it (ED at a molar ratio of 0.9–1.1:5.5 ± 0.3%, p < 0.001, Fig. 3B) as background levels were 1.64 ± 0.06%. The reduction in FRET signal due to SNAP25 cleavage was only 27%, whereas a reduction to background level was 78%. Compared with the 64% reduction in the case of syntaxin cleavage (Fig. 3B), this result suggests that only 27% of the tomosyn-syntaxin FRET signal reports on the ternary SNARE complex, whereas 51% reports on the syntaxin-tomosyn binary complex (22% is background). Hence, we found that tomosyn can be found in either a binary complex with syntaxin or a ternary complex with syntaxin and SNAP25.
To further straighten these results, we next tested the effect of Munc18 on the FRET between tomosyn and syntaxin and between tomosyn and SNAP25. Munc18 is essential for synaptic vesicle fusion and interacts with the neuronal SNARE proteins in at least two distinct modes. Munc18 interacts with the closed conformation of syntaxin, causing the inhibition of SNARE complex assembly, as well as with the assembled SNARE complex containing syntaxin in the open conformation (39). Tomosyn has been shown to compete with Munc18 for binding to syntaxin (4, 26), and therefore we hypothesized that if the tomosyn-syntaxin FRET signal results from a binary interaction, then addition of Munc18 would decrease the FRET signal. Results confirming our hypothesis are shown in Fig. 3D (left). Cotransfection of Munc18 with CFP-syntaxin and citrine-tomosyn resulted in a decrease in the FRET signal between tomosyn and syntaxin, but it did not completely abolish it (ED at molar ratio 0.9–1.1:2.48 ± 0.3%, p < 0.0001, Fig. 3D, left). This decrease in FRET remains significantly different from the background level (p < 0.05). These results indicate that Munc18 can out-compete with binding syntaxin and that most of the FRET signal results from the tomosyn-syntaxin binary interaction.
We next tested the effect of Munc18 overexpression on the FRET signal between tomosyn and SNAP25, which we demonstrate to represent mainly the ternary tomosyn-syntaxin-SNAP25. If indeed the FRET signal between tomosyn and SNAP25 is derived from a ternary interaction, then Munc18 will have no effect on it. It is unknown whether Munc18 can interact with the ternary tomosyn-syntaxin-SNAP25 complex. However, it interacts with the exocytotic SNARE complex as mentioned above. Coexpression of Munc18 with CFP-SNAP25 and citrine-tomosyn did not affect the FRET signal (ED at molar ratio 1:13.25 ± 1.9%, p > 0.05, Fig. 3D, right) suggesting that Munc18 does not disassemble the tomosyn-syntaxin-SNAP25 complex.
These results suggest that the change in the PM interaction mode shown in Fig. 1 for tomosyn Δ-loop 1 and tomosyn Δ-loop 3 can result from changes in their relative interactions with SNAP25 through the tomosyn-SNARE complex. For example, a shift from a ternary tomosyn-syntaxin-SNAP25 complex to a binary tomosyn-syntaxin complex can lead to faster diffusion on the PM for tomosyn's mutants.
Tomosyn Mutants Δ-Loop 1 and Δ-Loop 3 Display Lower Binding to SNAP25
Tomosyn mutants Δ-loop 1 and Δ-loop 3 lose their ability to inhibit exocytosis but show similar binding to syntaxin (10). Our finding that tomosyn can be found in a binary or ternary complex on the PM and that these mutants have different binding kinetics and Dm values led us to test whether these mutants have different affinities to SNAP25. We tested this hypothesis by applying two methods, imaging by proximity ligation assay (PLA) and GST pulldown of tomosyn.
For the PLA experiments, we transfected PC12 cells with EGFP-tomosyn or EGFP-fused Δ-loop mutants. The cells were fixed, labeled with primary antibodies, and stained to produce visible fluorescent signals where close proximity (<40 nm) between tomosyn mutants and endogenous syntaxin or tomosyn mutants and endogenous SNAP25 occurred. Confocal images were taken from the cells along the z axis, and projection images were produced showing signals from all planes of the cells. Representative cross-section images are displayed in Fig. 4A, and projection images are shown in Fig. 4B. Interestingly, interaction with syntaxin was labeled on the PM as well as in the cytosol, whereas interaction with SNAP25 seemed mostly membranal (Fig. 4A). PLA signals counted for mutants Δ-loop1 and Δ-loop3 to SNAP25 were statistically different (p < 0.01 and p < 0.05 respectively; Fig. 4C, left) representing fewer interactions between these mutants and SNAP25. However, for syntaxin, no differences were found in the number of PLA signals for all four tomosyn constructs (p > 0.05; Fig. 4C, right). These results suggest that tomosyn mutants Δ-loop1 and Δ-loop3 display lower proximity to SNAP25.
FIGURE 4.

Tomosyn mutants Δ-loop 1 and Δ-loop 3 display lower binding to SNAP25. PC12 cells expressing EGFP-labeled tomosyn or mutants were immunostained with PLA far-red reagent for identification of interactions with syntaxin and with SNAP25. A, cross-section of cells expressing EGFP-tomosyn. PLA interactions with syntaxin (left) and SNAP25 (right) are marked with purple spots. B, representative projection images of PLA signal between SNAP25 and tomosyn constructs. C, deletion mutants 1 and 3 display significantly lower numbers of PLA signal per cell with SNAP25 compared with the wild type tomosyn (p < 0.01; p < 0.05, respectively). No significant differences were found among tomosyn constructs in PLA signal per cell with syntaxin (p > 0.05). D, analysis showing in vitro GST-syntaxin or GST-SNAP25 pulldown of indicated EGFP-tagged tomosyn proteins. Recombinant GST-syntaxin or GST-SNAP25 was immobilized on glutathione-Sepharose beads and incubated with HEK293A cell lysates expressing EGFP-tomosyn wild type (wt) or the indicated tomosyn loop-deletion construct. Tomosyn immunoreactivity in each fraction was determined using anti-tomosyn antibody. Results show reduced binding of Δ-loops 1 and 3 to GST-SNAP25 compared with tomosyn wild type. No significant differences were found among tomosyn proteins binding to GST-syntaxin. *, p < 0.05; **, p < 0.01.
PLA's advantage is to demonstrate close proximity between the chosen proteins. However, the PLA signal appears in proximity of up to 40 nm, which is larger than a globular protein radius. To ensure that the tomosyn mutants Δ-loop 1 and Δ-loop 3 actually interact with SNAP25 to a lesser extent than does the tomosyn wild type, interaction between the proteins was measured. To further strengthen our findings, GST-pulldown assays were performed. GST-SNAP25 immobilized on glutathione-Sepharose beads was used to pull down each EGFP-Δ-loop mutant from HEK293 cell lysates and was then compared with the pulldown of wild type tomosyn. Neuronal SNARE proteins are not expressed in the HEK293 cell line (40), and therefore, this cell line provides a clean background upon which to test interactions with the various tomosyn deletion mutants. Immunoblots of the pulldown reactions were background-subtracted and normalized to totals (5%). Quantification showed that Δ-loop 1 and Δ-loop 3 mutants bind GST-SNAP25 to a lesser extent than the wild type tomosyn (Fig. 4D, left). Pulldown experiments of GST-syntaxin with tomosyn mutants showed no differences among those mutants and wild type tomosyn. Results were similar to those shown in Ref. 10; tomosyn mutants and wild type bound similarly to syntaxin (Fig. 4D, right). The pulldown results further strengthen the imaging analysis, which suggested that deletion of loop domains 1 and 3 decreases tomosyn binding to SNAP25. Binding to SNAP25 occurs within the tomosyn-SNARE complex and involves the binding of syntaxin, and this could be important for tomosyn's inhibition of exocytosis.
DISCUSSION
Tomosyn was discovered as a binding partner of syntaxin and as a negative regulator of neuronal exocytosis (4). However, the way that tomosyn participates in the inhibition of exocytosis remained unclear and is especially intriguing in light of its high molecular weight. Tomosyn is known to possess a SNARE motif, homologous to that of synaptobrevin, which can replace synaptobrevin in a nonfunctional tomosyn-SNARE complex (6, 41). However, this SNARE motif is not sufficient for its inhibitory effect. Indeed, later it was shown that the inhibitory function of tomosyn also depends on the integrity of the N-terminal WD40 domain (8). Recently, it was shown in vivo in Caenorhabditis elegans that neither tomosyn's SNARE motif by itself nor tomosyn lacking its SNARE motif can restore tomosyn's function, when expressed separately or together in tom-1 mutants (42). Yamamoto et al. (43) suggested that synaptotagmin binds tomosyn through tomosyn's N terminus and that this binding inhibits synaptotagmin from promoting exocytosis. However, it was also suggested that the N-terminal WD40 domain coordinates oligomerization of SNARE complexes that inhibit exocytosis (7). Therefore, tomosyn's mode of inhibition is not completely understood. In addition, the biophysical properties of tomosyn's interaction with the PM and its membranal distribution with respect to its membranal interacting partners are completely unknown.
To understand tomosyn's inhibition mechanism, we used several mutations in its key N-terminal domain to test its structure-function dependence and its membranal interactions shedding light on its general mode of action. As a first step, we characterized tomosyn's dynamics and distribution in relation to syntaxin, its main membranal binding partner. We used a model of tomosyn based on the structure of its homolog Sro7p, which showed that tomosyn's entire N terminus can fold into two β-propellers (9, 10). In proteins folded in this structure, the active site is often found in the cleft formed in the center of the propeller by loops connecting the successive four-sheet motifs (44, 45). We therefore focused on the three additional loops of tomosyn that do not exist in Sro7p, and we characterized the dynamics of their deletion mutants on the PM. We show that the Δ-loop 1 and Δ-loop 3 mutants exhibit faster membranal dynamics than the wild type. Because tomosyn binds the membrane through other proteins, the observed faster dynamics of the mutants on the PM suggest a change in the mutated proteins' interactions with their binding membranal partners, mainly syntaxin and SNAP25. Indeed, we found that Δ-loop 1 and Δ-loop 3 tomosyn mutants have less interaction with SNAP25, although the interaction with syntaxin remains the same. We show that the interaction of SNAP25 with tomosyn occurs only when tomosyn forms the inhibitory complex with syntaxin and SNAP25. Therefore, we suggest that Δ-loop 1 and Δ-loop 3 tomosyn mutants form less of these inhibitory complexes, and therefore the overall inhibitory function of tomosyn is reduced. These data suggest that loops 1 and 3 serve as essential regulatory elements of the protein, and in their absence, tomosyn cannot inhibit exocytosis (forms less tomosyn-syntaxin-SNAP25 complexes). Tomosyn's inhibitory function requires both its SNARE motif (forming the inhibitory complex with syntaxin and SNAP25) and its N terminus containing loops 1 and 3 regulating the formation of the complexes.
Using a new method to measure protein diffusion at the PM, we described tomosyn's biophysical properties on the PM. The diffusion coefficient of wild type tomosyn was similar to that of syntaxin in our system. This finding is in agreement with syntaxin's ability to localize tomosyn on the membrane and the previously published FRET signal between these two proteins, making syntaxin the main PM interactor of tomosyn (26). Although our measurements of syntaxin diffusion coefficient on the PM (0.03 μm2/s) resemble the published value (0.06 μm2/s, weighted average (17), and 0.025 μm2/s (27)), the slightly slower diffusion of tomosyn may stem from its presence in complexes either with syntaxin or syntaxin-SNAP-25 complexes. A recent paper by Ribrault et al. (16) found syntaxin diffuses in the synapse (0.07 μm2/s) but pauses occasionally to participate in fusion complexes. This finding can also add to the complexity of interpretation of diffusion data and suggest SNARE and SNARE-regulating proteins exhibit several rates of lateral diffusion depending on their presence as single molecules or multiprotein complexes. The use of an innovative tool for accurate quantification of diffusion rates (19) allowed us to acquire additional data on syntaxin and tomosyn dynamics and characterize the membranal binding and unbinding kinetics. The binding rates that we found (membranal unbinding rate of 0.139/s) resemble those of other nonintegral membrane proteins such as Ras mutants (19) and may reflect an equilibrium between cytosolic and PM-associated pools of tomosyn. As for synaptic protein-binding rates, tomosyn's off-rate (membranal unbinding rate, 0.14/s) is slower than the rate of complexin's dissociation from the SNARE complex (0.3/s (46, 47)) and faster than Munc18's dissociation rate from syntaxin (0.0085/s; (48)), reflecting the very high affinity interaction of Munc18 with syntaxin. The binding rate of tomosyn that we found reflects a mixture of dissociation/association constants of the tomosyn-SNARE complexes and tomosyn-syntaxin binary complexes revealed in this study. As discussed below, we assume that the dissociation/association of tomosyn from the binary tomosyn-syntaxin complex is slower than the one from the ternary complex, according to the measurements with tomosyn Δ-loop mutants.
Based on the Sro7p structure (9), we created a model of tomosyn's structure and three deletion mutants lacking three unstructured regions named loops1, 2, and 3 (10). We measured the mobility of these mutants and found faster diffusion coefficients and lower Kon and Koff (i.e. slower membranal association and dissociation rates) for loop deletion mutants 1 (Δ537–578 aa) and 3 (Δ897–917 aa of m-tomosyn-1). Interestingly, these mutants also exhibited loss of function with respect to the inhibition of exocytosis (10). The faster diffusion rates suggest that the slower diffusion component of tomosyn as part of the ternary complex with syntaxin and SNAP-25 is reduced. The diffusion of proteins is anticorrelated with their overall mass (Stokes-Einstein formula, for review see Ref. 49). One possibility is that the mutants form less syntaxin-SNAP25-tomosyn complexes that exhibit a slower diffusion rate and more syntaxin-tomosyn complexes, and therefore their overall diffusion coefficient is faster.
To understand the dynamics of tomosyn on the plasma membrane and the characteristics of its interactions with the membrane, it is required to characterize its membranal distribution and specifically with respect to its main binding partner, syntaxin. Therefore, we next focused on characterizing the endogenous distribution of tomosyn only at the membrane by isolating native membrane sheets from PC12 cells. Super-resolution imaging studies already indicated that syntaxin distributes nonhomogeneously on the plasma membrane forming nano-sized clusters (11, 13). Similarly, using super-resolution imaging by dSTORM, we found that more than 50% of tomosyn molecules are distributed into nano-sized clusters as well, resembling in their size and shape to syntaxin clusters, but differ markedly from SNAP25 clusters (11). Tomosyn is not anchored independently to the PM, but similarly to Munc18 and bound to the membrane through syntaxin. Munc18 was also found to be organized in clusters (50–52). Our results suggest that tomosyn, similarly to other syntaxin-binding proteins, distributes into nano-sized clusters. Next, we measured the relative distribution of syntaxin and tomosyn by dual-color dSTORM experiments. We measured the extent of colocalization between syntaxin and tomosyn and found that although 66% of tomosyn is colocalized with syntaxin entities, only 18% of syntaxin is colocalized with tomosyn entities. Our results demonstrate that indeed syntaxin is responsible for most of tomosyn's recruitment to the PM. The remaining tomosyn that is not colocalized with syntaxin may be anchored to the PM through interactions with other proteins, which have yet to be discovered.
Next, we aimed to examine the actual interaction of tomosyn with its partners. Using FRET and overexpression of BoNT-C or BoNT-E, we learned that tomosyn interacts with SNAP25 in a ternary complex and with syntaxin in both binary and ternary complexes. It is interesting to compare our data with a recently published paper (50) that mapped the colocalization between syntaxin, SNAP25, and Munc18. Upon deletion of syntaxin, the colocalization between Munc18 and SNAP25 was lost, similar to our experiment with syntaxin cleavage of BoNT-C resulting in a decrease in FRET between tomosyn and SNAP25. These experiments display the central role for syntaxin in the protein interaction cascade occurring upon exocytosis and emphasize the regulatory role of tomosyn as well as of Munc18 in syntaxin's modulation. The FRET results were supported by expressing Munc18 serving as a binding competitor to tomosyn. Tomosyn is known to compete with Munc18 for syntaxin's binding, both in vitro (4) and in situ (26). The differential regulation by Munc18 strongly suggests that the tomosyn-syntaxin FRET signal does not report on the ternary complex, i.e. it likely reports on the binary tomosyn-syntaxin complex.
Advances in super-resolution microscopy and new analysis tools enable single molecule localization with a localization certainty of 10–20 nm. However, the available tools cannot explicitly indicate whether the existence of two molecules per pixel (e.g. 20 × 20 nm pixel size) means that these two molecules interact with each other. FRET methods can be used to investigate the interaction between two molecules in the 2–10-nm range. However, correlation of dSTORM and FRET data is not straightforward. The dSTORM data suggest that some tomosyn molecules are distributed in clusters and some are nonclustered, similar to syntaxin, and about 66% of these molecules coincide in the same pixel as syntaxin. FRET data suggest that about 15% of the FRET pixels have a 1:1 stoichiometry, meaning one tomosyn molecule interacting with one syntaxin molecule. In addition, 85% show a stoichiometry that differs from 1:1. A possible explanation is that the 1:1 pixels represent the interaction of the nonclustered tomosyn or syntaxin, and the other ratios might represent interaction at the edges of the clusters, where the density of one of the proteins is higher. This is a possible explanation; however, to answer this question one would have to combine super-resolution imaging methods with FRET. We assume such combination will be available in the coming years.
Several lines of evidence in this paper support the notion that tomosyn loop-deletion mutants 1 and 3 have a weaker affinity for SNAP25 than the wild type protein as follows: they bind less SNAP25 in GST pulldown; they show reduced PLA signals with SNAP25; and they diffuse faster on the PM. We inferred that these mutants preferably bind syntaxin and not syntaxin bound to SNAP25 (i.e. the acceptor complex). Hattendorf et al. (9) showed that Sro7p, tomosyn's homolog in yeast, has an intramolecular autoinhibited interaction with Sec9 (SNAP25 homolog) that can be “activated” to allow the assembly of active t-SNARE complexes. Tomosyn also possesses the hypothesized autoinhibitory domain, termed tail (9). In our structural model of tomosyn, loops 1 and 3 face the same direction as the tail. The sequence following the tail is the SNARE motif of tomosyn. Indeed, if allosteric autoinhibition exists using this tail, it is reasonable to assume that loops 1 and 3 can regulate the interactions between the tail, the SNARE motif, and perhaps additional regions in the β-propellers.
A model representing this suggested mechanism is presented in Fig. 5. According to the model, tomosyn can form a binary complex with syntaxin, with or without loops 1 and 3 whose function is unknown. Tomosyn can also form a ternary complex with syntaxin and SNAP25, for which the presence of the loops is required. This complex contributes to tomosyn's inhibitory effect on exocytosis. Without the loops, tomosyn's ability to form tomosyn-syntaxin-SNAP25 complexes decreases, and the inhibition of exocytosis is attenuated. It is unknown whether Munc18 can bind the tomosyn-syntaxin-SNAP25 complex according to the data collected so far.
FIGURE 5.
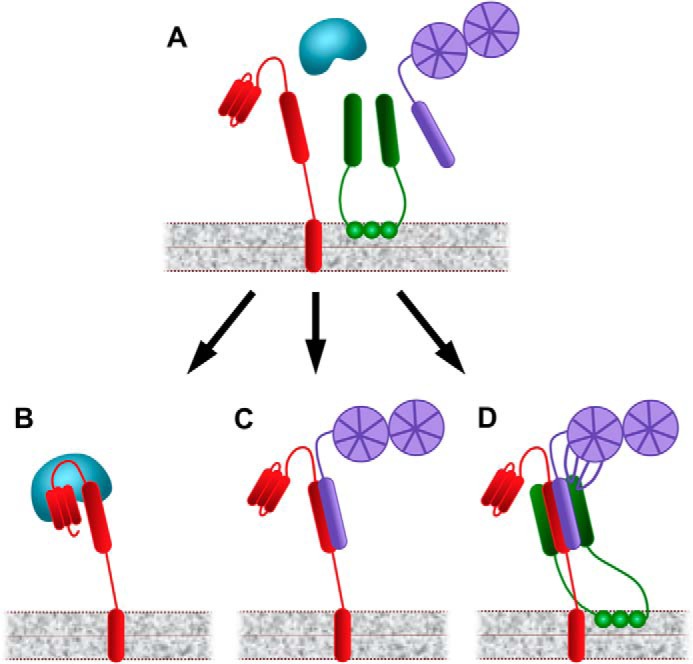
Model describing tomosyn's mode of inhibition. A, tomosyn (purple) is a cytosolic protein, targeted to the PM via syntaxin (red) and possibly other unknown proteins. At the membrane it interacts with syntaxin or with syntaxin and SNAP25 (green). B, syntaxin can also interact with Munc18 (blue) as a binary complex. C and D, tomosyn can interact with syntaxin as a dimer (for which it competes with Munc18) or with syntaxin and SNAP25 as a ternary complex. The tomosyn-syntaxin dimer does not require the tomosyn loops, and its function is unknown. The ternary complex is a nonfunctional SNARE complex, and tomosyn's inhibition of exocytosis occurs through this complex. The formation of this ternary complex requires tomosyn loops 1 and 3 but not 2. Loops 1 and 3 regulate tomosyn's participation in the ternary complex, possibly through interactions with tomosyn's tail or SNARE motif. Deleting loops 1 or 3 abolishes tomosyn's ability to inhibit exocytosis, and they are therefore important elements in tomosyn's function.
Our results demonstrate that tomosyn possesses regulatory elements upstream of the SNARE motif that affect its function and dynamics, which is in line with previous observations demonstrating the importance of tomosyn's intactness for its function. The information on the regulation of tomosyn's interaction with SNAP25, its dynamics, and distribution on the PM provides new directions and tools to further characterize its mode of action and role in the regulation of exocytosis.
Acknowledgments
We thank Prof. Reinhard Jahn for technical support in the membrane sheets experiments. We also thank Dr. Haguy Wolfenson for insightful input and assistance with the FRAP experiments and analysis.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1 NS053978 (to E. S. and U. A.). This work was also supported by Israel Science Foundation Grant 730/11 (to U. A.), United States-Israel Binational Science Foundation Grant 2009279 (to U. A.), and German-Israeli Foundation Grant 1125-145.1/2010 (to U. A. and M. S.).
- PM
- plasma membrane
- aa
- amino acid
- EGFP
- enhanced GFP
- FRAP
- fluorescence recovery after photobleaching
- CFP
- cyan fluorescent protein
- BoNT-C
- botulinum neurotoxin C
- PLA
- proximity ligation assay.
REFERENCES
- 1. Chen Y. A., Scales S. J., Scheller R. H. (2001) Sequential SNARE assembly underlies priming and triggering of exocytosis. Neuron 30, 161–170 [DOI] [PubMed] [Google Scholar]
- 2. Söllner T., Whiteheart S. W., Brunner M., Erdjument-Bromage H., Geromanos S., Tempst P., Rothman J. E. (1993) SNAP receptors implicated in vesicle targeting and fusion. Nature 362, 318–324 [DOI] [PubMed] [Google Scholar]
- 3. Chen Y. A., Scheller R. H. (2001) SNARE-mediated membrane fusion. Nat. Rev. 2, 98–106 [DOI] [PubMed] [Google Scholar]
- 4. Fujita Y., Shirataki H., Sakisaka T., Asakura T., Ohya T., Kotani H., Yokoyama S., Nishioka H., Matsuura Y., Mizoguchi A., Scheller R. H., Takai Y. (1998) Tomosyn: a syntaxin-1-binding protein that forms a novel complex in the neurotransmitter release process. Neuron 20, 905–915 [DOI] [PubMed] [Google Scholar]
- 5. Yizhar O., Matti U., Melamed R., Hagalili Y., Bruns D., Rettig J., Ashery U. (2004) Tomosyn inhibits priming of large dense-core vesicles in a calcium-dependent manner. Proc. Natl. Acad. Sci. U.S.A. 101, 2578–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hatsuzawa K., Lang T., Fasshauer D., Bruns D., Jahn R. (2003) The R-SNARE motif of tomosyn forms SNARE core complexes with syntaxin 1 and SNAP-25 and down-regulates exocytosis. J. Biol. Chem. 278, 31159–31166 [DOI] [PubMed] [Google Scholar]
- 7. Sakisaka T., Yamamoto Y., Mochida S., Nakamura M., Nishikawa K., Ishizaki H., Okamoto-Tanaka M., Miyoshi J., Fujiyoshi Y., Manabe T., Takai Y. (2008) Dual inhibition of SNARE complex formation by tomosyn ensures controlled neurotransmitter release. J. Cell Biol. 183, 323–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yizhar O., Lipstein N., Gladycheva S. E., Matti U., Ernst S. A., Rettig J., Stuenkel E. L., Ashery U. (2007) Multiple functional domains are involved in tomosyn regulation of exocytosis. J. Neurochem. 103, 604–616 [DOI] [PubMed] [Google Scholar]
- 9. Hattendorf D. A., Andreeva A., Gangar A., Brennwald P. J., Weis W. I. (2007) Structure of the yeast polarity protein Sro7 reveals a SNARE regulatory mechanism. Nature 446, 567–571 [DOI] [PubMed] [Google Scholar]
- 10. Williams A. L., Bielopolski N., Meroz D., Lam A. D., Passmore D. R., Ben-Tal N., Ernst S. A., Ashery U., Stuenkel E. L. (2011) Structural and functional analysis of tomosyn identifies domains important in exocytotic regulation. J. Biol. Chem. 286, 14542–14553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bar-On D., Wolter S., van de Linde S., Heilemann M., Nudelman G., Nachliel E., Gutman M., Sauer M., Ashery U. (2012) Super-resolution imaging reveals the internal architecture of nano-sized syntaxin clusters. J. Biol. Chem. 287, 27158–27167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Halemani N. D., Bethani I., Rizzoli S. O., Lang T. (2010) Structure and dynamics of a two-helix SNARE complex in live cells. Traffic 11, 394–404 [DOI] [PubMed] [Google Scholar]
- 13. Sieber J. J., Willig K. I., Kutzner C., Gerding-Reimers C., Harke B., Donnert G., Rammner B., Eggeling C., Hell S. W., Grubmüller H., Lang T. (2007) Anatomy and dynamics of a supramolecular membrane protein cluster. Science 317, 1072–1076 [DOI] [PubMed] [Google Scholar]
- 14. Bar-On D., Gutman M., Mezer A., Ashery U., Lang T., Nachliel E. (2009) Evaluation of the heterogeneous reactivity of the syntaxin molecules on the inner leaflet of the plasma membrane. J. Neurosci. 29, 12292–12301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ma C., Su L., Seven A. B., Xu Y., Rizo J. (2013) Reconstitution of the vital functions of Munc18 and Munc13 in neurotransmitter release. Science 339, 421–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ribrault C., Reingruber J., Petković M., Galli T., Ziv N. E., Holcman D., Triller A. (2011) Syntaxin1A lateral diffusion reveals transient and local SNARE interactions. J. Neurosci. 31, 17590–17602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Knowles M. K., Barg S., Wan L., Midorikawa M., Chen X., Almers W. (2010) Single secretory granules of live cells recruit syntaxin-1 and synaptosomal associated protein 25 (SNAP-25) in large copy numbers. Proc. Natl. Acad. Sci. U.S.A. 107, 20810–20815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bar-On D., Winter U., Nachliel E., Gutman M., Fasshauer D., Lang T., Ashery U. (2008) Imaging the assembly and disassembly kinetics of cis-SNARE complexes on native plasma membranes. FEBS Lett. 582, 3563–3568 [DOI] [PubMed] [Google Scholar]
- 19. Berkovich R., Wolfenson H., Eisenberg S., Ehrlich M., Weiss M., Klafter J., Henis Y. I., Urbakh M. (2011) Accurate quantification of diffusion and binding kinetics of nonintegral membrane proteins by FRAP. Traffic 12, 1648–1657 [DOI] [PubMed] [Google Scholar]
- 20. Hoppe A., Christensen K., Swanson J. A. (2002) Fluorescence resonance energy transfer-based stoichiometry in living cells. Biophys. J. 83, 3652–3664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu J., Ernst S. A., Gladycheva S. E., Lee Y. Y., Lentz S. I., Ho C. S., Li Q., Stuenkel E. L. (2004) Fluorescence resonance energy transfer reports properties of syntaxin1a interaction with Munc18–1 in vivo. J. Biol. Chem. 279, 55924–55936 [DOI] [PubMed] [Google Scholar]
- 22. Löschberger A., van de Linde S., Dabauvalle M. C., Rieger B., Heilemann M., Krohne G., Sauer M. (2012) Super-resolution imaging visualizes the eightfold symmetry of gp210 proteins around the nuclear pore complex and resolves the central channel with nanometer resolution. J. Cell Sci. 125, 570–575 [DOI] [PubMed] [Google Scholar]
- 23. Wolter S., Löschberger A., Holm T., Aufmkolk S., Dabauvalle M. C., van de Linde S., Sauer M. (2012) rapidSTORM: accurate, fast open-source software for localization microscopy. Nat. Methods 9, 1040–1041 [DOI] [PubMed] [Google Scholar]
- 24. van de Linde S., Löschberger A., Klein T., Heidbreder M., Wolter S., Heilemann M., Sauer M. (2011) Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nat. Protoc. 6, 991–1009 [DOI] [PubMed] [Google Scholar]
- 25. Bolte S., Cordelières F. P. (2006) A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232 [DOI] [PubMed] [Google Scholar]
- 26. Gladycheva S. E., Lam A. D., Liu J., D'Andrea-Merrins M., Yizhar O., Lentz S. I., Ashery U., Ernst S. A., Stuenkel E. L. (2007) Receptor-mediated regulation of tomosyn-syntaxin 1A interactions in bovine adrenal chromaffin cells. J. Biol. Chem. 282, 22887–22899 [DOI] [PubMed] [Google Scholar]
- 27. Zilly F. E., Halemani N. D., Walrafen D., Spitta L., Schreiber A., Jahn R., Lang T. (2011) Ca2+ induces clustering of membrane proteins in the plasma membrane via electrostatic interactions. EMBO J. 30, 1209–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Heilemann M., van de Linde S., Schüttpelz M., Kasper R., Seefeldt B., Mukherjee A., Tinnefeld P., Sauer M. (2008) Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew. Chem. Int. Ed. Engl. 47, 6172–6176 [DOI] [PubMed] [Google Scholar]
- 29. Lehman K., Rossi G., Adamo J. E., Brennwald P. (1999) Yeast homologues of tomosyn and lethal giant larvae function in exocytosis and are associated with the plasma membrane SNARE, Sec9. J. Cell Biol. 146, 125–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Widberg C. H., Bryant N. J., Girotti M., Rea S., James D. E. (2003) Tomosyn interacts with the t-SNAREs syntaxin4 and SNAP23 and plays a role in insulin-stimulated GLUT4 translocation. J. Biol. Chem. 278, 35093–35101 [DOI] [PubMed] [Google Scholar]
- 31. Schiavo G., Shone C. C., Bennett M. K., Scheller R. H., Montecucco C. (1995) botulinum neurotoxin type C cleaves a single Lys-Ala bond within the carboxyl-terminal region of syntaxins. J. Biol. Chem. 270, 10566–10570 [DOI] [PubMed] [Google Scholar]
- 32. Lam A. D., Tryoen-Toth P., Tsai B., Vitale N., Stuenkel E. L. (2008) SNARE-catalyzed fusion events are regulated by Syntaxin1A-lipid interactions. Mol. Biol. Cell 19, 485–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Foran P., Lawrence G. W., Shone C. C., Foster K. A., Dolly J. O. (1996) Botulinum neurotoxin C1 cleaves both syntaxin and SNAP-25 in intact and permeabilized chromaffin cells: correlation with its blockade of catecholamine release. Biochemistry 35, 2630–2636 [DOI] [PubMed] [Google Scholar]
- 34. Vaidyanathan V. V., Yoshino K., Jahnz M., Dörries C., Bade S., Nauenburg S., Niemann H., Binz T. (1999) Proteolysis of SNAP-25 isoforms by botulinum neurotoxin types A, C, and E: domains and amino acid residues controlling the formation of enzyme-substrate complexes and cleavage. J. Neurochem. 72, 327–337 [DOI] [PubMed] [Google Scholar]
- 35. Banerjee A., Kowalchyk J. A., DasGupta B. R., Martin T. F. (1996) SNAP-25 is required for a late postdocking step in Ca2+-dependent exocytosis. J. Biol. Chem. 271, 20227–20230 [DOI] [PubMed] [Google Scholar]
- 36. Chen Y. A., Scales S. J., Patel S. M., Doung Y. C., Scheller R. H. (1999) SNARE complex formation is triggered by Ca2+ and drives membrane fusion. Cell 97, 165–174 [DOI] [PubMed] [Google Scholar]
- 37. Hayashi T., McMahon H., Yamasaki S., Binz T., Hata Y., Südhof T. C., Niemann H. (1994) Synaptic vesicle membrane fusion complex: action of clostridial neurotoxins on assembly. EMBO J. 13, 5051–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aikawa Y., Lynch K. L., Boswell K. L., Martin T. F. (2006) A second SNARE role for exocytic SNAP25 in endosome fusion. Mol. Biol. Cell 17, 2113–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Burkhardt P., Hattendorf D. A., Weis W. I., Fasshauer D. (2008) Munc18a controls SNARE assembly through its interaction with the syntaxin N-peptide. EMBO J. 27, 923–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gladycheva S. E., Ho C. S., Lee Y. Y., Stuenkel E. L. (2004) Regulation of syntaxin1A-munc18 complex for SNARE pairing in HEK293 cells. J. Physiol. 558, 857–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Masuda E. S., Huang B. C., Fisher J. M., Luo Y., Scheller R. H. (1998) Tomosyn binds t-SNARE proteins via a VAMP-like coiled coil. Neuron 21, 479–480 [DOI] [PubMed] [Google Scholar]
- 42. Burdina A. O., Klosterman S. M., Shtessel L., Ahmed S., Richmond J. E. (2011) In vivo analysis of conserved C. elegans tomosyn domains. PLoS One 6, e26185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yamamoto Y., Mochida S., Miyazaki N., Kawai K., Fujikura K., Kurooka T., Iwasaki K., Sakisaka T. (2010) Tomosyn inhibits synaptotagmin-1-mediated step of Ca2+-dependent neurotransmitter release through its N-terminal WD40 repeats. J. Biol. Chem. 285, 40943–40955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Neer E. J., Smith T. F. (2000) A groovy new structure. Proc. Natl. Acad. Sci. U.S.A. 97, 960–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xia Z. X., Dai W. W., Xiong J. P., Hao Z. P., Davidson V. L., White S., Mathews F. S. (1992) The three-dimensional structures of methanol dehydrogenase from two methylotrophic bacteria at 2.6-A resolution. J. Biol. Chem. 267, 22289–22297 [PubMed] [Google Scholar]
- 46. Li Y., Augustine G. J., Weninger K. (2007) Kinetics of complexin binding to the SNARE complex: correcting single molecule FRET measurements for hidden events. Biophys. J. 93, 2178–2187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pabst S., Margittai M., Vainius D., Langen R., Jahn R., Fasshauer D. (2002) Rapid and selective binding to the synaptic SNARE complex suggests a modulatory role of complexins in neuroexocytosis. J. Biol. Chem. 277, 7838–7848 [DOI] [PubMed] [Google Scholar]
- 48. Ciufo L. F., Barclay J. W., Burgoyne R. D., Morgan A. (2005) Munc18-1 regulates early and late stages of exocytosis via syntaxin-independent protein interactions. Mol. Biol. Cell 16, 470–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lippincott-Schwartz J., Snapp E., Kenworthy A. (2001) Studying protein dynamics in living cells. Nat. Rev. 2, 444–456 [DOI] [PubMed] [Google Scholar]
- 50. Pertsinidis A., Mukherjee K., Sharma M., Pang Z. P., Park S. R., Zhang Y., Brunger A. T., Südhof T. C., Chu S. (2013) Ultrahigh-resolution imaging reveals formation of neuronal SNARE/Munc18 complexes in situ. Proc. Natl. Acad. Sci. U.S.A. 110, E2812–2820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zilly F. E., Sørensen J. B., Jahn R., Lang T. (2006) Munc18-bound syntaxin readily forms SNARE complexes with synaptobrevin in native plasma membranes. PLoS Biol. 4, e330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Smyth A. M., Yang L., Martin K. J., Hamilton C., Lu W., Cousin M. A., Rickman C., Duncan R. R. (2013) Munc18-1 protein molecules move between membrane molecular depots distinct from vesicle docking sites. J. Biol. Chem. 288, 5102–5113 [DOI] [PMC free article] [PubMed] [Google Scholar]