

Background: Leucine-rich repeat-containing 8 (LRRC8) proteins are involved in lymphocyte development and adipocyte differentiation.
Results: LRRC8D is required for the import of the antibiotic blasticidin S.
Conclusion: LRRC8D functions as a transporter of small molecules.
Significance: This suggests a mode of action for LRRC8 family members in lymphocyte development and adipocyte differentiation.
Keywords: Antibiotics, Drug Transport, Gene Knockout, Mass Spectrometry (MS), Membrane Protein, Protein-Protein Interaction, Somatic Cell Genetics, Blasticidin S, LRRC8D, Metabolomics
Abstract
Leucine-rich repeat-containing 8 (LRRC8) proteins have been identified as putative receptors involved in lymphocyte development and adipocyte differentiation. They remain poorly characterized, and no specific function has been assigned to them. There is no consensus on how this family of proteins might function because homology searches suggest that members of the LRRC8 family act not as plasma membrane receptors, but rather as channels that mediate cell-cell signaling. Here we provide experimental evidence that supports a role for LRRC8s in the transport of small molecules. We show that LRRC8D is a mammalian protein required for the import of the antibiotic blasticidin S. We characterize localization and topology of LRRC8A and LRRC8D and demonstrate that LRRC8D interacts with LRRC8A, LRRC8B, and LRRC8C. Given the suggested involvement in solute transport, our results support a model in which LRRC8s form one or more complexes that may mediate cell-cell communication by transporting small solutes.
Introduction
The leucine-rich repeat-containing 8 (LRRC8)2 family of proteins comprises five members, named LRRC8A, LRRC8B, LRRC8C, LRRC8D, and LRRC8E. Each member comprises four predicted transmembrane domains at the N terminus and up to 17 leucine-rich repeats (LRRs) at the C terminus (1, 2). Whereas LRRC8A and LRRC8C have been implicated in lymphocyte development and adipocyte differentiation, respectively, their mechanism of function remains unknown, and the other members of the family remain poorly characterized (3–5).
The founding member of the family, LRRC8A, was identified when white blood cells from a patient suffering from congenital agammaglobulinemia showed a balanced chromosomal translocation t(9;20)(q33.2;q12) that resulted in the expression of both a normal copy of LRRC8A and a truncated form of LRRC8A that lacks the last two and a half LRR domains (3). Forced expression of the truncated version of LRRC8A in murine bone marrow cells inhibited B cell development and confirmed that the patient's B cell deficiency was due to the mutation in LRRC8A. Given the dominant nature of the mutation and the presence of LRRs in the extracellular binding domain of diverse receptors (Toll-like receptors, follicle-stimulating hormone receptor, high affinity nerve growth factor receptor), LRRC8A was proposed to function as a cell surface receptor that oligomerizes upon binding an unknown ligand important for B cell development (6).
The only other functional studies on LRRC8s concern adipocyte development. LRRC8C, also known as FAD158, was identified as a gene whose expression is induced in the early stage of adipocyte differentiation (4). Knockdown and overexpression studies supported a role for LRRC8C in adipocyte differentiation. When LRRC8C−/− and wild-type mice were fed a high fat diet, LRRC8C−/− mice had significantly lower body weights and fat mass (5). How exactly LRRC8C is involved in adipocyte differentiation and the development of obesity in response to a high fat diet remains unclear.
A recent hypothesis paper, based exclusively on a bioinformatics approach, proposed that LRRC8s evolved from the combination of a pannexin and an LRR domain prior to the diversification of chordates (2). Sequence similarity and inferred homology between LRRC8s and pannexins would place the LRR domains of LRRC8s facing the cytoplasm rather than the outside of the cell, in contrast to the report by Sawada et al. (3).
Pannexins are a family of communication channels composed of four α-helical transmembrane domains. They are thought to function primarily in transporting intracellular molecules to the extracellular space (7). Whereas pannexins could participate in many biological processes, evidence is strongest for a role of pannexin 1 (Panx1) in the release of “find-me” signals, specifically ATP and UTP, by apoptotic cells to recruit phagocytes (7–9). Whereas LRRC8s, like pannexins, might form channels and participate in cell-cell communication, there has been no experimental evidence to support such a role.
Here we show that LRRC8D is required for the import of the antibiotic blasticidin S (BlaS) by mammalian cells and is the first protein implicated in this process, supporting a role for LRRC8s in solute transport. We further provide evidence that the LRR domains of LRRC8A and LRRC8D face the cytoplasm rather than extracellular space and that LRRC8D interacts with LRRC8A, LRRC8B, and LRRC8C.
EXPERIMENTAL PROCEDURES
Constructs
cDNA was obtained from either RAW 264.7 macrophages or KBM7 cells for use as a cloning template. Murine LRRC8A-myc, LRRC8A-HA, LRRC8B-V5, LRRC8C-FLAG, and LRRC8D-HA were cloned into LRCX vectors (Clontech). Human LRRC8A-myc, LRRC8B-V5, LRRC8C-FLAG and LRRC8D-HA, and LRRC8E-His6-T7 were cloned into pCDH vectors (System Biosciences). Human KEL-myc-LPETG-HA and human myc-GYPA-LPETG-HA were cloned into pCDH vectors. The HA-SEL1L and AUP1-GFP constructs were described previously in Ref. 10.
Isolation and Characterization of LRRC8D Mutant Clones
The isolation of mutant clones has been described previously (11). In brief, a subset of the survivors from the screen was FACS sorted into 96-well plates such that no more than a single cell was plated per well (12). Clones were expanded and then assayed for a gene trap insertion in the LRRC8D locus by PCR using primers 5′-TCTCCAAATCTCGGTGGAAC-3′ and 5′-CCAGACTAAACATCTCAGAACTCG-3′. To confirm that the cells were truly clonal and to confirm the absence of the wild-type DNA locus, a PCR was performed with primers 5′-GGATCTCTCTAGCTCTTTCTCTCC-3′ and 5′-CCAGACTAAACATCTCAGAACTCG-3′. The absence of the LRRC8D transcript in the isolated mutant clones was determined by RT-PCR. Total RNA was isolated using the RNeasy Mini kit (Qiagen) and reverse-transcribed using the SuperScriptTM III First-strand synthesis system (Invitrogen). Amplification of LRRC8D was performed using primers 5′-CGCCGTGGTTCCAGCCTCC-3′ and 5′-CGCATGCTGTCTCAGACAACGC-3′, and amplification of GAPDH was performed using primers 5′-GCCTCCTGCACCACCAACTGC-3′ and 5′-CCACTGACACGTTGGCAGTGGG-3′.
Antibodies
Anti-HA affinity matrix (Roche Applied Science 11815016001), anti-HA HRP (Roche Applied Science 12013819001), Alexa Fluor 488-conjugated anti-HA antibody (Invitrogen A-21287), anti-FLAG M2 affinity gel (Sigma A2220), anti-FLAG HRP (Sigma A8592), anti-myc-Sepharose beads (Cell Signaling 3400), anti-V5 antibody (Invitrogen R96025), anti-V5 HRP (Invitrogen R96125), anti-actin (BD Biosciences 612656), HRP-conjugated sheep anti-mouse IgG (GE Healthcare NXA931), anti-calnexin (Santa Cruz Biotechnology sc11397), goat anti-Rb Alexa Fluor 568 (Molecular Probes A11011), and Alexa Fluor 647 phalloidin (Invitrogen A22287) were used.
Cell Viability Assay
CellTiter-Glo Luminescent Cell Viability Assay (Promega) was used to quantify cell viability. 200,000 cells were seeded per well in clear bottom 96-well plates (BD Biosciences or Costar).
Transfection and Viral Transduction
Transfections were accomplished using Trans-IT 293 (Mirus) for 293T cells. Transductions were accomplished using the pPACKH1 HIV Lentivector Packaging kit (System Biosciences) according to its protocol.
Preparation of Cell Extracts for LC/MS Analysis
Four million wild-type reporter cells, LRRC8DGT1 cells, and LRRC8DGT2 cells were treated with varying concentrations of BlaS for 3 h in triplicate. Cells were washed twice in ice-cold Iscove's modified Dulbecco's medium and one time in ice-cold, filtered 0.9% NaCl. 200 μl of ice-cold extraction buffer (40% acetonitrile, 40% methanol, 20% water, 10 ng/ml phenylalanine-d8, 10 ng/ml valine-d8) was added to each cell pellet. Extracts were vortexed for 30 s and then spun at 14,000 rpm for 10 min at 4 °C in a table-top centrifuge. Extract supernatants were transferred to new Eppendorf tubes and stored at −80 °C until analysis. Extract solvents were obtained from Fisher Scientific and were Optima LC/MS grade; phenylalanine-d8 and valine-d8 were purchased from Cambridge Isotope Laboratories.
LC Analysis
Solvents were obtained from Fisher Scientific and were Optima LC/MS grade except where otherwise specified. An UltiMate 3000 UPLC system with autosampler (Dionex) was used for this study. Biological triplicate samples (typically 10 μl) were injected onto a Luna-NH2 2 × 150-mm (3-μm particle size) column (Phenomenex) equipped with an inline particulate filter. The mobile phases were 5 mm ammonium acetate, 0.2% ammonium hydroxide, pH 9.9 (mobile phase A) and acetonitrile (mobile phase B). The flow rate was 0.25 ml/min, and the column temperature was held at 15 °C. The column was eluted with a gradient from 90% to 10% mobile phase B over 20 min, held at 10% mobile phase B for 5 min, returned to 90% mobile phase B over 1 min, and re-equilibrated at 90% mobile phase B for 8 min.
MS Analysis
The UPLC system was coupled to a QExactive orbitrap mass spectrometer equipped with a HESI II probe (Thermo Fisher Scientific) operating in positive ion mode. The spray voltage was set to 3.9 kV, the heated capillary was held at 335 °C, and the HESI probe was held at 350 °C. The sheath gas flow was set to 35 units, the auxiliary gas flow was set to 7 units, and the sweep gas flow was set to 0 unit. External mass calibration was performed every 7 days. The MS data acquisition was performed by targeted Selected Ion Monitoring of the metabolites of interest (BlaS and phenylalanine-d8 as an internal standard), with the resolution set at 70,000, the AGC target at 5 × 104, the maximum injection time at 120 ms, and the isolation window at 1.0 m/z. Quantitation of the data was performed with XCalibur QuanBrowser 2.2 (Thermo Fisher Scientific) using a 5-ppm mass tolerance. Pure BlaS samples in extraction buffer (half-log serial dilution, 1 nm–1 μm) were analyzed to confirm chromatographic retention times and generate standard curves for quantitation of each analytical batch. The limit of detection in sample matrix was 5 nm, as determined by addition of increasing concentrations of BlaS to a pooled BlaS-free cell extract.
Immunofluorescence
Cells were washed with PBS, fixed with 4% paraformaldehyde in PBS, washed with PBS, incubated with a quenching solution containing 20 mm glycine and 50 mm NH4Cl in PBS, washed with PBS, permeabilized with saponin binding buffer (PBS containing 0.1% (v/v) saponin and 0.2% (w/v) BSA), incubated with primary antibodies for 1 h, washed with saponin binding buffer, incubated with secondary antibodies for 1 h, washed with saponin binding buffer, PBS, and then water before mounting on glass slides using ProLong Gold with DAPI (Invitrogen). Images were acquired with a PerkinElmer Life Sciences Ultraview Spinning Disk Confocal equipped with a Hammamatsu ORCA-ER CCD camera and analyzed with Volocity acquisition software.
Coimmunoprecipitation
Cells were washed in ice-cold PBS before lysing with Nonidet P-40 lysis buffer containing 50 mm Tris, pH 7.4, 150 mm NaCl, 0.5 mm EDTA, 1% (v/v) Nonidet P-40, and protease inhibitors (Roche Applied Science). Lysate concentrations were measured by Bradford Protein Assay (Bio-Rad) and normalized. Lysates were precleared by incubating them with protein G-agarose beads (Sigma P3296) for 1 h at 4 °C. Precleared lysates were then incubated with either anti-HA affinity matrix, anti-V5 antibody with protein G-agarose beads, anti-FLAG M2 affinity gel, or anti-myc-Sepharose beads for 2 h at 4 °C. Beads were washed three times with buffer containing 50 mm Tris, pH 7.4, 150 mm NaCl, 0.5 mm EDTA, and 0.1% (v/v) Nonidet P-40. Beads were then boiled in sample buffer, and proteins in the supernatant were resolved by SDS-PAGE.
Affinity Purification and Mass Spectrometry
After coimmunoprecipitation, eluted samples were separated by SDS-PAGE, and polypeptides were visualized by silver staining. Bands of interest were excised, digested with trypsin, and analyzed by tandem mass spectrometry.
Immunoblot Analysis
Proteins were separated by SDS-PAGE, transferred to nitrocellulose membranes, blocked with 5% (w/v) skim milk in PBS with 0.1% (v/v) Tween 20 and were probed with the appropriate primary antibodies. Membranes were washed with PBS with 0.1% (v/v) Tween 20 and were incubated with horseradish peroxidase-conjugated secondary antibodies if necessary. Proteins were visualized with an enhanced chemiluminescence detection reagent. Restore PLUS Western blot Stripping Buffer (Thermo) was used to strip the membranes between probing.
Silver Staining
Proteins were fixed in polyacrylamide gel by incubating the gel in 40% (v/v) ethanol, 10% (v/v) acetic acid. The gel was washed twice in 30% (v/v) ethanol and once in water. The gel was then sensitized in 0.02% (w/v) Na2S2O3, washed three times in water, incubated in ice-cold 0.1% (w/v) AgNO3, washed four more times in water, and developed in 3% (w/v) Na2CO3, 0.05% (v/v) formaldehyde. When protein bands became sufficiently visible, the gel was washed once in water, and the staining process was terminated by incubating the gel in 5% (v/v) acetic acid. The gel was then washed a final three times in water.
FACS Analysis of Protein Topology
Cells were washed in PBS and then resuspended in staining solution containing 1 μg/ml antibody and 3% inactivated fetal calf serum in PBS (PBS/IFS) and incubated in the dark at 4 °C for 30 min. Cells were washed in PBS/IFS and then fixed with PBS containing 4% (v/v) formaldehyde for 30 min at room temperature in the dark. The protocol for permeabilized cells was similar except cells were first fixed, washed with PBS, permeabilized with 0.5% (w/v) saponin in PBS/IFS, stained with 1 μg/ml antibody in 0.5% (w/v) saponin in PBS/IFS, and then washed with PBS/IFS.
Protease Digestion of Epitope-tagged Proteins in Microsomes
293T cells expressing epitope-tagged proteins were washed in ice-cold PBS and then incubated for 10 min in hypotonic buffer containing: 20 mm Hepes-KOH, pH 7.5, 5 mm KCl, 1.5 mm MgCl2, 1 mm DTT. Cells were then broken open with a Dounce homogenizer. NaCl was added to the lysates for a final concentration of 150 mm NaCl. Lysates were spun at 800 × g at 4 °C for 10 min. The supernatant was transferred to a new tube that was then spun at 10,000 × g at 4 °C for 10 min. Pellets were resuspended in buffer containing 50 mm Hepes-KOH, pH 7.5, 50 mm KOAc, 2 mm Mg(OAc)2, and 250 mm sucrose, and this microsome-containing mixture was equally aliquoted to tubes with or without Triton X-100 (final 1% v/v). Proteinase K (New England Biolabs P8102S) was then added to the samples at varying concentrations, and the samples were incubated on ice for 30 min. PMSF was added to deactivate proteinase K. Sample buffer was added, and the samples were boiled. Proteins were resolved by SDS-PAGE followed by immunoblot analysis.
RESULTS
LRRC8D-deficient Cells Are Resistant to BlaS
We identified LRRC8D and LRRC8A as hits in a haploid reporter genetic screen aimed at discovering inhibitors of NF-κB (12). We performed the screen with cells carrying a NF-κB transcriptional reporter that drives expression of the blasticidin S-resistance gene (BSR), so that cells in which the NF-κB pathway is activated would survive in the presence of BlaS (Fig. 1A).
FIGURE 1.
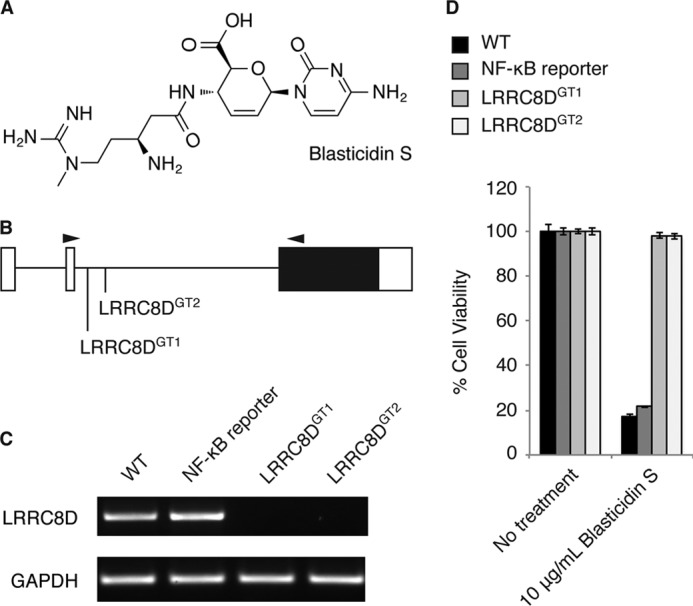
LRRC8D-deficient cells are resistant to blasticidin S. A, structure of BlaS. B, genomic locus of LRRC8D and the location of two gene trap insertion sites. White boxes denote the 5′- and 3′-untranslated regions, and the black box denotes the coding sequence, all of which is within exon 3. Arrowheads indicate the binding sites of primers used for RT-PCR analysis. C, RT-PCR analysis of WT KBM7 cells, WT NF-κB-BSR reporter cells, and two clonally derived cell lines containing gene trap insertions in LRRC8D as indicated in B. D, WT KBM7 cells, WT NF-κB reporter cells, LRRC8DGT1 cells, and LRRC8DGT2 cells treated with varying concentrations of BlaS. Cell viability was determined after 24 h of treatment using the CellTiter Glo assay, and results are plotted as percentage viability of treated cells compared with untreated cells. Results are mean ± S.E. (error bars) of triplicates and are representative of three independent experiments.
After insertional mutagenesis, reporter cells that survived in the presence of BlaS were selected, with the expectation that they would contain mutations in NF-κB inhibitors and thus show constitutive expression of the NF-κB-BSR reporter. Indeed, one of the significant hits in the screen, CYLD, is a known negative regulator of NF-κB. We confirmed that CYLD is required at steady state to prevent aberrant activation of NF-κB (12). To further characterize LRRC8D, we isolated two clonal cell lines from the screen, LRRC8DGT1 and LRRC8DGT2, that have gene trap insertion sites located in the intron between exon 2 and exon 3 (Fig. 1B). Neither LRRC8DGT1 nor LRRC8DGT2 cell line expresses full-length LRRC8D mRNA (Fig. 1C). Consistent with the design of the gene trap vector, this suggests that in LRRC8DGT1 and LRRC8DGT2 cells, exon 2 is not splicing to exon 3, but rather to the splice acceptor site in the gene trap, which terminates the transcript. Thus, none of the protein coding sequence for LRRC8D, fully contained in exon 3, is expressed in LRRC8DGT1 and LRRC8DGT2 cell lines.
We determined that both LRRC8DGT1 and LRRC8DGT2 cell lines are indeed resistant to BlaS (Fig. 1D). We hypothesized that LRRC8D deficiency results in constitutive activation of the NF-κB-BSR reporter and therefore confers BlaS resistance. To our surprise we were unable to demonstrate a defect in NF-κB regulation in LRRC8DGT1 and LRRC8DGT2 cells (data not shown). Previous haploid genetic screens with tunicamycin and 3-bromopyruvate as the selecting agents identified transporters for these small molecules (13, 14). We then considered the possibility that resistance of LRRC8DGT1 and LRRC8DGT2 cells to BlaS was attributable to defective import of the antibiotic rather than altered NF-κB signaling.
LRRC8D Mediates Transport of BlaS
How BlaS enters mammalian cells is not known, but given its polar nature (Fig. 1A), it is unlikely to cross the plasma membrane unaided. There is no easy path to the synthesis of radiolabeled BlaS that could then be used for cellular uptake experiments. We generated a biotinylated version of BlaS for that purpose, but found that it lacked biological activity when applied to intact cells and therefore could not be used as a BlaS analog to measure transport. We resorted to a LC/MS-based assay to directly measure BlaS levels in extracts from BlaS-exposed cells. To determine whether LRRC8D is required for BlaS import, wild-type cells, LRRC8DGT1 cells, and LRRC8DGT2 cells were incubated with varying concentrations of BlaS for 3 h. BlaS levels in extracts made from BlaS-treated cells were measured by targeted mass spectrometry. Whereas BlaS was easily detected in wild-type cells, it was either absent from, or present in only trace amounts in LRRC8DGT1 and LRRC8DGT2 cells, thus demonstrating that LRRC8D deficiency results in defective BlaS import (Fig. 2).
FIGURE 2.
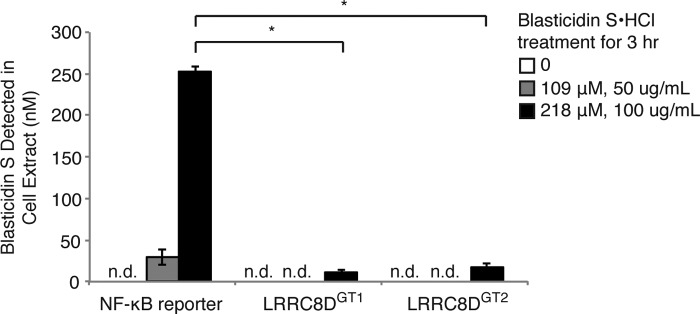
BlaS internalization is defective in LRRC8D-deficient cells. WT reporter cells, LRRC8DGT1 cells, and LRRC8DGT2 cells were treated with varying concentrations of BlaS for 3 h. Extracts of polar metabolites from the cells were analyzed for the presence of BlaS using LC/MS. Results are mean ± S.D. (error bars) of triplicates and are representative of three independent experiments. n.d., not detected. Statistical analysis was performed using the Welch test followed by a Bonferroni correction (*, p < 0.0001).
Localization of LRRC8A and LRRC8D
We next sought to determine where LRRC8A and LRRC8D are located in the cell. If they are transporters for BlaS and other molecules, one would expect to see them most likely at the plasma membrane. In HeLa cells stably expressing LRRC8A equipped with a C-terminal HA tag, LRRC8A-HA was present at the plasma membrane (Fig. 3). In HeLa cells that stably express LRRC8D-HA, LRRC8D-HA appeared less prominently at the plasma membrane whereas the majority colocalized with calnexin in the ER. Surface display of LRRC8D may require the involvement of a partner protein present in limiting amounts. Overexpression of LRRC8D on its own in the absence of such cofactors would then prevent a major portion of LRRC8D from reaching the plasma membrane.
FIGURE 3.
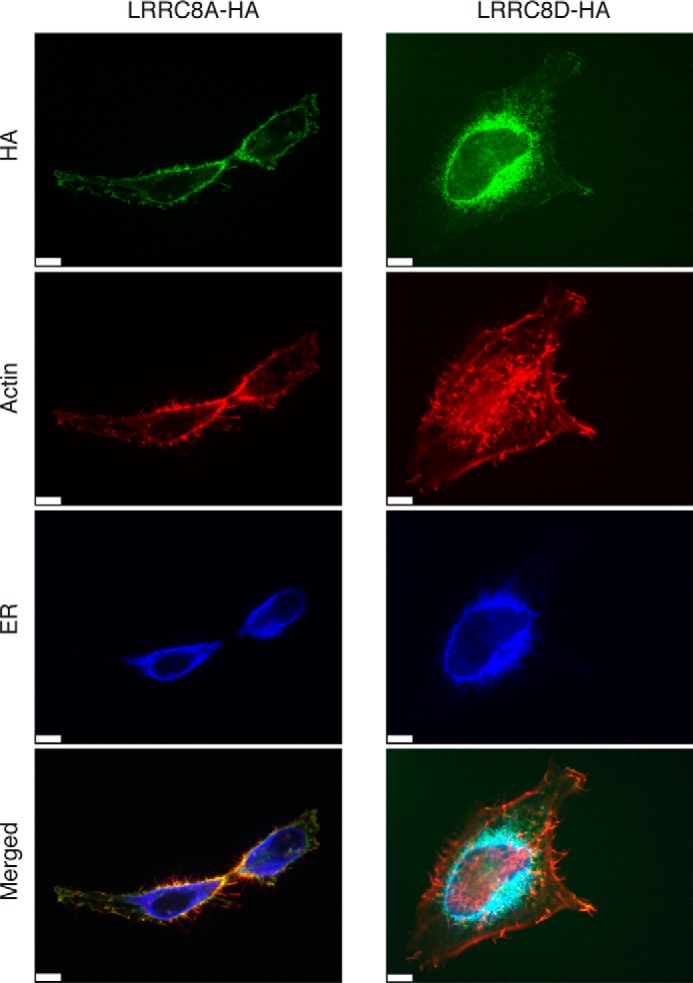
Localization of LRRC8A and LRRC8D. HeLa cells stably expressing human LRRC8A or LRRC8D C-terminally tagged with a HA epitope tag were stained for HA, the ER, and actin and examined by confocal microscopy. Images are representative of three independent experiments. Scale bars, 7 μm.
LRR Domains of LRRC8A and LRRC8D Face the Cytoplasm
LRR domains are widespread and often mediate protein-protein interactions (6). The LRR domain of LRRC8A was initially reported to face the outside of cells, but this assertion has recently been questioned based on sequence similarity between LRRC8s and pannexins, a family of communication channels (2, 3). If LRRC8s have the same topology as pannexins, then the LRR domains of LRRC8s would face the cytoplasm and have access to very different interaction partners than on the outside of cells.
To determine the topology of LRRC8A and LRRC8D we performed a flow cytometry experiment. HeLa cells that stably express either empty vector or C-terminally HA-tagged Kell (KEL-HA), glycophorin A (GYPA-HA), LRRC8A, or LRRC8D were incubated with Alexa Fluor 488-conjugated anti-HA in the absence or presence of the detergent saponin (Fig. 4, A and B). Kell, a mammalian type II transmembrane protein, and glycophorin A, a mammalian type I transmembrane protein, are expressed on the cell surface and were used as controls (15, 16). As expected, HA was detected on intact cells expressing KEL-HA. Permeabilization with saponin allowed detection of both the plasma membrane and intracellular populations of Kell. In contrast, for cells that express GYPA-HA, the HA tag could be detected only when the cells were permeabilized due to the cytosolic exposure of the tag. Similarly, for cells that express LRRC8A-HA and LRRC8D-HA, the HA tag could be detected only when the cells were permeabilized, suggesting that their LRR domains face the cytosol.
FIGURE 4.

Topology of LRRC8A and LRRC8D. A, schematic depicts the topology of KEL-HA and GYPA-HA at the plasma membrane. Also depicted are the predicted topology of LRRC8A-HA and LRRC8D-HA at the plasma membrane based upon results in B. B, HeLa cells stably expressing either KEL-HA, GYPA-HA, LRRC8A-HA, LRRC8D-HA, or empty vector were incubated with Alexa Fluor 488-conjugated anti-HA in the absence or presence of saponin. C, schematic depicts the experimental set-up in D, the topology of HA-SEL1L and AUP1-GFP in the ER, and the predicted topology of LRRC8D-HA based upon results in D. D, HA-SEL1L, AUP1-GFP, and LRRC8D-HA were transiently expressed in 293T cells. A microsome-containing fraction from these cells was exposed to proteinase K in the absence or presence of Triton X-100 followed by immunoblot analysis. Results are representative of two (B) and three (D) independent experiments.
Because the expression level of LRRC8D-HA was lower than that of LRRC8A-HA in the stable cell lines used for flow cytometry and because LRRC8D-HA was not as prominently displayed at the plasma membrane in our immunofluorescence experiments, we sought to verify the topology of LRRC8D biochemically with a protease protection assay (Fig. 4, C and D). We used SEL1L and AUP1, members of an ER complex required for the dislocation of misfolded proteins, as controls (10). The N terminus and bulk of SEL1L are located in the ER lumen, whereas AUP1 is a monotypic membrane protein that has both termini facing the cytosol (10, 17, 18). Microsome-containing fractions from 293T transfectants expressing HA-SEL1L, AUP1-GFP, and LRRC8D-HA were incubated with proteinase K in the absence or presence of Triton X-100. Proteinase K-treated samples were then analyzed by immunoblotting. Consistent with the known topology of SEL1L and AUP1, detection of HA-SEL1L was lost only upon addition of both detergent and proteinase K, whereas detection of AUP1-GFP was lost with the addition of proteinase K even in the absence of detergent. Similar to AUP1-GFP, detection of LRRC8D-HA was lost with the addition of proteinase K even in the absence of detergent, thus confirming that the LRR domain of LRRC8D faces the cytosol.
LRRC8D Interacts with Other Members of the LRRC8 Family
Given that the LRR domain of LRRC8s faces the cytosol rather than extracellular space, we hypothesized that the LRR domain could be mediating interactions with cytoplasmic proteins or molecules that affect the opening or closing of a channel of which the LRRC8s might be part. To identify potential interacting partners of LRRC8A and LRRC8D, lysates from RAW 264.7 macrophages that stably express murine LRRC8A-HA, LRRC8D-HA, or empty vector were incubated with anti-HA agarose beads and the immunoprecipitated proteins were identified by mass spectrometry (Table 1). The results indicate that LRRC8A and LRRC8D interact with each other as well as with LRRC8C. A similar experiment with KBM7 cells showed that LRRC8A and LRRC8D interact with LRRC8B as well (data not shown). We verified that LRRC8D interacts with LRRC8A, LRRC8B, and LRRC8C by coexpressing epitope-tagged versions of the proteins in 293T cells, immunoprecipitating for LRRC8A-Myc, LRRC8B-V5, and LRRC8C-FLAG and blotting for LRRC8D-HA in the coimmunoprecipitating fractions (Fig. 5).
TABLE 1.
Identification of potential LRRC8A and LRRC8D interacting partners
Lysates from RAW 264.7 macrophages stably expressing murine LRRC8A-HA, LRRC8D-HA, or empty vector were incubated with anti-HA agarose beads, and the immunoprecipitated proteins were identified by mass spectrometry. Displayed are proteins that were immunoprecipitated from both LRRC8A-HA- and LRRC8D-HA-containing lysates, but not from the empty vector control. Proteins are listed in order of the number of unique peptides identified.
| Immunoprecipitated protein | LRRC8D-HA |
LRRC8A-HA |
||
|---|---|---|---|---|
| No. unique peptides | % Coverage | No. unique peptides | % Coverage | |
| LRRC8D | 31 | 41 | 19 | 27 |
| LRRC8A | 15 | 20 | 32 | 40 |
| LRRC8C | 10 | 14 | 15 | 20 |
| Serine hydroxymethyltransferase 2 | 7 | 16 | 6 | 12 |
| Carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase | 7 | 4.2 | 3 | 2.7 |
| Heterogeneous nuclear ribonucleoprotein A/B | 6 | 18 | 4 | 14 |
| 60S acidic ribosomal protein P0 | 5 | 23 | 4 | 21 |
| ADP/ATP translocase 2 | 5 | 14 | 3 | 11 |
| Aldehyde dehydrogenase 2 | 4 | 10 | 7 | 17 |
| Aspartate aminotransferase, mitochondrial | 4 | 12 | 2 | 5.1 |
FIGURE 5.
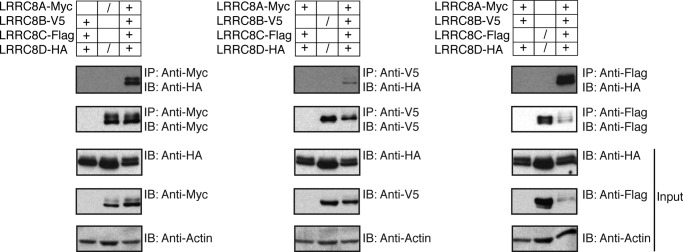
LRRC8D interacts with LRRC8A, B, and C. 293T cells were transiently transfected with murine LRRC8A-Myc, LRRC8B-V5, LRRC8C-FLAG, and LRRC8D-HA. + indicates that constructs were coexpressed in the same cells. / indicates that constructs were independently expressed in separate cells and the lysates were combined post-lysis as a control. Blots are representative of two independent experiments.
We demonstrated that the interaction between LRRC8s does not occur post-lysis by using transfectants that each express a single epitope-tagged LRRC8 member. Lysates from LRRC8A-Myc, LRRC8B-V5, and LRRC8C-FLAG transfectants were each combined with lysates from LRRC8D-HA transfectants. When LRRC8A-Myc, LRRC8B-V5, and LRRC8C-FLAG were immunoprecipitated from these combined lysates, LRRC8D-HA did not appear in the coimmunoprecipitating fractions. We did not verify the interaction of LRRC8D with the other proteins identified by mass spectrometry, as they were identified with modest peptide coverage. We have found that several of these hits occur as common contaminants in immunoprecipitation-mass spectrometry experiments.
DISCUSSION
Defective LRRC8D expression results in BlaS resistance due to a defect in BlaS import. Because import of a toxic molecule is not a function that would be selected for by evolution, it is likely that there are as yet undiscovered physiological substrates that are normally transported by LRRC8D or the complex of which it is a part. Because BlaS contains both a cytosine moiety and an arginine-like appendage, it is possible that nucleotides, amino acids, or short (modified) peptides could be substrates for LRRC8D. Pannexins, which display homology to LRRC8s, have often been linked to the release of ATP (7). Thus ATP and other second messengers could be candidate substrates as well.
The discovery that LRRC8D has a role in the transport of a small molecule suggests that all members of the LRRC8 family, which are on average 45.92% identical, may have a similar function (2). LRRC8A was initially proposed to be a cell surface receptor that initiates a signaling cascade in response to the binding of an extracellular ligand important for B cell development (3). In light of our results, it is possible that instead of acting as a receptor, LRRC8A may act as a selective communication channel that mediates the import of a growth and/or differentiation factor secreted by nearby cells to allow continuation of B cell development. A similar mechanism may be at play for LRRC8C in adipocyte differentiation.
Contrary to what was reporter earlier, the LRR domains of LRRC8A and LRRC8D face the cytoplasm (3). The topology inferred from our experiments is consistent with that of pannexins (2). It has been proposed that LRRC8s are the evolutionary result of the combination of a pannexin and a LRR domain (2). What could be the added benefit of the LRR domain? It is unclear whether LRRC8 channels are always open or if they selectively open when needed. Perhaps the LRR domain regulates the opening of the channel through an interaction with a protein or molecule that occurs in response to specific cellular needs. Alternatively, the LRR domain could bind to additional proteins that make up the scaffold of the transporter, or it could recruit proteins that interact with incoming or outgoing substrates.
We discovered that LRRC8D interacts in stable fashion with other members of the LRRC8 family. Obviously, proteins that interact transiently or only under certain ligand-stimulated conditions would have been missed in our experimental conditions. In addition to future searches for such interactors, it would also be interesting to identify interactions that depend upon the LRR domain by comparing immunoprecipitates from full-length LRRC8s and LRRC8s missing their LRR domain. Panx1 oligomerizes into hexamers whereas Panx2 is thought to assemble into heptamers or octamers (7). Some intermixing between Panx1, Panx2, and Panx3 has been observed, but the biological significance of this is currently unknown. It will be important to determine in future studies whether LRRC8s interact with each other in a single complex or if there are multiple complexes of distinct stoichiometry and composition.
While this work was under review, two recent publications established a plausible role for LRRC8A as a volume-regulated anion channel (19, 20). The proposed topology reported in our work is entirely consistent with that proposed by Voss et al. (20).
Together our results provide clear experimental evidence for a functional link between members of the LRRC8 family and pannexins and shed new light on how LRRC8s could be mediating lymphocyte development and adipocyte differentiation.
Acknowledgments
We are grateful to Lenka Kundrat and the laboratory of Harvey Lodish for providing plasmids containing KEL and GYPA, respectively; Eric Spooner and Wendy Salmon for providing protein mass spectrometry and confocal microscopy core facility services, respectively; and Jan Carette and Thijn Brummelkamp for advice on isolating the LRRC8D-deficient cell lines.
Footnotes
- LRRC8
- leucine-rich repeat-containing 8
- BlaS
- blasticidin S
- BSR
- blasticidin S-resistance
- ER
- endoplasmic reticulum
- IFS
- inactivated fetal calf serum
- LRR
- leucine-rich repeat.
REFERENCES
- 1. Smits G., Kajava A. V. (2004) LRRC8 extracellular domain is composed of 17 leucine-rich repeats. Mol. Immunol. 41, 561–562 [DOI] [PubMed] [Google Scholar]
- 2. Abascal F., Zardoya R. (2012) LRRC8 proteins share a common ancestor with pannexins, and may form hexameric channels involved in cell-cell communication. Bioessays 34, 551–560 [DOI] [PubMed] [Google Scholar]
- 3. Sawada A., Takihara Y., Kim J. Y., Matsuda-Hashii Y., Tokimasa S., Fujisaki H., Kubota K., Endo H., Onodera T., Ohta H., Ozono K., Hara J. (2003) A congenital mutation of the novel gene LRRC8 causes agammaglobulinemia in humans. J. Clin. Invest. 112, 1707–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tominaga K., Kondo C., Kagata T., Hishida T., Nishizuka M., Imagawa M. (2004) The novel gene fad158, having a transmembrane domain and leucine-rich repeat, stimulates adipocyte differentiation. J. Biol. Chem. 279, 34840–34848 [DOI] [PubMed] [Google Scholar]
- 5. Hayashi T., Nozaki Y., Nishizuka M., Ikawa M., Osada S., Imagawa M. (2011) Factor for adipocyte differentiation 158 gene disruption prevents the body weight gain and insulin resistance induced by a high-fat diet. Biol. Pharm. Bull. 34, 1257–1263 [DOI] [PubMed] [Google Scholar]
- 6. Bella J., Hindle K. L., McEwan P. A., Lovell S. C. (2008) The leucine-rich repeat structure. Cell. Mol. Life Sci. 65, 2307–2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Penuela S., Gehi R., Laird D. W. (2013) The biochemistry and function of pannexin channels. Biochim. Biophys. Acta 1828, 15–22 [DOI] [PubMed] [Google Scholar]
- 8. Chekeni F. B., Elliott M. R., Sandilos J. K., Walk S. F., Kinchen J. M., Lazarowski E. R., Armstrong A. J., Penuela S., Laird D. W., Salvesen G. S., Isakson B. E., Bayliss D. A., Ravichandran K. S. (2010) Pannexin 1 channels mediate “find-me” signal release and membrane permeability during apoptosis. Nature 467, 863–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Qu Y., Misaghi S., Newton K., Gilmour L. L., Louie S., Cupp J. E., Dubyak G. R., Hackos D., Dixit V. M. (2011) Pannexin-1 is required for ATP release during apoptosis but not for inflammasome activation. J. Immunol. 186, 6553–6561 [DOI] [PubMed] [Google Scholar]
- 10. Mueller B., Klemm E. J., Spooner E., Claessen J. H., Ploegh H. L. (2008) SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc. Natl. Acad. Sci. U.S.A. 105, 12325–12330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guimaraes C. P., Carette J. E., Varadarajan M., Antos J., Popp M. W., Spooner E., Brummelkamp T. R., Ploegh H. L. (2011) Identification of host cell factors required for intoxication through use of modified cholera toxin. J. Cell Biol. 195, 751–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee C. C., Carette J. E., Brummelkamp T. R., Ploegh H. L. (2013) A reporter screen in a human haploid cell line identifies CYLD as a constitutive inhibitor of NF-κB. PLoS One 8, e70339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Birsoy K., Wang T., Possemato R., Yilmaz O. H., Koch C. E., Chen W. W., Hutchins A. W., Gultekin Y., Peterson T. R., Carette J. E., Brummelkamp T. R., Clish C. B., Sabatini D. M. (2013) MCT1-mediated transport of a toxic molecule is an effective strategy for targeting glycolytic tumors. Nat. Genet. 45, 104–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reiling J. H., Clish C. B., Carette J. E., Varadarajan M., Brummelkamp T. R., Sabatini D. M. (2011) A haploid genetic screen identifies the major facilitator domain containing 2A (MFSD2A) transporter as a key mediator in the response to tunicamycin. Proc. Natl. Acad. Sci. U.S.A. 108, 11756–11765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Redman C. M., Lee S. (1995) The Kell blood group system. Transfus. Clin. Biol. 2, 243–249 [DOI] [PubMed] [Google Scholar]
- 16. Chasis J. A., Mohandas N. (1992) Red blood cell glycophorins. Blood 80, 1869–1879 [PubMed] [Google Scholar]
- 17. Klemm E. J., Spooner E., Ploegh H. L. (2011) Dual role of ancient ubiquitous protein 1 (AUP1) in lipid droplet accumulation and endoplasmic reticulum (ER) protein quality control. J. Biol. Chem. 286, 37602–37614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Spandl J., Lohmann D., Kuerschner L., Moessinger C., Thiele C. (2011) Ancient ubiquitous protein 1 (AUP1) localizes to lipid droplets and binds the E2 ubiquitin conjugase G2 (Ube2g2) via its G2 binding region. J. Biol. Chem. 286, 5599–5606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Qiu Z., Dubin A. E., Mathur J., Tu B., Reddy K., Miraglia L. J., Reinhardt J., Orth A. P., Patapoutian A. (2014) SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell 157, 447–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Voss F. K., Ullrich F., Münch J., Lazarow K., Lutter D., Mah N., Andrade-Navarro M. A., Kries, von J. P., Stauber T., Jentsch T. J. (2014) Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science, in press [DOI] [PubMed] [Google Scholar]