

Background: Dbl is a GEF that activates the small GTPases RhoA, Cdc42, and Rac1. The events that regulate Dbl activity are unknown.
Results: Growth factors lead to phosphorylation of Dbl Tyr510 and stimulate its GEF activity.
Conclusion: Dbl activation cycle is tightly regulated by the activity of tyrosine kinases and phosphatases.
Significance: Understanding signaling pathways enhances our understanding of their roles in health and disease states.
Keywords: Guanine Nucleotide Exchange Factor (GEF), Phosphorylation, Phosphotyrosine, Rho GTPases, Small GTPase
Abstract
Rho GTPases are molecular “switches” that cycle between “on” (GTP-bound) and “off” (GDP-bound) states and regulate numerous cellular activities such as gene expression, protein synthesis, cytoskeletal rearrangements, and metabolic responses. Dysregulation of GTPases is a key feature of many diseases, especially cancers. Guanine nucleotide exchange factors (GEFs) of the Dbl family are activated by mitogenic cell surface receptors and activate the Rho family GTPases Cdc42, Rac1, and RhoA. The molecular mechanisms that regulate GEFs from the Dbl family are poorly understood. Our studies reveal that Dbl is phosphorylated on tyrosine residues upon stimulation by growth factors and that this event is critical for the regulated activation of the GEF. These findings uncover a novel layer of complexity in the physiological regulation of this protein.
Introduction
The Dbl family of guanine nucleotide exchange factors (GEFs)2 comprises >80 members that facilitate the activation of the small GTPases Rac1, Cdc42, and/or RhoA. Dbl was the first identified mammalian GEF and as such is considered to be the prototypic member of the family (1). Two highly conserved domains characterize Dbl family GEFs: a unique Dbl homology domain and a pleckstrin homology domain. The Dbl homology domain is the minimal unit necessary for GEF activity that binds the substrate GTPase and catalyzes the exchange of GDP for GTP in its binding pocket (2). Following GEF-mediated activation, the bound GTP is hydrolyzed, and the GTPase returns to its quiescent state. The pleckstrin homology domain is thought to mediate proper localization of the GEF to specific subcellular compartments (3–5). In response to growth factor stimulation, Dbl-mediated GTPase signaling regulates numerous cellular activities such as cytoskeletal rearrangements, gene expression, and vesicular trafficking, thereby promoting cell proliferation (6–10).
Because GTPases control multiple and diverse aspects of cellular physiology, proper regulation of GEF activities is crucial for normal cellular function. Indeed, mutations that cause GEF overexpression or gain of function result in persistent activation of downstream pathways and lead to aberrant cell growth that manifest in developmental disorders and cancers. For example, mutations in Fgd1 cause the X-linked developmental disorder, Aarskog-Scott syndrome (11), and chromosomal rearrangements of the Bcr gene contribute to leukemogenesis (12). Mutations in Plekhg4 are associated with heritable autosomal spinocerebellar ataxia (13, 14). Overexpression of P-rex1, Ect2, and Tiam1 has been noted in cancers of the breast, lung, and colon, respectively (15–20), and overexpression of Dbl was reported in sarcomas and tumors of neuroectodermal origins (21–24). As our appreciation for the involvement of Dbl family GEFs in disease grows, the need for understanding the mechanisms that regulate these proteins increases.
The upstream events that regulate the GEF activity of Dbl are poorly understood. Dbl is thought to exist in an inhibited state, which is activated by growth factor receptors; however, the molecular mechanisms that underlie the inhibited state and its release are incompletely understood. Some studies proposed an intramolecular autoinhibited state brought about by interactions between the amino and carboxyl termini of Dbl (25). Other studies demonstrated that association with the molecular chaperones Hsc70 and Hsp90 regulate Dbl activity by controlling its degradation by the proteasome (26, 27). The mechanisms by which growth factor stimulation leads to release of Dbl inhibition are completely unknown.
In this study, we aimed to understand the upstream events that relieve Dbl autoinhibition and stimulate its GEF activity. We show that a key feature of the activation cycle of Dbl involves reversible, growth factor-sensitive, tyrosine phosphorylation.
MATERIALS AND METHODS
Cell Culture
COS7 cells and HEK293T cells were cultured in DMEM supplemented with 10% FBS (Hyclone) in 5% CO2 at 37 °C. NIH3T3 cells were cultured in DMEM supplemented with 10% calf serum (Hyclone). Parental and EGFR-expressing CHO cells were a generous gift from Dr. Cathleen Carlin, Case Western Reserve University School of Medicine and were cultured in minimum essential mediumα supplemented with 10% FBS (Hyclone).
Molecular Constructs
Dbl constructs encoded the reading frame encoding the full-length protein (residues 1–925) in the pCMV6 vector, or the pCEFL-GST vector (for focus formation experiments). Recombinant GTPases were expressed in Escherichia coli as glutathione S-transferase fusions and purified as described previously (28). Grb2 cDNA (HA-tagged in the pCGN vector) was a generous gift from Dr. Dafna Bar-Sagi, New York University School of Medicine. All transfections were done using PolyFect (Qiagen).
Chemicals
EGF (Sigma) was dissolved in serum-free DMEM and used at a final concentration of 50 nm. 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2, EMD Millipore) was dissolved in dimethyl sulfoxide and used at a final concentration of 1 or 10 μm, as was Iressa (gefitnib, Selleck Biochemicals). The phosphatase inhibitor-activated sodium orthovanadate (Sigma) was added during the last 10 min to a final concentration of 1 mm.
Immunoprecipitations from Cell Lysates
Cells were lysed in 1 ml of 20 mm Hepes, pH 7.4, 1 mm EDTA, 150 mm NaCl, 1% Igepal, 20 mm NaF, 20 mm β-glycerophosphate, 1 mm vanadate, 200 μm phenylmethylsulfonyl fluoride, and 10 μg/ml each of leupeptin and aprotinin. Lysates were spun at 14,000 rpm for 20 min, and cleared supernatant was combined with anti-Dbl antibody (Santa Cruz Biotechnology, sc-89, 1:100 dilution) or anti-HA antibody (Covance, MMS-101R, 1:100). The lysate-antibody mixture was mixed at 4 °C for 1 h before the addition of 50 μl of protein A beads (Millipore) and mixing at 4 °C for 2.5 h. Beads were washed three times with lysis buffer and spun at 3,500 rpm for 3 min. Precipitated beads were boiled in 2.5× Lamelli buffer, and the supernatant was resolved by 8–12% SDS-PAGE. Tyrosine phosphorylation was visualized using anti-phosphotyrosine antibody (4G10-platinum, Millipore, 1:1000 dilution).
Immunoprecipitations from Mouse Brain
Whole brain extracts from 10-day-old mice were homogenized manually in 1 ml of 150 mm NaCl, 1 mm dithiothreitol, 1 mm EDTA, 150 mm NaCl, 25 mm Tris-HCl (pH 8.0), 0.25 mm phenylmethylsulfonyl fluoride, and 10 μg/ml each of leupeptin and aprotinin. The homogenate was cleared by centrifugation at 14,000 rpm for 45 min at 4 °C, followed by immunoprecipitation with Dbl antibody as described above.
Site-directed Mutagenesis
Site-directed mutagenesis was performed according to the manufacturer's instructions (QuikChange XL II, Agilent).
Starvation and Stimulation of CHO Cells
Five hours prior to lysis, cells were switched to MEMα containing 0.5% FBS. After 3 h, cells were placed on ice for 2 h and treated as indicated (EGF, Iressa, or PP2). Cells were then incubated at 37 °C for 15 min, lysed, and subjected to immunoprecipitations as described above.
Grb2-Dbl Association
COS7 cells were co-transfected with HA-Grb2 and Dbl constructs, harvested in 50 mm HEPES, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 10 mm pyrophosphate, 10 mm glycerophosphate, 50 mm NaF, 1 mm vanadate, 200 μm phenylmethylsulfonyl fluoride, and 10 μg/ml each leupeptin and aprotinin, and centrifuged at 14,000 rpm for 20 min. Association was visualized by anti-HA immunoblotting of anti-Dbl immunoprecipitates.
GEF-GTPase Association Assays
These were designed based on the established high affinity of GEFs to their nucleotide-free form of their cognate GTPases (29–31). Wild-type GTPases and their respective nucleotide-free mutants (Cdc42(T17N) or RhoA(T19N)) were expressed in E. coli as GST fusions and purified as described previously (32). The purified GTPases were immobilized on glutathione agarose and washed extensively with cell lysis buffer supplemented with 50 μm GDP, 10 mm MgCl2 (wild-type GTPase), or 10 mm EDTA (Asn17/Asn19 GTPase). Dbl-transfected cells were lysed in the same lysis buffer, incubated with immobilized GTPases at 4 °C for 2 h, and washed three times in the respective buffers, prior to SDS-PAGE and anti-Dbl immunoblotting.
GTPase Activation Assays
To measure GEF activity, the fraction of endogenous GTPase that is in the active (GTP-bound) state was determined using an established selective precipitation approach (33–35). Briefly, HEK293T cells were transfected with Dbl cDNA, serum-starved for 31 h, and lysed in 20 mm HEPES (pH 7.4), 150 mm NaCl, 1% Nonidet P-40, 10% glycerol, 10 mm MgCl2, 1 mm EDTA, 0.2 mm phenylmethylsulfonyl fluoride, and 10 μg/ml each of leupeptin and aprotinin. Lysates were spun at 14,000 rpm for 20 min, and cleared supernatant was combined with ∼20 μg of immobilized GST-fused GTPase-binding domain of rhotekin and rotated at 4 °C for 2 h (34). Beads were washed three times in lysis buffer and resolved on SDS-PAGE. The amount of activated (bead-associated) GTPase was visualized with anti-RhoA (Cytoskeleton, 1:200 dilution) immunoblotting and compared with the total GTPase levels as determined by anti-RhoA immunoblotting of whole cell lysates.
NFκB Transcriptional Activation Assays
NIH3T3 cells were plated in triplicate in a 24-well plate and co-transfected with the NFκB response element fused to the luciferase reporter construct (pGL3B-HIV1-luc), the β-galactosidase-expressing pCH110 (Pharmacia), and Dbl cDNA. Thirty-six hours after transfection, cells were washed in serum-free medium and starved for 17 h prior to harvesting cell lysates and measuring luciferase and β-galactosidase activities. Luciferase expression was normalized to β-galactosidase activity to account for differences in transfection efficiency.
Focus Formation Assays
The indicated constructs were used to transfect subconfluent NIH3T3 cells in 10-cm plates. After 48 h, media was replaced, and cells were refed every other day. Approximately 2 weeks after transfection, cells were fixed with methanol and stained with crystal violet, and foci were manually scored.
Fluorescence Microscopy
NIH3T3 cells were seeded on glass coverslips and transfected with the indicated constructs (opr empty pCMV6 vector as control) using PolyFect according to the manufacturer's instructions (Qiagen). Thirty-one hours post-transfection, cells were serum-starved for 17 h, fixed with 3.7% paraformaldehyde, permeabilized with 0.2% Triton X-100, stained with the indicated reagents, and imaged using the Leica TCS SP2 microscope. Dbl proteins were visualized using Santa Cruz Biotechnology sc-89 antibody and a secondary Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen) antibody; the actin cytoskeleton was visualized with Texas Red-conjugated phalloidin (Invitrogen).
RESULTS
Tyrosine Phosphorylation of Dbl
In a search for post-translational events that may regulate the GEF activity of Dbl, we found that anti-Dbl immunoprecipitates give a strong signal on anti-phosphotyrosine immunoblots. Importantly, the signal was only visible in the presence of the tyrosine phosphatase inhibitor sodium orthovanadate (Fig. 1A). The physiological relevance of this finding is evident by the observation that tyrosine phosphorylation of Dbl occurs in intact tissue where the protein is expressed, such as mouse brain (Fig. 1B). Moreover, we found that when serum-deprived COS7 cells are treated with serum, Dbl phosphorylation transiently increases after 2–5 min and then tapers off (Fig. 1C). These observations show that Dbl is phosphorylated on tyrosine residue(s) and that this modification is a regulated event, dependent on growth factors. To begin to delineate the upstream events that regulate Dbl phosphorylation, we used pharmacological inhibitors of two major intracellular tyrosine kinases: Iressa (gefitinib), which inhibits the EGF receptor tyrosine kinase, and the inhibitor of Src family tyrosine kinases, PP2. To confirm these findings we examined whether expression of the EGFR kinase affects the phosphorylation of Dbl. In CHO cells that stably overexpress the EGFR kinase, stimulation with EGF caused a marked phosphorylation of Dbl (Fig. 1D). Moreover, modification was inhibited by treatment with either Iressa or PP2. Importantly, Dbl expression, but not phosphorylation, was observed in the parental CHO cell line, which does not express EGFR (Fig. 1D). Taken together, these findings indicate that phosphorylation of Dbl is mediated by mitogenic stimulation through the activated EGF receptor and a Src family kinase. Furthermore, the data suggest that in this pathway, the Src family kinase is downstream of EGFR.
FIGURE 1.

Tyrosine phosphorylation of Dbl. Anti-Dbl immunoprecipitates from transfected COS7 cells (A) or whole mouse brains (B) were immunoblotted (IB) with anti-phosphotyrosine (PY) antibodies. C, regulation of Dbl phosphorylation by serum. Serum-starved Dbl-transfected COS7 cells were stimulated with 10% FBS for the indicated duration, and tyrosine phosphorylation was examined as described in A. D, regulation of Dbl phosphorylation by EGF receptor and Src family kinases. Dbl-transfected EGFR expressing and parental CHO cells were starved, stimulated with EGF (50 nm), and treated with 1 or 10 μm Iressa or 1 or 10 μm PP2. Starvation and stimulation conditions are as described under “Materials and Methods.” All blots are representative of at least three independent experiments.
Site(s) of Tyrosine Phosphorylation in Dbl
We utilized a bioinformatics approach to identify the tyrosine residue(s) that are phosphorylated in Dbl. The NetPhos2.0 algorithm (36) identified seven residues in Dbl that reside within tyrosine phosphorylation consensus sequences. These putative phosphorylation sites are dispersed throughout the different domains of the protein (Tyr217, Tyr510, Tyr553, Tyr749, Tyr780, Tyr787, and Tyr904; see Fig. 2A). We focused our attention to Tyr510, due to its position in a high-scoring putative phosphorylation sequence in the catalytic Dbl homology domain. Using site-directed mutagenesis, we substituted Tyr510 of Dbl to a phenylalanine and observed a dramatic decrease (∼80%) in the tyrosine phosphorylation intensity of the protein (Fig. 2B). The fact that some tyrosine phosphorylation persists in the Y510F mutant indicates that sites of tyrosine phosphorylation other than Tyr510 exist in the protein. In attempting to identify such residues, we found that mutation of Tyr787 did not affect the residual phosphorylation signal observed in Dbl(Y510F) (data not shown).
FIGURE 2.
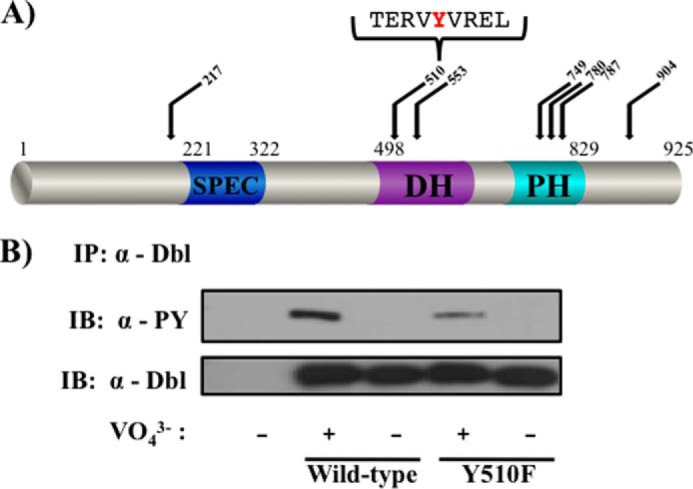
Tyr510 is an important site of phosphorylation in Dbl. A, domain structure of the Dbl protein. Of the 29 tyrosines of Dbl, the top-scoring putative phosphorylation sites are indicated, as well as the immediate sequence surrounding Tyr510. B, tyrosine phosphorylation of Dbl(Y510F) was examined as in Fig. 1A. Data are representative of three independent experiments. PY, phosphotyrosine; IB, immunoblot; DH, Dbl homology domain; PH, pleckstrin homology domain; SPEC, spectrin.
Regulation of Molecular Interactions of Dbl by Tyr510 Phosphorylation
To understand the molecular consequences of tyrosine phosphorylation in Dbl, we examined its ability to associate with established effectors. Grb2 is an adapter molecule that contains an Src homology 2 domain that binds phosphorylated tyrosine residues and an Src homology 3 domain that binds proline-rich sequences (37). Due to this dual-recognition capacity, Grb2 often functions as a “bridging” adapter, allowing it to recruit multiple proteins to their cellular site of activation, as seen in the activation of Ras, where Grb2 recruits Son of Sevenless to the tyrosine-phosphorylated EGF receptor (38, 39). Because Grb2 was recently reported to be associated with Dbl (40), we sought to evaluate the role of Tyr510 phosphorylation in this interaction. As seen in Fig. 3A, substitution of the Tyr510 of Dbl with a non-phosphorylatable phenylalanine residue greatly diminished Grb2 binding. Furthermore, the association between Grb2 and Dbl required the presence of the tyrosine phosphatase inhibitor sodium orthovanadate (Fig. 3B). Together, these data strongly suggest that phosphorylation of the Tyr510 of Dbl is critical for its binding to Grb2.
FIGURE 3.
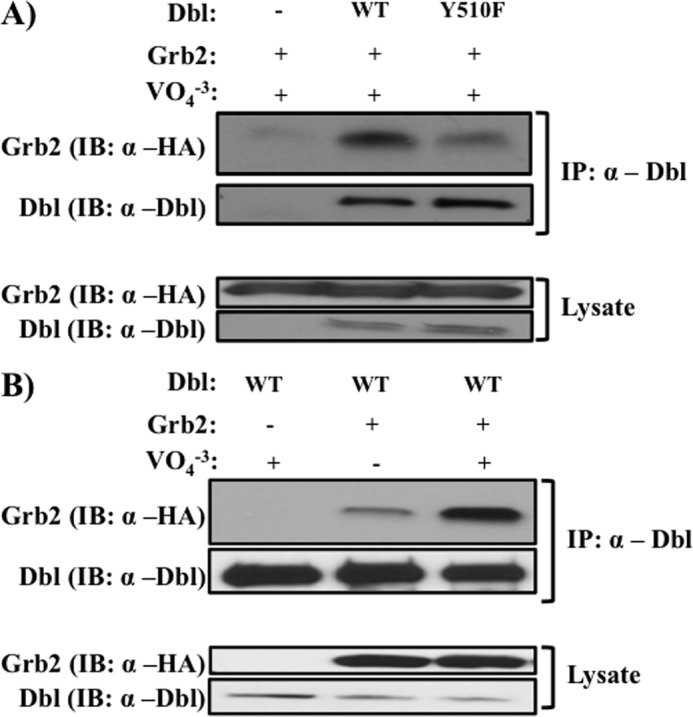
Role of Tyr510 in binding of Dbl to Grb2. COS7 cells were co-transfected with indicated constructs, and association was evaluated as described under “Materials and Methods.” A, association of Grb2 with Dbl or Dbl (Y510F). B, association of Grb2 with Dbl is dependent on tyrosine phosphorylation. Experiments were done as described in A, in the presence or absence of the tyrosine phosphate inhibitor sodium orthovanadate (1 mm). Top panels, presence of Grb2 and Dbl in anti-Dbl immunoprecipitates. Bottom panels, protein expression in cell lysates. Data are representative of three independent experiments. IB, immunoblot.
Next, we asked whether tyrosine phosphorylation of Dbl is necessary for association of the GEF with its substrate GTPase. Cells expressing Dbl (or its phospho-defective mutant) were lysed and precipitated with immobilized recombinant GTPases that have been purified in either the GDP-bound form (GST-fused wild-type GTPases) or the nucleotide-free form (GST-RhoA(T19N) or GST-Cdc42(T17N) mutants). The rationale for this design is based on the notion that the highest affinity of GEFs to their GTPase is observed in the nucleotide-free form of the latter (29–31). As expected, we observed a strong preference in the association of Dbl with the nucleotide-free forms of RhoA and Cdc42, as compared with the GDP-bound GTPases (compare lanes 1 and 2 in Fig. 4, A and B). Importantly, we observed that the affinity of the nucleotide-free GTPase to the GEF is dramatically reduced when phosphorylation of Tyr510 of Dbl is abolished by mutation to phenylalanine (Fig. 4, A and B). These findings indicate that that phosphorylation of Tyr510 is required for Dbl to be able to bind substrate Rho GTPases.
FIGURE 4.
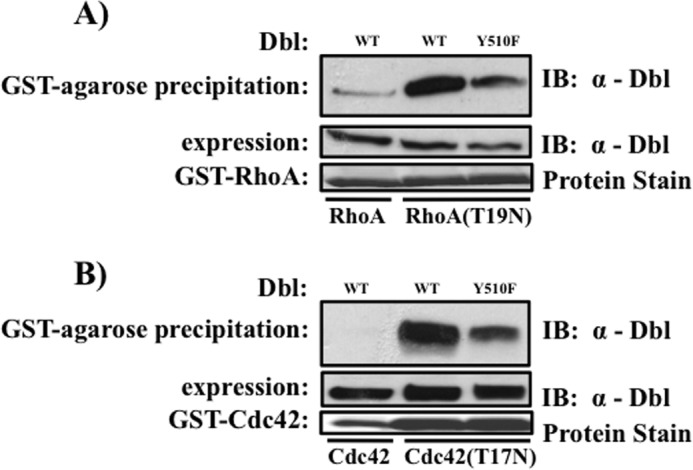
Role of Tyr510 in association of Dbl with substrate GTPases. GST fusion proteins of the indicated RhoA (A) and Cdc42 (B) variants were overexpressed in E. coli, purified on glutathione affinity chromatography, and their nucleotide content manipulated as described under “Materials and Methods.” Top panels, GEF presence in affinity precipitates. Middle panels, Dbl expression in respective cell lysates. Lower panels, GST-GTPases “baits” were visualized by Ponceau Red staining of the blotted membranes. Data are representative of three independent experiments. IB, immunoblot.
Phosphorylation of Tyr510 Is Critical for GEF Activity of Dbl
To address the role of Tyr510 phosphorylation in Dbl activity as a GEF, we examined its ability to stimulate GTP binding by a substrate GTPase. Because both Cdc42 and Rac1 are phosphorylated on Tyr64 (41),3 whereas RhoA is not (Fig. 5A), we chose to focus on the latter GTPase. This allowed us to evaluate the effect of kinase inhibitors on Dbl actions without complication arising from the effect of the inhibitors on the substrate GTPase. We treated Dbl-transfected cells with Iressa and measured the GEF activity of Dbl by selectively precipitating GTP-bound form of RhoA ((33–35); see “Materials and Methods”). We found that treatment with Iressa all but abolished the ability of Dbl to activate RhoA (Fig. 5B), indicating that EGFR-mediated tyrosine phosphorylation of Dbl is essential for its activity as a GEF. Importantly, substitution of Tyr510 to phenylalanine abolished the GEF activity of Dbl (Fig. 5C).
FIGURE 5.
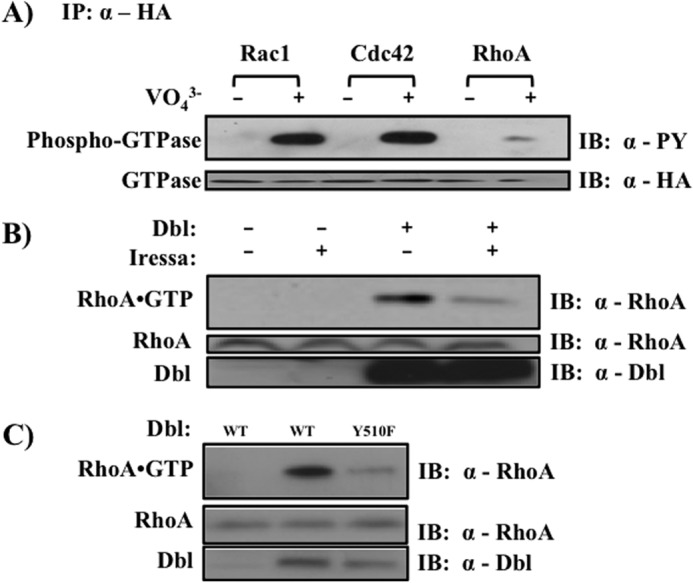
Tyrosine phosphorylation of Dbl is necessary for proper activation of RhoA. A, tyrosine phosphorylation of Cdc42, Rac1, and RhoA. COS7 cells were transiently transfected with the indicated construct and treated with orthovanadate where indicated. Tyrosine phosphorylation was examined in anti-HA immunoprecipitates. B, effect of tyrosine kinase inhibition on the GEF activity of Dbl. HEK293T cells were transiently transfected with the indicated constructs and serum-starved for 24 h. Thirty minutes prior to lysis, cells were treated with 10 μm Iressa, and the fraction of RhoA in the GTP-bound state was measured as described under “Materials and Methods.” Top panel, GTP-bound RhoA visualized by anti-RhoA immunoblotting after affinity precipitation. Lower panel, expression levels of the indicated proteins in cell lysates. C, role of Tyr510 phosphorylation in Dbl-stimulated nucleotide exchange. HEK293T cells were transiently transfected with indicated Dbl constructs, and RhoA activation was measured as described in B. Each panel is representative of three independent experiments. IB, immunoblot; PY, phosphotyrosine.
Next, we examined the role of Tyr510 phosphorylation on further downstream signaling events. We found that the Y510F mutant of Dbl abolished transcriptional activation of NFkB (Fig. 6A), a downstream effector of RhoA, Rac1, and Cdc42 (42–44). These results further confirm that tyrosine phosphorylation of Dbl is necessary to activate the Rho family small GTPases. Lastly, we examined at the role of Tyr510 phosphorylation on the signature biological activity of Dbl: transformation of cultured murine fibroblasts (45–47). We examined the outcome of Dbl overexpression on contact inhibition by quantifying the number of three-dimensional foci formed in confluent NIH3T3 cells transfected with Dbl cDNAs. Y510F mutation of Dbl caused a marked reduction in the focus-formation activity of Dbl (Fig. 6B).
FIGURE 6.
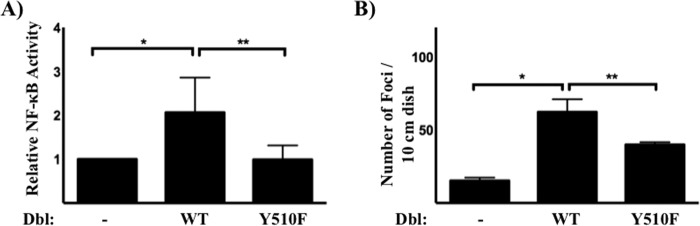
Role of Tyr510 phosphorylation in Dbl-induced downstream activities. A, transcriptional effects. NIH3T3 cells were co-transfected with the indicated Dbl construct, an NFκB reporter construct, and a β-galactosidase expressing vector. NF-κB transcriptional activity was measured as described under “Materials and Methods.” Shown are average values of three independent transfections, and statistical significance is indicated by asterix (p < 0.05 compared with the vector control (*) or to Dbl (**) as determined by Student's t test. B, loss of contact inhibition. Triplicate plates of NIH3T3 cells were transfected with the indicated Dbl cDNAs and monitored for foci formation as described under “Materials and Methods.” *, p < 0.05 compared with the empty vector control; **, p < 0.05 compared with wild-type Dbl, as determined by a Student's t test (statistical significance). Data shown are representative of three independent experiments.
One of the best studied outcomes of GEF-induced stimulation of Rho GTPases is rearrangement of the actin cytoskeleton. Thus, it has been established that activation of Cdc42 stimulates filopodia formation, whereas GTP-binding by RhoA and Rac1 enhances formation of stress fibers and lamellipodia, respectively (48–50). Accordingly, ectopic expression of GEFs in cultured fibroblasts elicits the cytoskeletal pattern typical of their substrate GTPase and provides a convenient read-out of their signaling state (51). We therefor examined the role of Tyr510 phosphorylation in the ability of Dbl to induce these cytoskeletal changes in cultured NIH3T3 cells. Fig. 7 shows fluorescence micrographs of cells that express wild-type Dbl, the mutated variant Dbl(Y510F), or a control vector. We observed that expression of wild-type Dbl lead to enhanced formation of lamellipodia, filopodia, and stress fibers, as reported previously (Fig. 7) (51). The intracellular distribution pattern of the Dbl protein in these cells was diffuse throughout the cytosol with strong perinuclear and membrane localization. In contrast, the intracellular localization pattern of Dbl(Y510F) was markedly different, typified by punctate distribution throughout the cytosol. Additionally, cells that expressed the mutant GEF were smaller, rounder, and did not show the cytoskeletal reorganization induced by the wild-type protein. Rather, actin structures in mutant-expressing cells exhibited aberrant organization of the actin cytoskeleton, in a pattern that resembled “immature beginning” stress fibers, lamellopodia, and filopodia. These findings indicate that phosphorylation of Tyr510 is critical for proper regulation of cellular architecture. The observation that expression of the Dbl(Y510F) mutant has an evident yet altered impact on cytoskeletal reorganization suggest that other residues in Dbl are also important for this activity.
FIGURE 7.
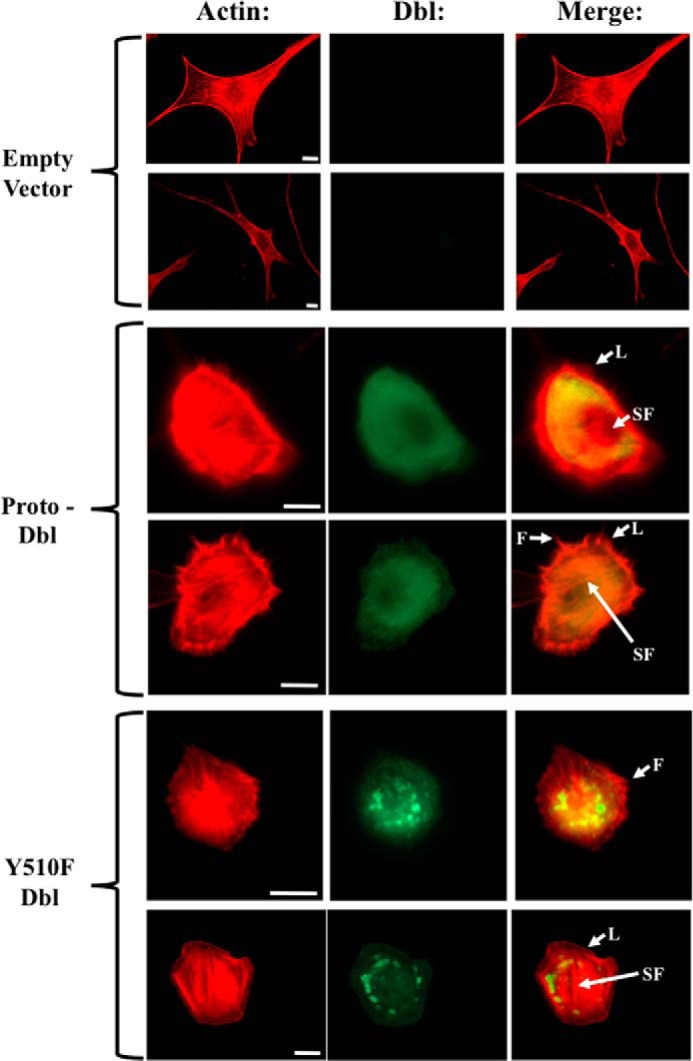
Tyrosine phosphorylation of Tyr510 regulates Dbl intracellular localization and activity. Cells expressing wild-type Dbl, Dbl(Y510F), or control vector, were immunostained with anti-Dbl-antibody (green) and Texas Red-conjugated phalloidin (red) and visualized by fluorescence microscopy. Arrowheads indicate stress fibers (SF), lamellipodia (L), or filopodia (F). Scale bar, 10 μm.
DISCUSSION
Members of the Dbl family GEFs regulate signaling pathways that emanate from their downstream targets, the Rho GTPases. As such, they control numerous intracellular processes and are critical for mediating the effects of diverse extracellular stimuli (8, 10, 52–54). Underscoring the fundamental biological importance of Dbl family members is that improper regulation of their GEF activities is associated with pathological states, including cancer, developmental and neurological disorders, and viral pathogenesis (8). To date, great progress has been made in understanding the molecular mechanisms that govern small GTPase signaling and the downstream pathways that they regulate. However, our understanding of the upstream events that control and regulate GEF actions is very limited. Especially enigmatic are the events that couple GEFs to growth factor receptors and the molecular mechanisms that control their activation/deactivation cycle.
A number of studies indicate that both intramolecular and intermolecular interactions maintain Dbl family GEFs in an inhibited basal state that is relieved upon stimulation. Members of the Vav family of GEFs are basally autoinhibited via intramolecular interactions between the amino-terminal calponin homology domain and the Dbl homology domain that hinders access of Rho GTPases to the catalytic site. Stimulation of the T cell receptor leads to the phosphorylation of amino-terminal tyrosines by a recruited kinase, structural relaxation of the autoinhibited structure, and stimulation of nucleotide exchange on substrate GTPases (55, 56). Similarly, autoinhibition in Ngef and Wgef, is maintained by the C-terminal SH3 domain and an N-terminal polyproline region, which is released by phosphorylation of amino-terminal tyrosines (57). In the case of PDZ-RhoGEF, leukemia-associated RhoGEF, and p115, the autoinhibited state is disrupted by binding of the Gα12/13 to the RGS domains of these GEFs (58–60).
Dbl was initially discovered as a potent fibroblast transforming sequence, the activity of which is stimulated upon truncation of the amino terminus of the protein (1, 7, 9, 25). Dbl has been shown to activate RhoA, Rac1, and Cdc42 and to be expressed in the central nervous system, testis, as well as in neuroectodermal cancers and Ewing sarcomas (21–24). Upstream regulation of Dbl involves signaling concepts utilized in multiple other GEFs. The protein appears to be autoinhibited by interactions between its amino-terminal proto-oncogenic sequence, and the carboxyl-terminal half of the protein (25). In addition, Dbl is regulated by interactions with the molecular chaperones Hsc70 and Hsp90 that control the rate at which the protein is degraded by the proteasome (26, 27). Here, we show that Dbl is also regulated by tyrosine phosphorylation of Tyr510. The modification is stimulated by growth factors, reversible, and essential for the transient conversion of Dbl to an active GEF. Our working hypothesis regarding the role of tyrosine phosphorylation in Dbl actions is depicted in Fig. 8. According to this model, receptor stimulation leads to phosphorylation of the Tyr510 of Dbl by an as yet unidentified kinase, followed by association of Grb2 with the phosphorylated residue and recruitment of another kinase(s) that phosphorylates additional tyrosines. Our data strongly suggest that Dbl phosphorylation occurs through the actions of both the EGFR kinase and a member of the Src family. Our observations are in line with a model in which EGFR kinase (kinase 1 in Fig. 8) is upstream of a Src family member (kinase 2 in Fig. 8), which directly phosphorylates Dbl. Once Dbl is fully phosphorylated, it is likely to be in a conformation that allows productive interactions with the substrate GTPase. Following substrate activation, the GEF undergoes phosphatase-mediated dephosphorylation and transitions to its quiescent state.
FIGURE 8.

Proposed model of Dbl regulation by tyrosine phosphorylation. In quiescent cells, both intramolecular and intermolecular interactions maintain Dbl in an inactive state (step 5). Growth factor stimulation leads to phosphorylated of Tyr510 (step 1), leading to the recruitment of Grb2 (step 2) and other kinases (step 3) necessary for the phosphorylation of other phosphorylation sites. Full phosphorylation relaxes the autoinhibited structure of Dbl to one that allows high-affinity binding of substrate GTPases and facilitation of nucleotide exchange (step 4). DH, Dbl homology domain; PH, pleckstrin homology domain; N, N terminus; C, C terminus.
A number of important questions remain unanswered. First, the identity of the specific kinase(s) that phosphorylate Dbl is yet to be determined. Similarly, the phosphorylation site(s) that are modified in addition to Tyr510 and their impact on protein function are not known presently. Perhaps most interesting is the functional relationship that may exist between the different modes of regulation of Dbl, i.e. whether tyrosine phosphorylation affects the proteasomal degradation of the protein or its association with cytoskeletal elements such as ERM proteins (61). Interestingly, the intracellular distribution pattern of the Dbl(Y510F) mutant bears a striking resemblance to that of wild-type Dbl upon treatment with 17-AAG, an inhibitor of the chaperone Hsp90 (55, 56). Previous studies in our laboratory have shown that Dbl associates with the molecular chaperones Hsc70 and Hsp90 and the ubiquitin ligase CHIP. These molecules regulate the levels, the localization, and the activity of Dbl. In the inactive state, association with these molecules renders Dbl a short-lived, aggregation-prone protein (26, 27). However, the mechanisms by which these inhibitory interactions are relieved have not been elucidated. The findings that the distribution pattern of Dbl(Y510F) resembles aggresomes (xxx) raises the intriguing possibility that tyrosine phosphorylation regulates the degradation rate of Dbl.
Additionally, a stable complex between Dbl and the lipid kinase PI3KC2β was reported (40), raising the possibility that a localized change in membranous phosphoinositide levels comprises an additional level of regulation. It is possible that regulated phosphorylation is coupled to, or coordinated with, the interaction of Dbl with this lipid kinase or its catalytic products. These questions open the door for exciting future research.
This work was supported by Award CA082391 from the National Institutes of Health.
M. Gupta, X. Qi, V. Thakur, and D. Manor, unpublished results.
- GEF
- guanine nucleotide exchange factor
- EGFR
- EGF receptor
- PP2
- 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine.
REFERENCES
- 1. Srivastava S. K., Wheelock R. H., Aaronson S. A., Eva A. (1986) Identification of the protein encoded by the human diffuse B-cell lymphoma (dbl) oncogene. Proc. Natl. Acad. Sci. U.S.A. 83, 8868–8872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hart M. J., Eva A., Evans T., Aaronson S. A., Cerione R. A. (1991) Catalysis of guanine nucleotide exchange on the Cdc42Hs protein by the dbl oncogene product. Nature 354, 311–314 [DOI] [PubMed] [Google Scholar]
- 3. Haslam R. J., Koide H. B., Hemmings B. A. (1993) Pleckstrin domain homology. Nature 363, 309–310 [DOI] [PubMed] [Google Scholar]
- 4. Russo C., Gao Y., Mancini P., Vanni C., Porotto M., Falasca M., Torrisi M. R., Zheng Y., Eva A. (2001) Modulation of oncogenic DBL activity by phosphoinositol phosphate binding to pleckstrin homology domain. J. Biol. Chem. 276, 19524–19531 [DOI] [PubMed] [Google Scholar]
- 5. Zheng Y., Zangrilli D., Cerione R. A., Eva A. (1996) The pleckstrin homology domain mediates transformation by oncogenic Dbl through specific membrane targeting. J. Biol. Chem. 271, 19017–19020 [DOI] [PubMed] [Google Scholar]
- 6. Cerione R. A., Zheng Y. (1996) The Dbl family of oncogenes. Curr. Opin. Cell Biol. 8, 216–222 [DOI] [PubMed] [Google Scholar]
- 7. Zheng Y. (2001) Dbl family guanine nucleotide exchange factors. Trends Biochem. Sci. 26, 724–732 [DOI] [PubMed] [Google Scholar]
- 8. Rossman K. L., Der C. J., Sondek J. (2005) GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 6, 167–180 [DOI] [PubMed] [Google Scholar]
- 9. Whitehead I. P., Campbell S., Rossman K. L., Der C. J. (1997) Dbl family proteins. Biochim. Biophys. Acta 1332, F1–F23 [DOI] [PubMed] [Google Scholar]
- 10. Erickson J. W., Cerione R. A. (2001) Multiple roles for Cdc42 in cell regulation. Curr. Opin. Cell Biol. 13, 153–157 [DOI] [PubMed] [Google Scholar]
- 11. Pasteris N. G., Cadle A., Logie L. J., Porteous M. E., Schwartz C. E., Stevenson R. E., Glover T. W., Wilroy R. S., Gorski J. L. (1994) Isolation and characterization of the faciogenital dysplasia (Aarskog-Scott syndrome) gene: a putative Rho/Rac guanine nucleotide exchange factor. Cell 79, 669–678 [DOI] [PubMed] [Google Scholar]
- 12. Groffen J., Stephenson J. R., Heisterkamp N., de Klein A., Bartram C. R., Grosveld G. (1984) Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell 36, 93–99 [DOI] [PubMed] [Google Scholar]
- 13. Ishikawa K., Mizusawa H. (2006) On autosomal dominant cerebellar ataxia (ADCA) other than polyglutamine diseases, with special reference to chromosome 16q22.1-linked ADCA. Neuropathology 26, 352–360 [DOI] [PubMed] [Google Scholar]
- 14. Ishikawa K., Toru S., Tsunemi T., Li M., Kobayashi K., Yokota T., Amino T., Owada K., Fujigasaki H., Sakamoto M., Tomimitsu H., Takashima M., Kumagai J., Noguchi Y., Kawashima Y., Ohkoshi N., Ishida G., Gomyoda M., Yoshida M., Hashizume Y., Saito Y., Murayama S., Yamanouchi H., Mizutani T., Kondo I., Toda T., Mizusawa H. (2005) An autosomal dominant cerebellar ataxia linked to chromosome 16q22.1 is associated with a single-nucleotide substitution in the 5′ untranslated region of the gene encoding a protein with spectrin repeat and rho guanine-nucleotide exchange-factor domains. Am. J. Hum. Genet. 77, 280–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hynes N. E., Gattelli A. (2011) P-Rex1, a guanine exchange factor that is overexpressed in breast cancer, is a convergence node for ErbB and CXCR4 signaling. Mol. Cell 41, 5–7 [DOI] [PubMed] [Google Scholar]
- 16. Justilien V., Jameison L., Der C. J., Rossman K. L., Fields A. P. Oncogenic activity of Ect2 is regulated through protein kinase C ι-mediated phosphorylation. J. Biol. Chem. 286, 8149–8157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fields A. P., Justilien V. The guanine nucleotide exchange factor (GEF) Ect2 is an oncogene in human cancer. Adv. Enzyme Regul. 50, 190–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cottonham C. L., Kaneko S., Xu L. miR-21 and miR-31 converge on TIAM1 to regulate migration and invasion of colon carcinoma cells. J. Biol. Chem. 285, 35293–35302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Minard M. E., Ellis L. M., Gallick G. E. (2006) Tiam1 regulates cell adhesion, migration and apoptosis in colon tumor cells. Clin. Exp. Metastasis 23, 301–313 [DOI] [PubMed] [Google Scholar]
- 20. Minard M. E., Herynk M. H., Collard J. G., Gallick G. E. (2005) The guanine nucleotide exchange factor Tiam1 increases colon carcinoma growth at metastatic sites in an orthotopic nude mouse model. Oncogene 24, 2568–2573 [DOI] [PubMed] [Google Scholar]
- 21. Vecchio G., Cavazzana A. O., Triche T. J., Ron D., Reynolds C. P., Eva A. (1989) Expression of the dbl proto-oncogene in Ewing's sarcomas. Oncogene 4, 897–900 [PubMed] [Google Scholar]
- 22. Colucci-D'Amato G. L., De Franciscis V., Rosati R., Mauro A., Bulfone A., Eva A., Vecchio G. (1990) Preferential expression of the dbl proto-oncogene in some neuroectodermal tumors. J. Neurosurg. Sci. 34, 187–188 [PubMed] [Google Scholar]
- 23. Navarro S., Pellín A., Noguera R., Díaz M. P., Tsokos M., Triche T. J., Llombart-Bosch A. (1993) dbl oncogene expression in childhood tumors and tumor cell lines. Diagn. Mol. Pathol. 2, 158–162 [PubMed] [Google Scholar]
- 24. Velasco J. A., Ramsamooj P., Thraves P. J., Eva A., Dritschilo A., Notario V. (1995) Co-regulated expression of dbl and poly(ADP-ribose) polymerase in Ewing's sarcoma cells and dbl-transformed NIH3T3 fibroblasts. Oncogene 10, 2253–2258 [PubMed] [Google Scholar]
- 25. Bi F., Debreceni B., Zhu K., Salani B., Eva A., Zheng Y. (2001) Autoinhibition mechanism of proto-Dbl. Mol. Cell Biol. 21, 1463–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kamynina E., Kauppinen K., Duan F., Muakkassa N., Manor D. (2007) Regulation of proto-oncogenic dbl by chaperone-controlled, ubiquitin-mediated degradation. Mol. Cell Biol. 27, 1809–1822; PMC1820456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kauppinen K. P., Duan F., Wels J. I., Manor D. (2005) Regulation of the Dbl proto-oncogene by heat shock cognate protein 70 (Hsc70). J. Biol. Chem. 280, 21638–21644 [DOI] [PubMed] [Google Scholar]
- 28. Leonard D. A., Evans T., Hart M., Cerione R. A., Manor D. (1994) Investigation of the GTP-binding/GTPase cycle of Cdc42Hs using fluorescence spectroscopy. Biochemistry 33, 12323–12328 [DOI] [PubMed] [Google Scholar]
- 29. Hart M. J., Eva A., Zangrilli D., Aaronson S. A., Evans T., Cerione R. A., Zheng Y. (1994) Cellular transformation and guanine nucleotide exchange activity are catalyzed by a common domain on the dbl oncogene product. J. Biol. Chem. 269, 62–65 [PubMed] [Google Scholar]
- 30. Bourne H. R., Sanders D. A., McCormick F. (1990) The GTPase superfamily: a conserved switch for diverse cell functions. Nature 348, 125–132 [DOI] [PubMed] [Google Scholar]
- 31. Bourne H. R., Sanders D. A., McCormick F. (1991) The GTPase superfamily: conserved structure and molecular mechanism. Nature 349, 117–127 [DOI] [PubMed] [Google Scholar]
- 32. Wu W. J., Leonard D. A., A-Cerione R., Manor D. (1997) Interaction between Cdc42Hs and RhoGDI is mediated through the Rho insert region. J. Biol. Chem. 272, 26153–26158 [DOI] [PubMed] [Google Scholar]
- 33. Bagrodia S., Dérijard B., Davis R. J., Cerione R. A. (1995) Cdc42 and PAK-mediated signaling leads to Jun kinase and p38 mitogen-activated protein kinase activation. J. Biol. Chem. 270, 27995–27998 [DOI] [PubMed] [Google Scholar]
- 34. Ren X. D., Schwartz M. A. (2000) Determination of GTP loading on Rho. Methods Enzymol. 325, 264–272 [DOI] [PubMed] [Google Scholar]
- 35. Taylor S. J., Shalloway D. (1996) Cell cycle-dependent activation of Ras. Curr. Biol. 6, 1621–1627 [DOI] [PubMed] [Google Scholar]
- 36. Blom N., Sicheritz-Pontén T., Gupta R., Gammeltoft S., Brunak S. (2004) Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4, 1633–1649 [DOI] [PubMed] [Google Scholar]
- 37. Downward J. (1994) The GRB2/Sem-5 adaptor protein. FEBS Lett. 338, 113–117 [DOI] [PubMed] [Google Scholar]
- 38. Chardin P., Camonis J. H., Gale N. W., van Aelst L., Schlessinger J., Wigler M. H., Bar-Sagi D. (1993) Human Sos1: a guanine nucleotide exchange factor for Ras that binds to GRB2. Science 260, 1338–1343 [DOI] [PubMed] [Google Scholar]
- 39. Gale N. W., Kaplan S., Lowenstein E. J., Schlessinger J., Bar-Sagi D. (1993) Grb2 mediates the EGF-dependent activation of guanine nucleotide exchange on Ras. Nature 363, 88–92 [DOI] [PubMed] [Google Scholar]
- 40. Blajecka K., Marinov M., Leitner L., Uth K., Posern G., Arcaro A. Phosphoinositide 3-kinase C2β regulates RhoA and the actin cytoskeleton through an interaction with Dbl. PLoS One 7, e44945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tu S., Wu W. J., Wang J., Cerione R. A. (2003) Epidermal growth factor-dependent regulation of Cdc42 is mediated by the Src tyrosine kinase. J. Biol. Chem. 278, 49293–49300 [DOI] [PubMed] [Google Scholar]
- 42. Cammarano M. S., Minden A. (2001) Dbl and the Rho GTPases activate NF κB by I κB kinase (IKK)-dependent and IKK-independent pathways. J. Biol. Chem. 276, 25876–25882 [DOI] [PubMed] [Google Scholar]
- 43. Montaner S., Perona R., Saniger L., Lacal J. C. (1998) Multiple signalling pathways lead to the activation of the nuclear factor κB by the Rho family of GTPases. J. Biol. Chem. 273, 12779–12785 [DOI] [PubMed] [Google Scholar]
- 44. Whitehead I. P., Lambert Q. T., Glaven J. A., Abe K., Rossman K. L., Mahon G. M., Trzaskos J. M., Kay R., Campbell S. L., Der C. J. (1999) Dependence of Dbl and Dbs transformation on MEK and NF-κB activation. Mol. Cell Biol. 19, 7759–7770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ron D., Tronick S. R., Aaronson S. A., Eva A. (1988) Molecular cloning and characterization of the human Dbl proto-oncogene: evidence that its overexpression is sufficient to transform NIH/3T3 cells. EMBO J. 7, 2465–2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ron D., Zannini M., Lewis M., Wickner R. B., Hunt L. T., Graziani G., Tronick S. R., Aaronson S. A., Eva A. (1991) A region of proto-dbl essential for its transforming activity shows sequence similarity to a yeast cell cycle gene, CDC24, and the human breakpoint cluster gene, bcr. New Biol. 3, 372–379 [PubMed] [Google Scholar]
- 47. Khosravi-Far R., Chrzanowska-Wodnicka M., Solski P. A., Eva A., Burridge K., Der C. J. (1994) Dbl and Vav mediate transformation via mitogen-activated protein kinase pathways that are distinct from those activated by oncogenic Ras. Mol. Cell Biol. 14, 6848–6857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hall A. (1992) Ras-related GTPases and the cytoskeleton. Mol. Biol. Cell 3, 475–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ridley A. J., Hall A. (1992) Distinct patterns of actin organization regulated by the small GTP-binding proteins Rac and Rho. Cold Spring Harb. Symp. Quant Biol. 57, 661–671 [DOI] [PubMed] [Google Scholar]
- 50. Nobes C. D., Hall A. (1995) Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81, 53–62 [DOI] [PubMed] [Google Scholar]
- 51. Lin R., Cerione R. A., Manor D. (1999) Specific contributions of the small GTPases Rho, Rac, and Cdc42 to Dbl transformation. J. Biol. Chem. 274, 23633–23641 [DOI] [PubMed] [Google Scholar]
- 52. Rossman K. L., Sondek J. (2005) Larger than Dbl: new structural insights into RhoA activation. Trends Biochem. Sci. 30, 163–165 [DOI] [PubMed] [Google Scholar]
- 53. Cerione R. A. (2004) Cdc42: new roads to travel. Trends Cell Biol. 14, 127–132 [DOI] [PubMed] [Google Scholar]
- 54. Wu W. J., Erickson J. W., Lin R., Cerione R. A. (2000) The γ-subunit of the coatomer complex binds Cdc42 to mediate transformation. Nature 405, 800–804 [DOI] [PubMed] [Google Scholar]
- 55. Katzav S. (2009) Vav1: a hematopoietic signal transduction molecule involved in human malignancies. Int. J. Biochem. Cell Biol. 41, 1245–1248 [DOI] [PubMed] [Google Scholar]
- 56. Tybulewicz V. L. (2005) Vav-family proteins in T-cell signalling. Curr. Opin. Immunol. 17, 267–274 [DOI] [PubMed] [Google Scholar]
- 57. Yohe M. E., Rossman K., Sondek J. (2008) Role of the C-terminal SH3 domain and N-terminal tyrosine phosphorylation in regulation of Tim and related Dbl-family proteins. Biochemistry 47, 6827–6839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zheng M., Cierpicki T., Momotani K., Artamonov M. V., Derewenda U., Bushweller J. H., Somlyo A. V., Derewenda Z. S. (2009) On the mechanism of autoinhibition of the RhoA-specific nucleotide exchange factor PDZRhoGEF. BMC Struct. Biol. 9, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chen Z., Guo L., Sprang S. R., Sternweis P. C. (2011) Modulation of a GEF switch: autoinhibition of the intrinsic guanine nucleotide exchange activity of p115-RhoGEF. Protein Sci. 20, 107–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Suzuki N., Tsumoto K., Hajicek N., Daigo K., Tokita R., Minami S., Kodama T., Hamakubo T., Kozasa T. (2009) Activation of leukemia-associated RhoGEF by Gα13 with significant conformational rearrangements in the interface. J. Biol. Chem. 284, 5000–5009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Vanni C., Parodi A., Mancini P., Visco V., Ottaviano C., Torrisi M. R., Eva A. (2004) Phosphorylation-independent membrane relocalization of ezrin following association with Dbl in vivo. Oncogene 23, 4098–4106 [DOI] [PubMed] [Google Scholar]