

Background: AS101, a novel small tellurium compound, is a potent anti-inflammatory/anti-apoptotic immunomodulator.
Results: AS101 prevents leukocyte migration and protects the mucosal barrier in murine colitis.
Conclusion: AS101 exerts multifunctional anti-inflammatory/anti-apoptotic activity in experimental inflammatory bowel diseases.
Significance: This is the first report showing significant effect of tellurium-based compound for the treatment of inflammatory bowel diseases.
Keywords: Cytokine, Inflammation, Inflammatory Bowel Disease (IBD), Innate Immunity, Intestine, AS101, DSS, Interleukin 17, Interleukin 1beta, Immunomodulator
Abstract
Inflammatory bowel diseases (IBDs) are a group of idiopathic, chronic immune-mediated diseases characterized by an aberrant immune response, including imbalances of inflammatory cytokine production and activated innate and adaptive immunity. Selective blockade of leukocyte migration into the gut is a promising strategy for the treatment of IBD. This study explored the effect of the immunomodulating tellurium compound ammonium trichloro (dioxoethylene-o,o′) tellurate (AS101) on dextran sodium sulfate (DSS)-induced murine colitis. Both oral and intraperitoneal administration of AS101 significantly reduced clinical manifestations of IBD. Colonic inflammatory cytokine levels (IL-17 and IL-1β) were significantly down-regulated by AS101 treatment, whereas IFN-γ was not affected. Neutrophil and α4β7+ macrophage migration into the tissue was inhibited by AS101 treatment. Adhesion of mesenteric lymph node cells to mucosal addressin cell adhesion molecule (MAdCAM-1), the ligand for α4β7 integrin, was blocked by AS101 treatment both in vitro and in vivo. DSS-induced destruction of colonic epithelial barrier/integrity was prevented by AS101, via up-regulation of colonic glial-derived neurotrophic factor, which was found previously to regulate the intestinal epithelial barrier through activation of the PI3K/AKT pathway. Indeed, the up-regulation of glial-derived neurotrophic factor by AS101 was associated with increased levels of colonic pAKT and BCL-2 and decreased levels of BAX. Furthermore, AS101 treatment reduced colonic permeability to Evans blue and decreased colonic TUNEL+ cells. Our data revealed multifunctional activities of AS101 in the DSS-induced colitis model via anti-inflammatory and anti-apoptotic properties. We suggest that treatment with the small, nontoxic molecule AS101 may be an effective early therapeutic approach for controlling human IBD.
Introduction
Crohn disease and ulcerative colitis are the two major forms of chronic inflammatory bowel disease (IBD).2 Studies in humans and in experimental models of IBD have revealed impaired mucosal barrier function, activated innate immunity, altered production of Th1 and Th2 cytokines, and the activation of CD4+ T cells in the pathogenesis of IBD (1–3). T regulatory cells play an essential role in the maintenance of intestinal immune homeostasis and were also shown to be capable of preventing and reversing established colitis (4). The etiologies of these diseases remain unclear. The most widely held hypothesis regarding the pathogenesis of IBD is that aberrant activation or defective down-regulation of innate immunity and overly aggressive acquired (T cell) immune responses to a subset of commensal enteric bacteria develop in genetically susceptible hosts, and environmental factors precipitate the onset or reactivation of disease (1, 2, 5).
Cytokines provide key signals in the intestinal immune system and were shown to have a central role in the pathophysiology of IBD (6). It is evident now that both innate cell populations (e.g. γδ T cells, LTi-like cells, and monocytes/macrophages) and adaptive T cells (Th17 cells) can produce IL-17, which has a crucial role in inflammatory and autoimmune diseases (7–10). IL-17 was found to be elevated in peripheral blood and intestinal tissue of IBD patients (11, 12). In addition to IL-17, IL-1 is important in the pathogenesis of IBD because of its pro-inflammatory activities. Peripheral blood mononuclear cells obtained from patients with Crohn disease were shown to produce high levels of IL-1 in vitro compared with normal control cells (13), and enhanced secretion of IL-1β was shown in colonic biopsies from patients with inflammatory bowel disease (12, 14). Based on these findings, several cytokine-based therapies have been developed and tested for the treatment of IBD patients (6, 15).
Macrophages have been implicated in the pathogenesis of a variety of chronic and autoimmune diseases including IBD. The classically activated macrophages are key producers of many cytokines (e.g. IL-1, IL-6, and TNFα) and reactive metabolites of oxygen and nitrogen (e.g. nitric oxide) that have been implicated in the development of IBD (16, 17).
Feeding mice with polymeric dextran sodium sulfate (DSS) in their drinking water induces acute or chronic colitis characterized by bloody diarrhea, ulceration, loss of body weight, and infiltration with granulocytes or mononuclear cells, reflecting IBD symptoms (18). This widely used murine colitis model is helpful for studying the effect of novel therapeutic agents for the management of IBD (19).
Integrins are transmembrane cell adhesion receptors composed of noncovalently associated α and β subunits that bind to cell surface ligands, soluble ligands, and extracellular matrix proteins (20). These adhesive interactions are essential for leukocyte recirculation, migration into inflammatory sites, recognition of foreign antigens, and survival and proliferation (21–23). Circulating T cells that bear α4β7 integrins bind to mucosal addressin cell adhesion molecule (MAdCAM), which is constitutively expressed on high endothelial venules of Peyer's patches and mesenteric lymph nodes (MLNs), as well as on postcapillary venules of gut lamina propia (LP) (2, 24). MAdCAM-1 expression is up-regulated on inflamed venules in chronic inflammatory diseases such as in IBD, diabetes, chronic relapsing experimental autoimmune encephalomyelitis, etc. Of note, α4β7 integrin is also expressed on stimulated monocytes and macrophages (25). Monoclonal antibody to integrin-α4 (natalizumab), which binds both integrin-α4β7 (the MAdCAM-1 ligand) and integrin-α4β1 (the vascular cell adhesion molecule-1 ligand), is effective in treating Crohn disease (26), whereas a humanized anti-integrin-α4β7 antibody has been used to treat active ulcerative colitis (27).
The neurotrophin glial-derived neurotrophic factor (GDNF) has a strong effect on neurite outgrowth and differentiation and is able to protect neurons from apoptosis under various conditions. Interestingly, it has been shown that ablation of enteric glia leads to a fulminant hemorrhagic jejunoileitis in mice (28), suggesting a central role of the enteric glia in the maintenance of gut mucosal integrity. GDNF is up-regulated in IBD and has strong anti-apoptotic properties in colonic epithelial cells via activation of MAPK and PI3K/AKT pathways (29).
AS101 is a small nontoxic tellurium IV compound (Fig. 1) currently being evaluated in phase II clinical trials in psoriasis patients (unpublished data) and in prevention of bone marrow toxicity induced by chemotherapy in cancer patients (30). This compound is a potent immunomodulator (in vitro and in vivo) with a variety of potential therapeutic applications (31–33). Accumulated evidence suggests that much of the biological activity of tellurium compounds is directly related to their specific chemical interactions with endogenous thiols. If the reacting thiol is a cysteine residue, the reaction product may alter the biological activity of the target protein (34). The Te(IV)-thiol chemical bond may lead to conformational change or disulfide bond formation, possibly resulting in a loss of biological activity, if the thiol residue is essential for a particular function. Indeed, we demonstrated that AS101 specifically inactivates cysteine proteases while exhibiting no effect on the other families of serine-, aspartic-, and metallo-proteases, in good agreement with the predictions of its unique Te (IV)-thiol chemistry. Furthermore, the proteolytic activity of the inactivated cysteine proteases could be recovered by reducing agents such as NaBH4, further supporting the suggestion that the inactivation process involves oxidation of the catalytic thiol to a disulfide (34). In light of its chemical activities, AS101 has been shown to exert anti-inflammatory and anti-apoptotic effects in different in vivo models (35–38), through inhibition of caspase 1, 3, and 8 activities and by its ability to induce GDNF production (38, 39).
FIGURE 1.
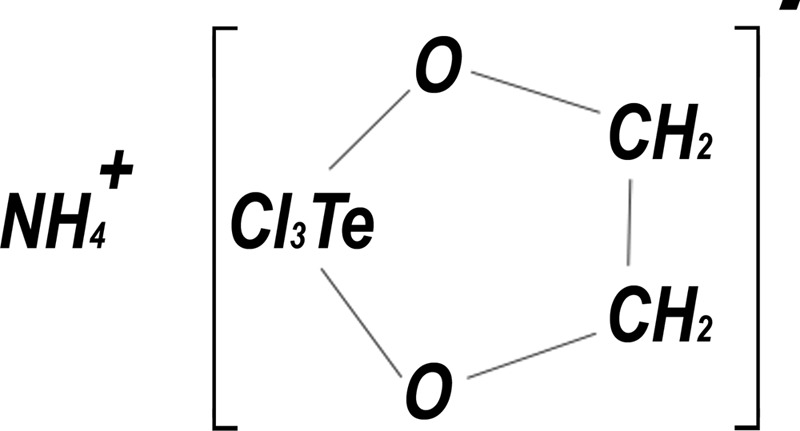
The chemical structure of AS101.
In the present study, we tested the effect of AS101 on a DSS-induced murine colitis model. Our findings revealed anti-inflammatory and anti-apoptotic properties of AS101 through the down-regulation of colonic inflammatory cytokine levels, prevention of inflammatory cell migration into the tissue, and inhibition of colonic apoptotic processes.
EXPERIMENTAL PROCEDURES
Mice
Male C57BL/6 mice, 8–12 weeks old, were purchased from Harlan Laboratories (Jerusalem, Israel). The mice were kept in a specific pathogen-free environment and fed standard pellet diet and tap water. The mice were allowed to acclimate for 7 days before the experiments. Animal experiments were performed in accordance with institutional protocols, and experiments were approved by the institutional animal care and use committee.
Induction of DSS Colitis
The mice were divided into groups with identical average body weight. DSS (molecular weight, 36,000–50,000; MP Biomedicals, Eschwege, Germany) was added to tap water at a concentration of 3.5% (w/v). The mice were given DSS solution for 7 days, followed by 5 days of regular water. Fresh DSS solution was prepared every other day.
AS101
AS101 was supplied in a solution of PBS (pH 7.4) and maintained at 4 °C. Before use, AS101 was diluted in PBS, and the indicated doses were used.
AS101 Treatment
AS101 was administrated either by intraperitoneal injection or by oral administration. In the injection treatment, mice were given DSS and injected daily intraperitoneally with PBS (DSS+PBS) or with AS101 (10 μg/mouse per injection) either concomitantly with the initiation of DSS supplementation (DSS+AS(d0)) or 2 days later (DSS+AS(d2)). The control mice received regular tap water only and were injected daily with intraperitoneal injections of PBS (PBS). For the oral treatment, the mice were given DSS and were treated by oral administration of PBS (DSS+PBS) or AS101 (100 μg/mouse) given concomitantly with DSS (DSS+AS(d0)) by 18 gauge feeding needle. Control mice received oral administration of PBS alone (PBS).
Assessment of DSS Colitis Severity
The mice were monitored for body weight, stool consistency, rectal bleeding, and fecal occult blood. Fecal occult blood was examined by Hemoccult test (Beckman Coulter Inc, Fullerton CA). Bleeding score included the fecal occult blood test, and visible blood from the rectum and was graded as 0 = negative, 2 = positive by Hemoccult, and 3 = visible blood. Fecal pellets were given a stool consistency score of 0 = normal fecal pellet, 1 = slightly loose stool, 2 = loose stool, and 3 = diarrhea. At necropsy, the colons were cut out and measured to determine colon length. 1 cm of the distal colon was taken for histology or immunohistochemical staining. Another 1–2 cm of the distal colon was used for protein analysis.
Histology
Tissue sections from the distal colon were fixed in 4% buffered formaldehyde. Paraffin-embedded sections were stained with hematoxylin and eosin. Histopathology scoring was performed by a pathologist in a blinded fashion and graded according to the following parameters: severity of inflammation (0–3; none, slight, moderate, and severe), depth of injury (0–3; none, mucosal, mucosal and submucosal, and transmural), and crypt damage (0–4; none, basal damage, severe basal damage, only surface epithelium intact, and loss of entire crypt and epithelium). The score of all the parameters was summed to a maximum score of 10 per mouse and calculated as an average histopathology score per group. Histopathology scoring was performed on sections prepared from the experiment testing AS101 treatment by intraperitoneal injection.
Immunohistochemistry
Immunohistochemical detection of IL-17 and CD4 was performed on paraffin-embedded slides of distal colon sections. Deparaffinized sections were incubated overnight (4 °C) with rabbit polyclonal antibody against human IL-17 (Santa Cruz) or mouse monoclonal antibody against mouse CD4 (Abcam, Cambridge, MA). Subsequently, incubation with secondary antibodies and substrate addition was performed using a commercial staining kit (Vectastain ABC kit) following the manufacturer's instructions. Sections were counterstained with hematoxylin. For in situ detection of apoptosis, the terminal deoxynucleotidyltransferase mediated dUTP nick end-labeling (TUNEL) method (Apoptag kit; Millipore) was used.
Immunofluorescence
Paraffin tissue sections were deparaffinized and rehydrated followed by heat-induced antigen retrieval using boiling 0.01 m citrate buffer (pH 6.0) for 10 min in a microwave. The sections were then blocked for 30 min using a blocking buffer (1% BSA and 0.5% Triton in PBS). Double staining was performed at 4 °C overnight using a mixture of mouse anti-CD68 (Abcam) and rabbit anti-ITGB7 (Proteintech) primary antibodies. Secondary antibodies were a mixture of Alexa 594 (anti-mouse) and Alexa 488 (anti-rabbit) conjugates (Molecular Probes). Nuclei were counterstained using Hoechst.
Tissue Myeloperoxidase (MPO) Activity
MPO was measured in tissue from distal colon. Samples were rinsed with cold PBS, blotted dry, and immediately frozen in liquid nitrogen. Samples were stored at −80 °C until assayed for MPO activity using the ο-dianisidine method (40). Tissue samples were thawed and weighed. The samples were then suspended (5% w/v) in 50 mm potassium buffer phosphate (pH 6) containing 0.5% hexadecyltrimethylammonium bromide (0.1 g/20 ml of potassium phosphate) and homogenized. Samples were sonicated for 30 s (15 watts) and then centrifuged at 13,000 rpm for 25 min at 4 °C. The reaction was initiated by incubating the supernatant (20 μl) at 20 °C for 20 min with 50 mm potassium phosphate buffer (180 μl) containing 1 mm ο-dianisidine dihydrochloride and 5 × 10−4% H2O2. The change in the absorbance was read every 30 s by a microplate reader (Bio-Rad). MPO activity was expressed as the amount of enzyme necessary to produce a 1 unit change in the absorbance per min per g of tissue.
Quantification of Cytokine and GDNF Levels
An IL-17/IL-1β ELISA kit (R&D Systems) and IFN-γ ELISA kit (Diaclone) were used for quantitative cytokine measurements in mouse colonic lysates. GDNF concentrations in colonic lysates were measured by a commercial GDNF ELISA kit according to the manufacturer's instructions (GDNF Emax ImmunoAssay system kit; Promega).
Protein Isolation and Western Blotting
Colon tissues were suspended in ice-cold lysis buffer containing 50 mm Tris (pH 7.5), 150 mm NaCl, 10% glycerol, 1% Triton X-100, 1 mm EDTA, 1 mm PMSF, 1 mm sodium vanadate, 0.1% protease inhibitor mixture (Calbiochem) for 30 min on ice and centrifuged at 13,000 rpm for 20 min. Tissue lysates were boiled for 5 min and electrophoresed on SDS-PAGE, and membranes were incubated with anti-Bax, anti-BCL2 (Santa-Cruz Biotechnology), anti-pAKT (Cell Signaling), anti-iNOS (Santa-Cruz Biotechnology), and anti-Actin (Sigma). Blots were developed using horseradish peroxidase-conjugated secondary antibodies and the ECL detection system (Pierce).
Adhesion Assay of MLN Cells to MAdCAM-1
A 96-well plate was coated with 80 μl/well of 5μg/ml recombinant mouse MAdCAM-1 Fc chimera (R&D Systems). For the in vitro assay, control wells were coated with 2.5% BSA (Sigma). The plate was incubated overnight at 4 °C. Then the wells were washed two times with 150–200 μl of PBS and blocked with 80 μl of 2.5% BSA for 1 h in the incubator at 37 °C. The wells were washed again two times with 150–200 μl of PBS. Next, 0.5 × 106 MLN cells/100 μl from five DSS-treated mice were seeded onto each well, and 0.1, 0.5, or 1 μg/ml AS101 was added into MAdCAM-1-coated wells. For the in vivo experiment, 0.5 × 106 MLN cells/100 μl from each group (n = 5) were seeded on the MAdCAM-1-coated wells. After 2 h, the wells were washed twice with 150–200 μl of PBS to remove the unbound cells. Then each well was loaded with 100 μl of RPMI + 50 μl of tetrazolium salt, XTT solution (Biological Industries), and the plate was incubated overnight in the incubator at 37 °C. Then the plate was read in a microplate reader (Bio-Rad) at a wavelength of 450 nm.
Measurement of Colonic Mucosal Permeability
Colonic mucosal permeability was assayed using a modification of the method described by Kitajima et al. (41). The mice were sacrificed, and the colon of each mouse was removed. The dissected colon was opened longitudinally and stained with 0.1 ml of 0.02% (w/v) Evans blue (Sigma) in PBS. After 120 min of exposure to Evans blue, the colon was rinsed three times in 6 mm acetylcysteine to remove any unabsorbed dye. The colon was then dried, weighed, and incubated with 1 ml of formamide for 12 h to elute the Evans blue. Colorimetric measurements of the solvent were performed in a microplate reader (Bio-Rad) at a wavelength of 655 nm, and permeability was calculated as μg of Evans blue/mg of colonic tissue based on the standard curve of Evans blue in formamide.
Isolation of Lamina Propia Mononuclear Cells from the Colon
The cells from intestinal LP were isolated as described previously (42) with some modifications. Briefly, the colons were flushed with PBS/BSA buffer, cut into small pieces, and incubated twice in PBS/FCS buffer containing 5 mm EDTA at 37 °C for 15 min in a shaking incubator. Then the LP cells were isolated by digesting intestinal tissue with collagenase type IV (Sigma-Aldrich) in RPMI/FCS/15 mm Hepes buffer for 1 h at 37 °C in a shaking incubator. After 1 h, the released LP cells were passed through a cell strainer (100 μm) and kept in PBS/BSA/EDTA buffer at 4 °C. Then the cells were further purified using Percoll (GE Healthcare Life Sciences), counted, and stained with relevant antibodies for subsequent flow cytometry analysis.
Flow Cytometry Analysis
Tregs LP lymphocytes were detected using the following antibodies: anti-mouse CD3-Pacific Blue, anti-mouse CD4-FITC, anti-mouse CD25-PE, and anti-mouse/rat Foxp3-APC (eBioscience). Briefly, LP cells were preblocked with anti-mouse CD16/CD32 FC blocker for 20 min at 4 °C, washed with FACS buffer, and stained with anti-mouse indicated antibodies for 30 min at 4 °C. The cells were washed again and incubated overnight with fixation/permeabilization reagent (eBioscience). Then cells were washed with permeabilization buffer and stained with anti-mouse/rat Foxp3 antibody for 30 min at 4 °C. The cells were analyzed using flow cytometry (FACSCanto II; Becton Dickinson). Data sets were analyzed using FlowJo software (Tree Star, Inc.). Tregs were designed as CD3+ CD4+ CD25+ Foxp3+ cells.
Statistical Analysis
The results are expressed as means ± S.E. p < 0.05 was considered statistically significant. Differences in clinical symptoms between groups were analyzed using ANOVA repeated measures. Differences between groups in colon length, cytokine levels, histopathology score, adhesion of MLN, and colonic permeability to Evans blue were analyzed using one-way ANOVA test. Differences between groups in IL-1β levels, MPO activity, and GDNF levels were analyzed using Student's t test.
RESULTS
AS101 Reduces Clinical Symptoms of DSS-induced Murine Colitis
Initially, we tested the effect of AS101 on clinical symptoms of diseased mice, reflecting manifestations of human IBD. Because the severity of clinical symptoms in our protocol (e.g. intestinal bleeding) started to increase 2 days after DSS administration, together with the fact that the onset of the inflammatory and apoptotic process in the colon of DSS-treated mice starts 24 h after DSS administration (43, 44), we decided to check the effect of AS101 compound given 2 days after DSS administration.
During the first 5 days of DSS administration, there were no differences in the total average body weight between the groups. Then the average body weight of the DSS+PBS group started to decline significantly from day 6 until the end of the experiment (p < 0.01). AS101, given concomitantly with DSS or starting 2 days later, significantly restored the decreased body weight seen in DSS+PBS-treated mice (Fig. 2A). Furthermore, both AS101-treated groups revealed a significantly reduced bleeding score (Fig. 2B) and stool consistency score (Fig. 2C) versus the DSS+PBS group over the course of the experiment. Colon length, which was greatly reduced in diseased mice, was significantly normalized in DSS+AS101-treated groups (Fig. 2, D and E). Importantly, in the oral treatment experiment, we found the similar efficacy in reducing clinical symptoms (Fig. 3). AS101, given concomitantly with DSS, significantly normalized the decreased body weight seen in DSS+PBS-treated mice (Fig. 3A) and reduced bleeding score versus the DSS+PBS group (Fig. 3B). Colonic shortening induced by DSS was also significantly restored by AS101 treatment (Fig. 3, C and D). These results demonstrate that AS101 reduces disease manifestations of DSS-induced murine colitis.
FIGURE 2.

AS101 injection reduces clinical symptoms of DSS-induced murine colitis. Colitis was induced by addition of 3.5% DSS to the drinking water of C57Bl/6 mice. AS101 (10 μg/mouse) was injected daily intraperitoneally from day 0 (DSS+AS(d0)) or from 2 days after DSS administration (DSS+AS(d2)). A, AS101 restores decreased body weight induced by colitis. Total average body weight of each group was monitored. *, p < 0.01 decrease versus other groups (analyzed on days 6–11 by ANOVA repeated measures). B, AS101 reduces intestinal bleeding induced by colitis. Intestinal bleeding was scored by Hemoccult test for the presence of occult blood and by visual observation of rectal bleeding. *, p < 0.001 increase versus other groups (analyzed by ANOVA repeated measures). C, AS101 reverses colitis-induced elevation of stool morphology score. *, p < 0.001 increase versus other groups (analyzed by ANOVA repeated measures). D and E, AS101 restores colon length shortened by colitis. Colons were removed and measured at necropsy (day 12). *, p < 0.001 increase versus DSS+PBS group (analyzed by one-way ANOVA test). The results shown are means ± S.E. (n = 4 for PBS; n = 8 for DSS+PBS; n = 6 for DSS+AS(d0); n = 10 for DSS+AS(d2)). For colon length, n = 7 for PBS; n = 18 for DSS+PBS; n = 17 for DSS+AS(d0); n = 10 for DSS+AS(d2).
FIGURE 3.

Oral administration of AS101 reduces clinical symptoms of DSS-induced murine colitis. Colitis was induced by addition of 3.5% DSS to the drinking water of C57Bl/6 mice. The mice were treated orally with daily administration of AS101 (100 μg/mouse), starting at day 0 (DSS+AS(d0)). The results shown are means ± S.E. of 10 mice per group. A, AS101 restores decreased body weight induced by colitis. Total body weight average of each group was monitored. *, p < 0.01 decrease versus other groups (analyzed by ANOVA repeated measures). B, AS101 reduces intestinal bleeding induced by colitis. Intestinal bleeding was scored by Hemoccult test for the presence of occult blood and by visual observation of rectal bleeding. *, p < 0.05 increase versus other groups (analyzed by ANOVA repeated measures). C and D, AS101 normalizes colon length shortened by colitis. Colons were removed and measured at necropsy (day 12). *, p < 0.001 increase versus DSS+PBS group (analyzed by one-way ANOVA test).
The Anti-inflammatory Role of AS101 on DSS-induced Murine Colitis
We next examined the immunomodulatory effect of AS101 on this model. The pro-inflammatory cytokines IL-17, IFN-γ, and IL-1β, which have a central role in the pathophysiology of IBD (6, 11–14, 45–49), were measured in the colon. The DSS+PBS group exhibited elevated levels of IL-17 as compared with control mice. Treatment with AS101, either at or after disease onset, significantly reduced colon IL-17 levels (Fig. 4A). To validate IL-17 inhibition upon AS101 treatment, we performed immunohistochemical analysis on paraffin-embedded tissue sections. As shown in Fig. 4B, IL-17 was significantly up-regulated in DSS+PBS-treated mice as compared with control mice. The colons of DSS+AS101-treated mice exhibited normal levels of IL-17, similar to those in control mice. Additionally, colonic IL-1β was significantly reduced by oral treatment of AS101 as compared with diseased mice (Fig. 4C). IFN-γ was up-regulated in the DSS+PBS group as compared with the control mice, although AS101 treatment, either at or after disease onset, did not significantly affect its level (data not shown).
FIGURE 4.

Treatment with AS101 reduces colonic levels of inflammatory cytokines. Colitis was induced by addition of 3.5% DSS to the drinking water of C57Bl/6 mice. AS101 (10 μg/mouse per injection) was injected daily intraperitoneally from day 0 (DSS+AS(d0)) or from 2 days after DSS administration (DSS+AS(d2)). At necropsy (day 12), 1–2 cm of the distal colon was removed and homogenized. Cytokine levels were measured by ELISA. The results shown are means ± S.E. A, treatment with AS101 either at or after disease onset significantly reduced colon IL-17 levels. *, p < 0.01 decrease versus DSS+PBS group (analyzed by one-way ANOVA test) (n = 7 for PBS; n = 12 for DSS+PBS or DSS+AS(d0); n = 10 for DSS+AS(d2)). B, immunohistochemical detection of IL-17 levels in distal colon. At necropsy (day 7), 1–2 cm of the distal colon was removed and fixed in 4% buffered formaldehyde. Immunohistochemical analysis was performed in paraffin-embedded slides. The pictures shown are representative of three mice per group. Scale bars indicate 80 μm. C, colonic IL-1β is significantly reduced by oral administration of AS101. *, p < 0.02 decrease versus DSS+PBS group (analyzed by Student's t test). The results shown are means ± S.E. of four mice per group.
Histopathological examination of the distal colon, at day 12, revealed granulated tissue exhibiting destruction of the crypt structure in DSS+PBS-treated mice. Furthermore, massive mononuclear cell (lymphocytes and macrophages) infiltration into the mucosa and submucosa was found. In comparison, the colons of DSS+AS101-treated mice (either by injection or oral treatment) exhibited normal appearance as in control mice (Fig. 5, A and B). Histopathology score, which includes severity of inflammation and tissue damage examination, was found to be significantly higher in DSS+PBS group versus the control mice. In comparison, the average histopathology score of DSS+AS101-treated mice was significantly reduced and similar to that of control animals (Fig. 5C).
FIGURE 5.
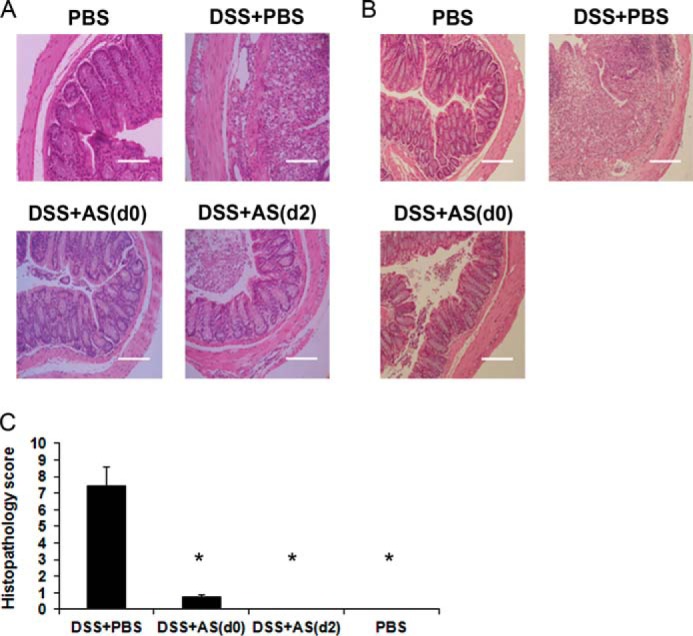
Histopathological examination of the distal colon. Colitis was induced by addition of 3.5% DSS to the drinking water of C57Bl/6 mice. At necropsy (day 12), 1–2 cm of the distal colon was removed and fixed in 4% buffered formaldehyde. Paraffin-embedded sections were stained with hematoxylin and eosin. A, histopathological examination of colons from AS101 injection treatment. AS101 (10 μg/mouse per injection) was injected intraperitoneally daily from day 0 or starting 2 days after DSS administration. The pictures shown are representative of three mice per group. Scale bars indicate 80 μm. B, histopathological examination of colons following oral AS101 treatment. The mice were treated orally with daily administration of AS101 (100 μg/mouse) starting at day 0. The pictures shown are representative of three mice per group. Scale bars indicate 160 μm. C, AS101 treatment (injection) significantly reduces histopathology score. Histopathology score was determined as described under “Experimental Procedures.” *, p < 0.01 decrease versus DSS+PBS group. The results shown are means ± S.E. (n = 2 for PBS or DSS+AS(d2), n = 7 for DSS+PBS, n = 8 for DSS+AS(d0)).
Histological features after 7 days of DSS administration showed destruction of the crypt structure and massive neutrophil infiltration into the mucosa and submucosa of DSS+PBS-treated mice. In comparison, the colons of DSS+AS101-treated mice exhibited a normal appearance (Fig. 6A). To confirm neutrophil infiltration into the colon, we examined colonic MPO activity, which directly reflects the level of neutrophil infiltration in tissues (40). High levels of MPO activity were measured in DSS+PBS-treated mice. MPO activity in AS101-treated mice was significantly reduced to normal levels, similar to those observed in control mice (Fig. 6B).
FIGURE 6.
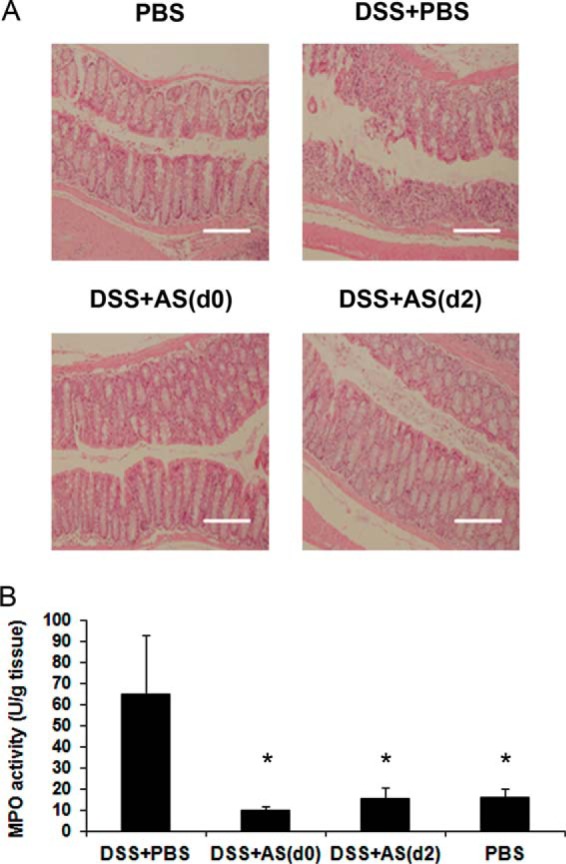
AS101 prevents neutrophil migration into the colon. Colitis was induced by addition of 3.5% DSS to the drinking water of C57Bl/6 mice. AS101 (10 μg/mouse per injection) was injected from day 0 or starting 2 days after DSS administration. At necropsy (day 7), 2 cm of the distal colon was removed and cut longitudinally. One section was fixed in 4% buffered formaldehyde and stained with hematoxylin and eosin. The other section was kept at −80 °C until assay for MPO activity, as described under “Experimental Procedures.” A, histopathological examination of the distal colon. The pictures shown are representative of three mice per group. Scale bars indicate 160 μm. B, MPO activity in the distal colon. *, p < 0.05 decrease versus DSS+PBS group (analyzed by Students t test). The results shown are means ± S.E. (n = 8 for PBS; n = 7 for DSS+PBS; n = 8 for DSS+AS(d0); n = 9 for DSS+AS(d2)).
Recently, we found in our lab that AS101 inhibits the activity of VLA-4 (α4β1 integrin) by redox modulation based on its Te-thiol interaction. Considering the clinical importance of blockade of the gut homing receptor α4β7 in human IBD patients (27), we wished to examine whether AS101 may prevent the migration of α4β7+ innate cells into the tissue of DSS-treated mice. Immunofluorescence double staining using anti-CD68 and anti-α4β7 antibodies indicated increased colocalization of α4β7+ and CD68+ cells in the colons of DSS-treated mice in comparison to control mice. However, AS101 treatment inhibited the infiltration of α4β7+ CD68+ cells into the colon of DSS-treated mice (Fig. 7A).
FIGURE 7.

AS101 prevents the migration of α4β7+ macrophages into the colon. A, colitis was induced by addition of 3.5% DSS to the drinking water of C57Bl/6 mice. AS101 (10 μg/mouse per injection) was injected daily starting from day 0. At necropsy (day 12), 1–2 cm of the distal colon was removed and fixed in 4% buffered formaldehyde. Paraffin tissue sections were prepared, and slides were stained with anti-CD68 (red), anti-α4β7 (green), and Hoechst (blue). White circles indicate cells of ∼20-μm diameter (estimated diameter of macrophages) which were double-stained for CD68 and α4β7 (yellow). Representative pictures are shown (magnification, ×40). Scale bars indicate 40 μm. B, down-regulation of colonic iNOS levels by AS101 treatment. Proteins in colonic tissue homogenates were separated by SDS-PAGE and subjected to immunoblotting with antibodies to iNOS and actin-HRP as a control (n = 5 for PBS; n = 8 for DSS+PBS or DSS+AS(d0) groups).
Expression of MAdCAM-1 (the ligand for α4β7 integrin) is restricted to gut-associated lymphoid tissues, and its expression dramatically increases in IBD (24, 50). To determine the effect of AS101 on the functional activity of α4β7 integrin or its ligand, MAdCAM-1, we isolated MLN cells from DSS-treated mice and incubated them on MAdCAM-coated plates. As shown in Fig. 8A, MLN cells from DSS-treated mice revealed significantly increased adhesion to MAdCAM-1 versus BSA. In comparison, the addition of 0.5 or 1 μg/ml of AS101 in vitro significantly reduced the adhesion of MLN cells to MAdCAM-1. To verify this property of AS101 in vivo, we isolated MLN cells from all groups at the end of the experiment (day 10) and found that MLN cells from DSS-treated group revealed increased adhesion to MAdCAM-1 versus the control mice. In comparison, MLN cells from the AS101-treated group revealed the same adhesion to MAdCAM-1 as the control (Fig. 8B). These results demonstrated the ability of AS101 to interrupt the interaction between α4β7 and MAdCAM-1 and therefore to block diapedesis of innate inflammatory effectors cells from gut-associated lymphoid tissue or the blood into the tissue itself.
FIGURE 8.

AS101 prevents the adhesion of MLN cells to MAdCAM-1 both in vitro and in vivo. Colitis was induced by addition of 2% DSS to the drinking water of C57Bl/6 mice. A, MLN cells from DSS+PBS group (n = 5) were seeded for 2 h on MAdCAM-1 or 2.5% BSA-coated wells. 0.1–1 μg/ml AS101 was added into MAdCAM-1-coated wells. After 2 h, the wells were washed, and XTT assays were performed to evaluate the bound cells. #, p < 0.01 increase versus BSA group; *, p < 0.05 decrease versus MAdCAM group analyzed by one way ANOVA test. The results shown are means ± S.E. B, AS101 (10 μg/mouse) was injected daily from day 0. At day 10, MLN cells were isolated from all groups (n = 5) and seeded for 2 h on MAdCAM-1-coated plate. After 2 h, the wells were washed, and XTT assay was performed to quantitate the bound cells. #, p < 0.01 increase versus PBS group; *, p < 0.01 decrease versus DSS+PBS group analyzed by one way ANOVA test. The results shown are means ± S.E.
Several reports implicated reactive oxygen and nitrogen metabolites in the initiation and/or progression of IBD. Macrophages, together with neutrophils, may contribute to intestinal damage by releasing reactive metabolites of oxygen and nitrogen (51). As shown in Fig. 7B, high levels of iNOS were detected in the colon of DSS-treated mice as compared with the control. In comparison, the iNOS level of AS101-treated mice was reduced to normal, as in the control mice.
Interestingly, we found increased infiltration of CD4 positive cells into the mucosa of DSS-treated mice 7 days after DSS administration, which represents the acute phase of colitis. In comparison, the DSS+AS101-treated groups contained basal levels of CD4 positive cells that were similar to those of the control group (Fig. 9A). To find the effect of AS101 on colonic T regulatory cells, we isolated colonic lamina propia lymphocyte (LPL) and found that AS101 treatment increases the percentage of CD4+CD25+Foxp3+ T regulatory population versus the DSS+PBS-treated group (Fig. 9B). Collectively, these results indicate anti-inflammatory activity of AS101 in this model, as manifested by the down-regulation of colonic inflammatory cytokine levels and prevention of inflammatory cell migration into the tissue.
FIGURE 9.

AS101 prevents the migration of CD4+ cells into the colon and increases colonic CD4+CD25+Foxp3+ T regulatory cells. A, colitis was induced by addition of 3.5% DSS to the drinking water of C57Bl/6 mice. AS101 (10 μg/mouse per injection) was injected daily intraperitoneally from day 0 (DSS+AS(d0)) or from 2 days after DSS administration (DSS+AS(d2)). At necropsy (day 7), 1–2 cm of the distal colon was removed and fixed in 4% buffered formaldehyde. Immunohistochemical analysis was performed in paraffin-embedded slides. The pictures shown are representative of three mice per group. Scale bars indicate 160 μm. B, lamina propia lymphocytes cells were isolated from the colons of treated groups and stained for CD4+CD25+Foxp3+ T regulatory cells as described under “Experimental Procedures.” Dot plots of each group showing CD25+FOXP3+ LPL cells. (Plots are gated on CD3+CD4+ LPL cells.) Each plot is representative of three experiments.
Colonic Epithelial Barrier Function Is Preserved by AS101 Treatment
The neurotrophin, GDNF, was shown to have a central role in protecting mucosal barrier function in experimental IBD and in IBD patients via up-regulation of PI3K/AKT pathway (29, 52). Previously, we showed that AS101 induces GDNF production both in vitro and in vivo (38, 39). Here, we show that the DSS-treated group revealed significant down-regulation of colonic GDNF relative to the PBS group. In comparison, AS101 treatment, starting 2 days after initiation of disease, significantly increased colonic GDNF levels versus the DSS+PBS group (Fig. 10A). This result was correlated with up-regulation of colonic pAKT and BCL-2 and down-regulation of BAX following AS101 treatment versus the DSS+PBS group (Fig. 10B).
FIGURE 10.

AS101 treatment restores DSS-induced destruction of colonic epithelial barrier integrity. Colitis was induced by addition of 3.5% DSS to the drinking water of C57Bl/6 mice. At necropsy (day 12), 1–2 cm of the distal colon was removed and homogenized. A, AS101 treatment increases GDNF levels. GDNF was measured in colonic lysates. #, p < 0.05 increases versus DSS+PBS group; *, p < 0.02 increase versus DSS+PBS group (analyzed by Student's t test). The results shown are means ± S.E. (n = 3 for PBS; n = 6 for DSS+PBS; and n = 9 for DSS+AS(d2)). B, up-regulation of pAKT and BCL-2 and down-regulation of Bax upon AS101 treatment. Proteins in tissue homogenates were separated by SDS-PAGE and subjected to immunoblotting with antibodies to BCL2, pAKT, Bax, and actin-HRP as a control (n = 4 in each group). C, AS101 decreases colonic TUNEL positive cells. At necropsy (day 7), 1–2 cm of the distal colon was removed and fixed in 4% buffered formaldehyde. The pictures shown are representative of three mice per group. Scale bars indicate 80 μm. D, AS101 normalizes the DSS-induced permeability of colonic epithelial cells. Colonic permeability to Evans blue was performed as described under “Experimental Procedures.” #, p < 0.01 increase versus PBS group; *, p < 0.01 decrease versus DSS+PBS group analyzed by one way ANOVA. The results shown are means ± S.E. (n = 5 for PBS; n = 7 for DSS+PBS; and n = 8 for DSS+AS(d0)).
To validate the anti-apoptotic effect of AS101, we performed a TUNEL assay and found increased apoptotic cells in the surface epithelium of DSS-treated mice. In comparison, AS101-treated groups revealed almost no staining, similar to the control group (Fig. 10C). To verify AS101-mediated protection of mucosal integrity, we used the colonic Evans blue staining assay and found significantly increased colonic permeability to Evans blue in the DSS-treated group relative to the control group. However, intraperitoneal administration of AS101 significantly normalized the increased colonic permeability seen in DSS-treated mice (Fig. 10D). These results demonstrate the contribution of AS101 to reducing apoptosis in the colon and in maintaining epithelial barrier/integrity.
DISCUSSION
The gut specific homing receptor, α4β7 integrin, and its ligand, MAdCAM-1, have emerged as anti-adhesion therapeutic targets in IBD because of their central role in leukocyte trafficking from blood to the intestinal tissue (15, 24, 27). In rodents and humans, MAdCAM-1 is constitutively expressed by mucosal endothelial cells of PPs, MLNs, and LP of the small and large intestine (50). Its expression is up-regulated on inflamed venules in chronic inflammatory diseases such as in IBD, diabetes, chronic relapsing experimental autoimmune encephalomyelitis, and other pathologies. Monoclonal antibody to integrin-α4 (natalizumab), which binds both integrin-α4β7 (the MAdCAM1 ligand) and integrin-α4β1 (the vascular cell adhesion molecule-1 ligand), is effective in treating Crohn disease (26), whereas a humanized anti-integrin-α4β7 antibody has been used to treat active ulcerative colitis (27). Therefore, interrupting the α4β7-MAdCAM-1 interaction in the MLN upon AS101 treatment (both in vitro and in vivo) and the ability of AS101 to block the migration of α4β7+ inflammatory cells into the colon further demonstrates the clinical importance of this interaction.
Accumulating data have suggested the role of IL-17/Th17 in inflammatory and autoimmune diseases in general, and in IBD in particular (10, 47, 48, 53). IL-17 and Th17 cells were both found to be elevated in peripheral blood and intestinal tissue of IBD patients (11, 12). The activation of the IL-23/IL-17 axis was found to play a fundamental role in the pathogenesis of IBD (47). Experimental IBD revealed that during innate immune-mediated intestinal inflammation, increased IL-17 expression is observed in neutrophils and monocytic cells isolated from the intestinal lamina propia (54). Of note, in human IBD patients, the increased expression of IL-17 in the inflamed mucosa was attributed to both CD68+ monocytes/macrophages and T cells (11). We assume that the reduction of colonic IL-17 by AS101 treatment may be a consequence of the inhibition of the migration of CD68+ or CD4+ (like Th17) cells into the colon of DSS-treated mice by AS101.
The DSS colitis model is particularly useful to study the contribution of innate immune mechanism of colitis. Interestingly, we found enhanced CD4+ cells in the lamina propia of the diseased mice, 7 days after DSS administration, which represents the acute colitis phase. Our results correlate with recent studies indicating that CD4+ T cells become activated in the periphery in the DSS-induced colitis model and home to the colon during the first 3–7 days of DSS administration (55, 56). Furthermore, Hall et al. (43) showed that adaptive immune populations such as CD3+ and B cells are also activated in the late acute phase of DSS-induced colitis. We show that AS101 treatment, starting either at or after disease onset, prevents the infiltration of CD4+ cells into the colon (Fig. 9). This inhibition of CD4+ T cell migration into the colon by AS101 treatment probably prevents the aggravation of the disease by the adaptive immunity system as seen in AS101-treated groups, 12 days after DSS administration, which represents the progression to chronic inflammation phase. Because T- and B-cell deficient C.B-17scid or Rag1−/− mice also develop severe colitis (57), the adaptive immune system does not play a major part, at least in the acute phase in this model, and appears to be a secondary response, enhancing the inflammatory process. Therefore, we assume that the main activity of AS101 in our model is mediated through the blocking of innate cell (macrophages and neutrophils) migration into the colon and the consequent prevention of the initiation/development of the inflammatory process.
IL-1β is produced predominantly by stimulated macrophages and monocytes, and its enhanced production has been detected both in experimental IBD and in IBD patients (12–14, 45). Our data show that colonic IL-1β is significantly down-regulated by oral AS101 treatment (Fig. 4C). We previously showed that AS101 is a novel inhibitor of caspase-1 and that it reduces IL-1β levels both in vitro and in vivo (35, 36). Of note, Siegmund et al. (49) found that caspase-1 KO mice exhibit a reduction in clinical manifestations, histology score, and colonic cytokine production in acute or chronic DSS-induced colitis. Recently, IL-1 was shown to be a critical factor for the induction of innate IL-17-producing cells (7, 58) and in the induction of IL-17-producing antigen-specific T cells in experimental autoimmune encephalomyelitis (59). It should be noted that IL-17 can promote IL-1β production in autoimmunity or synergize with IL-1β to promote production of other cytokines and chemokines (60, 61). Therefore, our findings demonstrate the clinical importance of inhibition of these two deleterious cytokines (IL-1β and IL-17) by AS101 treatment. Of note, inflammatory cytokines such as IL-1β and TNF-α can induce expression of MAdCAM both in vitro and in vivo and thereby enhance the inflammatory cycle in the gut (62, 63). Thus, the anti-inflammatory activity of AS101 in our model through the down-regulation of colonic cytokines such as IL-1β may negatively regulate the function of MAdCAM in recruitment of leukocytes into the intestine. Recently it was found that IL-1β attenuates CD4+CD25+FoxP3+ regulatory T cell function (64). In light of this, the down-regulation of IL-1β by oral AS101 administration may increase the frequency of colonic CD4+CD25+FoxP3+ regulatory T cells in AS101-treated group as shown in Fig. 9B.
We also showed reduced MPO levels in the colon in response to AS101. These results suggest that AS101 reduces the degree of neutrophil infiltration in the tissue. IL-17 has been shown to induce chemokines (CXCL-8 and CXCL-1) and growth factors (G-CSF and GM-CSF) leading to augmented neutrophil accumulation as well as granulopoiesis (53). Thus, IL-17 inhibition (Fig. 4, A and B) by AS101 may explain the blockade of neutrophil accumulation in the colon. Collectively, it appears that AS101 mediates its anti-inflammatory activity by attenuating innate immune function by preventing of colonic diapedesis of innate cells (neutrophils and macrophages) and by down-regulating inflammatory cytokines (IL-1β and IL-17).
It is now becoming evident that aberrant epithelial barrier function plays a central role in the pathophysiology of IBD (2, 5). With increasing evidence of the involvement of the innate immune system and the intestinal epithelium in IBD, the therapeutic paradigm is also shifting from mere immunosuppression to the reinforcement of the intestinal barrier (65). In light of this, the findings that ablation of enteric glia leads to a fulminant hemorrhagic jejunoileitis (28) and that enterocolitis is also inducible by autoimmune targeting of glial cells (66) led to the understanding of the complex network of neurotrophins and neurofactors such as GDNF as an important players in the maintenance of mucosal integrity. GDNF was found to be up-regulated both in experimental IBD and in IBD patients. GDNF has strong anti-apoptotic activities in colonic epithelial cells, which depends on activation of MAPK and PI3K/AKT pathways (29, 52). One form of epithelial cell injury in inflamed colonic mucosa in ulcerative colitis is reported to involve apoptosis of these cells (67). DSS-induced colitis as a widely used model of ulcerative colitis has been shown to cause marked apoptosis of colonic epithelial cells and up-regulation of proapoptotic proteins like Fas/FasL and Bax (68). Our results show that the diseased mice revealed reduced levels of colonic GDNF versus control animals. We assume that this reduction is probably due to the strong DSS-induced destruction of the colonic tissue as seen in the histological staining of DSS+PBS group (Fig. 5, A and B). Importantly, AS101, which has been previously shown by us to induce GDNF production both in vitro and in vivo (38, 39), significantly increased colonic GDNF levels as compared with the diseased mice. This increase in GDNF levels was accompanied by up-regulation of colonic pAKT and BCL-2 and down-regulation of BAX, indicating the anti-apoptotic properties of GDNF. Furthermore, TUNEL staining revealed almost no staining in the AS101-treated groups as compared with the DSS+PBS group (Fig. 10C). Together with the finding that AS101 normalized the increased colonic permeability seen in DSS-treated mice, these results demonstrate the contribution of AS101 to reducing the apoptosis process in the colon and in protection of mucosal barrier integrity as seen in histological sections of DSS+AS101-treated groups, compared with DSS+PBS mice (Figs. 5, A and B, and 6A).
Most of the current medical treatments for IBD rely on nonspecific, anti-inflammatory, and immunosuppressive drugs. These treatments can cause severe side effects, including cytotoxicity and increased susceptibility to opportunistic infections (15, 69). Acute, subchronic and chronic toxicity studies of AS101 were conducted in rats and dogs and demonstrated that AS101 has a wide safety margin with severe toxicity observed only at doses 60–120 times higher than the equivalent doses proposed for clinical studies (70). At present, no severe adverse toxicological effects have been seen in the course of clinical trials (70, 71).
Our results reveal multifunctional effects of the nontoxic, tellurium compound AS101 in the DSS-induced colitis model via anti-inflammatory and anti-apoptotic activities, through the down-regulation of colonic cytokine levels (IL-17 and IL-1β) and by the blockade of leukocyte (neutrophils and macrophages) migration into the colon. AS101 mediates these activities at least in part by blocking the α4β7-MAdCAM-1 interaction. Furthermore, AS101 treatment protects epithelial barrier function by induction of the anti-apoptotic pathway, GDNF/AKT.
Several trials have shown that early treatment of Crohn disease with immunomodulators and anti-TNFα agents leads to a superior clinical outcome, including healing of the mucosa, compared with standard therapy alone (72, 73). Therefore, we suggest that the nontoxic, tellurium compound AS101, may be an effective early treatment for the management of IBD patients.
This work was supported in part by the Dr. Tovi Comet-Walerstein Cancer Research Chair, by the Dave and Florence Muskovitz Chair in Cancer Research, and the Jaime Lusinchi Research Institute in Applied Sciences.
- IBD
- inflammatory bowel diseases
- DSS
- dextran sodium sulfate
- MPO
- myeloperoxidase
- TUNEL
- terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling
- MAdCAM
- mucosal addressin cell adhesion molecule
- GDNF
- glial-derived neurotrophic factor
- iNOS
- inducible nitric-oxide synthase
- MLN
- mesenteric lymph node
- ANOVA
- analysis of variance.
REFERENCES
- 1. Podolsky D. K. (2002) Inflammatory bowel disease. N. Engl. J. Med. 347, 417–429 [DOI] [PubMed] [Google Scholar]
- 2. Sartor R. B. (2006) Mechanisms of disease: pathogenesis of Crohn's disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 3, 390–407 [DOI] [PubMed] [Google Scholar]
- 3. Blumberg R. S., Saubermann L. J., Strober W. (1999) Animal models of mucosal inflammation and their relation to human inflammatory bowel disease. Curr. Opin. Immunol. 11, 648–656 [DOI] [PubMed] [Google Scholar]
- 4. Mottet C., Uhlig H. H., Powrie F. (2003) Cutting edge: cure of colitis by CD4+CD25+ regulatory T cells. J. Immunol. 170, 3939–3943 [DOI] [PubMed] [Google Scholar]
- 5. Strober W., Fuss I., Mannon P. (2007) The fundamental basis of inflammatory bowel disease. J. Clin. Invest. 117, 514–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sanchez-Munoz F., Dominguez-Lopez A., Yamamoto-Furusho J. K. (2008) Role of cytokines in inflammatory bowel disease. World J. Gastroenterol. 14, 4280–4288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cua D. J., Tato C. M. (2010) Innate IL-17-producing cells: the sentinels of the immune system. Nat. Rev. Immunol. 10, 479–489 [DOI] [PubMed] [Google Scholar]
- 8. Kanai T., Mikami Y., Sujino T., Hisamatsu T., Hibi T. (2012) RORγt-dependent IL-17A-producing cells in the pathogenesis of intestinal inflammation. Mucosal Immunol. 5, 240–247 [DOI] [PubMed] [Google Scholar]
- 9. Mangan P. R., Harrington L. E., O'Quinn D. B., Helms W. S., Bullard D. C., Elson C. O., Hatton R. D., Wahl S. M., Schoeb T. R., Weaver C. T. (2006) Transforming growth factor-β induces development of the T(H)17 lineage. Nature 441, 231–234 [DOI] [PubMed] [Google Scholar]
- 10. Steinman L. (2010) Mixed results with modulation of TH-17 cells in human autoimmune diseases. Nat. Immunol. 11, 41–44 [DOI] [PubMed] [Google Scholar]
- 11. Fujino S., Andoh A., Bamba S., Ogawa A., Hata K., Araki Y., Bamba T., Fujiyama Y. (2003) Increased expression of interleukin 17 in inflammatory bowel disease. Gut 52, 65–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eastaff-Leung N., Mabarrack N., Barbour A., Cummins A., Barry S. (2010) Foxp3+ regulatory T cells, Th17 effector cells, and cytokine environment in inflammatory bowel disease. J. Clin. Immunol. 30, 80–89 [DOI] [PubMed] [Google Scholar]
- 13. Satsangi J., Wolstencroft R. A., Cason J., Ainley C. C., Dumonde D. C., Thompson R. P. (1987) Interleukin 1 in Crohn's disease. Clin. Exp. Immunol. 67, 594–605 [PMC free article] [PubMed] [Google Scholar]
- 14. Reinecker H. C., Steffen M., Witthoeft T., Pflueger I., Schreiber S., MacDermott R. P., Raedler A. (1993) Enhanced secretion of tumour necrosis factor-α, IL-6, and IL-1β by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn's disease. Clin. Exp. Immunol. 94, 174–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rutgeerts P., Vermeire S., Van Assche G. (2009) Biological therapies for inflammatory bowel diseases. Gastroenterology 136, 1182–1197 [DOI] [PubMed] [Google Scholar]
- 16. Mosser D. M., Edwards J. P. (2008) Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wynn T. A., Chawla A., Pollard J. W. (2013) Macrophage biology in development, homeostasis and disease. Nature 496, 445–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Okayasu I., Hatakeyama S., Yamada M., Ohkusa T., Inagaki Y., Nakaya R. (1990) A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 98, 694–702 [DOI] [PubMed] [Google Scholar]
- 19. Wirtz S., Neurath M. F. (2007) Mouse models of inflammatory bowel disease. Adv. Drug Deliv. Rev. 59, 1073–1083 [DOI] [PubMed] [Google Scholar]
- 20. Hynes R. O. (1992) Integrins: versatility, modulation, and signaling in cell adhesion. Cell 69, 11–25 [DOI] [PubMed] [Google Scholar]
- 21. Hynes R. O. (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110, 673–687 [DOI] [PubMed] [Google Scholar]
- 22. Kinashi T. (2005) Intracellular signalling controlling integrin activation in lymphocytes. Nat. Rev. Immunol. 5, 546–559 [DOI] [PubMed] [Google Scholar]
- 23. Pribila J. T., Quale A. C., Mueller K. L., Shimizu Y. (2004) Integrins and T cell-mediated immunity. Annu. Rev. Immunol. 22, 157–180 [DOI] [PubMed] [Google Scholar]
- 24. Gorfu G., Rivera-Nieves J., Ley K. (2009) Role of β7 integrins in intestinal lymphocyte homing and retention. Curr. Mol. Med. 9, 836–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang Y., Harrison J. E., Print C. G., Lehnert K., Sammar M., Lazarovits A., Krissansen G. W. (1996) Interaction of monocytoid cells with the mucosal addressin MAdCAM-1 via the integrins VLA-4 and LPAM-1. Immunol. Cell Biol. 74, 383–393 [DOI] [PubMed] [Google Scholar]
- 26. Ghosh S., Goldin E., Gordon F. H., Malchow H. A., Rask-Madsen J., Rutgeerts P., Vyhnálek P., Zádorová Z., Palmer T., Donoghue S., and Natalizumab Pan-European Study Group (2003) Natalizumab for active Crohn's disease. N. Engl. J. Med. 348, 24–32 [DOI] [PubMed] [Google Scholar]
- 27. Feagan B. G., Greenberg G. R., Wild G., Fedorak R. N., Paré P., McDonald J. W., Dubé R., Cohen A., Steinhart A. H., Landau S., Aguzzi R. A., Fox I. H., Vandervoort M. K. (2005) Treatment of ulcerative colitis with a humanized antibody to the α4β7 integrin. N. Engl. J. Med. 352, 2499–2507 [DOI] [PubMed] [Google Scholar]
- 28. Bush T. G., Savidge T. C., Freeman T. C., Cox H. J., Campbell E. A., Mucke L., Johnson M. H., Sofroniew M. V. (1998) Fulminant jejuno-ileitis following ablation of enteric glia in adult transgenic mice. Cell 93, 189–201 [DOI] [PubMed] [Google Scholar]
- 29. Steinkamp M., Geerling I., Seufferlein T., von Boyen G., Egger B., Grossmann J., Ludwig L., Adler G., Reinshagen M. (2003) Glial-derived neurotrophic factor regulates apoptosis in colonic epithelial cells. Gastroenterology 124, 1748–1757 [DOI] [PubMed] [Google Scholar]
- 30. Sredni B., Albeck M., Tichler T., Shani A., Shapira J., Bruderman I., Catane R., Kaufman B., Kalechman Y. (1995) Bone marrow-sparing and prevention of alopecia by AS101 in non-small-cell lung cancer patients treated with carboplatin and etoposide. J. Clin. Oncol. 13, 2342–2353 [DOI] [PubMed] [Google Scholar]
- 31. Sredni B., Caspi R. R., Klein A., Kalechman Y., Danziger Y., Ben Ya'akov M., Tamari T., Shalit F., Albeck M. (1987) A new immunomodulating compound (AS-101) with potential therapeutic application. Nature 330, 173–176 [DOI] [PubMed] [Google Scholar]
- 32. Sredni B., Xu R. H., Albeck M., Gafter U., Gal R., Shani A., Tichler T., Shapira J., Bruderman I., Catane R., Kaufman B., Whisnant J. K., Mettinger K. L., Kalechman Y. (1996) The protective role of the immunomodulator AS101 against chemotherapy-induced alopecia studies on human and animal models. Int. J. Cancer 65, 97–103 [DOI] [PubMed] [Google Scholar]
- 33. Sredni B., Gal R., Cohen I. J., Dazard J. E., Givol D., Gafter U., Motro B., Eliyahu S., Albeck M., Lander H. M., Kalechman Y. (2004) Hair growth induction by the Tellurium immunomodulator AS101: association with delayed terminal differentiation of follicular keratinocytes and ras-dependent up-regulation of KGF expression. FASEB J. 18, 400–402 [DOI] [PubMed] [Google Scholar]
- 34. Albeck A., Weitman H., Sredni B., Albeck M. (1998) Tellurium compounds: Selective inhibition of cysteine proteases and model reaction with thiols. Inorg. Chem. 37, 1704–1712 [Google Scholar]
- 35. Brodsky M., Yosef S., Galit R., Albeck M., Longo D. L., Albeck A., Sredni B. (2007) The synthetic tellurium compound, AS101, is a novel inhibitor of IL-1β converting enzyme. J. Interferon Cytokine Res. 27, 453–462 [DOI] [PubMed] [Google Scholar]
- 36. Brodsky M., Hirsh S., Albeck M., Sredni B. (2009) Resolution of inflammation-related apoptotic processes by the synthetic tellurium compound, AS101 following liver injury. J. Hepatol. 51, 491–503 [DOI] [PubMed] [Google Scholar]
- 37. Brodsky M., Halpert G., Albeck M., Sredni B. (2010) The anti-inflammatory effects of the tellurium redox modulating compound, AS101, are associated with regulation of NFkappaB signaling pathway and nitric oxide induction in macrophages. J. Inflamm. (Lond.) 7, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sredni B., Geffen-Aricha R., Duan W., Albeck M., Shalit F., Lander H. M., Kinor N., Sagi O., Albeck A., Yosef S., Brodsky M., Sredni-Kenigsbuch D., Sonino T., Longo D. L., Mattson M. P., Yadid G. (2007) Multifunctional tellurium molecule protects and restores dopaminergic neurons in Parkinson's disease models. FASEB J. 21, 1870–1883 [DOI] [PubMed] [Google Scholar]
- 39. Okun E., Saida H., Albeck M., Sredni B., Avtalion R. R. (2006) Upregulation of carp GDNF mRNA by the immunomodulator AS101. Dev. Comp. Immunol. 30, 441–446 [DOI] [PubMed] [Google Scholar]
- 40. Krawisz J. E., Sharon P., Stenson W. F. (1984) Quantitative assay for acute intestinal inflammation based on myeloperoxidase activity. Assessment of inflammation in rat and hamster models. Gastroenterology 87, 1344–1350 [PubMed] [Google Scholar]
- 41. Kitajima S., Takuma S., Morimoto M. (1999) Changes in colonic mucosal permeability in mouse colitis induced with dextran sulfate sodium. Exp. Anim. 48, 137–143 [DOI] [PubMed] [Google Scholar]
- 42. Weigmann B., Tubbe I., Seidel D., Nicolaev A., Becker C., Neurath M. F. (2007) Isolation and subsequent analysis of murine lamina propria mononuclear cells from colonic tissue. Nat. Protoc. 2, 2307–2311 [DOI] [PubMed] [Google Scholar]
- 43. Hall L. J., Faivre E., Quinlan A., Shanahan F., Nally K., Melgar S. (2011) Induction and activation of adaptive immune populations during acute and chronic phases of a murine model of experimental colitis. Dig. Dis. Sci. 56, 79–89 [DOI] [PubMed] [Google Scholar]
- 44. Qiu W., Wu B., Wang X., Buchanan M. E., Regueiro M. D., Hartman D. J., Schoen R. E., Yu J., Zhang L. (2011) PUMA-mediated intestinal epithelial apoptosis contributes to ulcerative colitis in humans and mice. J. Clin. Invest. 121, 1722–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Acosta-Rodriguez E. V., Napolitani G., Lanzavecchia A., Sallusto F. (2007) Interleukins 1β and 6 but not transforming growth factor-β are essential for the differentiation of interleukin 17-producing human T helper cells. Nat. Immunol. 8, 942–949 [DOI] [PubMed] [Google Scholar]
- 46. Fuss I. J., Neurath M., Boirivant M., Klein J. S., de la Motte C., Strong S. A., Fiocchi C., Strober W. (1996) Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn's disease LP cells manifest increased secretion of IFN-γ, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J. Immunol. 157, 1261–1270 [PubMed] [Google Scholar]
- 47. Hölttä V., Klemetti P., Sipponen T., Westerholm-Ormio M., Kociubinski G., Salo H., Räsänen L., Kolho K. L., Färkkilä M., Savilahti E., Vaarala O. (2008) IL-23/IL-17 immunity as a hallmark of Crohn's disease. Inflamm. Bowel Dis. 14, 1175–1184 [DOI] [PubMed] [Google Scholar]
- 48. Kleinschek M. A., Boniface K., Sadekova S., Grein J., Murphy E. E., Turner S. P., Raskin L., Desai B., Faubion W. A., de Waal Malefyt R., Pierce R. H., McClanahan T., Kastelein R. A. (2009) Circulating and gut-resident human Th17 cells express CD161 and promote intestinal inflammation. J. Exp. Med. 206, 525–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Siegmund B., Lehr H. A., Fantuzzi G., Dinarello C. A. (2001) IL-1β-converting enzyme (caspase-1) in intestinal inflammation. Proc. Natl. Acad. Sci. U.S.A. 98, 13249–13254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Briskin M., Winsor-Hines D., Shyjan A., Cochran N., Bloom S., Wilson J., McEvoy L. M., Butcher E. C., Kassam N., Mackay C. R., Newman W., Ringler D. J. (1997) Human mucosal addressin cell adhesion molecule-1 is preferentially expressed in intestinal tract and associated lymphoid tissue. Am. J. Pathol. 151, 97–110 [PMC free article] [PubMed] [Google Scholar]
- 51. Krieglstein C. F., Cerwinka W. H., Laroux F. S., Salter J. W., Russell J. M., Schuermann G., Grisham M. B., Ross C. R., Granger D. N. (2001) Regulation of murine intestinal inflammation by reactive metabolites of oxygen and nitrogen: divergent roles of superoxide and nitric oxide. J. Exp. Med. 194, 1207–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang D. K., He F. Q., Li T. K., Pang X. H., Cui de J., Xie Q., Huang X. L., Gan H. T. (2010) Glial-derived neurotrophic factor regulates intestinal epithelial barrier function and inflammation and is therapeutic for murine colitis. J. Pathol. 222, 213–222 [DOI] [PubMed] [Google Scholar]
- 53. Kolls J. K., Lindén A. (2004) Interleukin-17 family members and inflammation. Immunity 21, 467–476 [DOI] [PubMed] [Google Scholar]
- 54. Hue S., Ahern P., Buonocore S., Kullberg M. C., Cua D. J., McKenzie B. S., Powrie F., Maloy K. J. (2006) Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J. Exp. Med. 203, 2473–2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sund M., Xu L. L., Rahman A., Qian B. F., Hammarström M. L., Danielsson A. (2005) Reduced susceptibility to dextran sulphate sodium-induced colitis in the interleukin-2 heterozygous (IL-2) mouse. Immunology 114, 554–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Da Silva A. P., Pollett A., Rittling S. R., Denhardt D. T., Sodek J., Zohar R. (2006) Exacerbated tissue destruction in DSS-induced acute colitis of OPN-null mice is associated with downregulation of TNF-α expression and non-programmed cell death. J. Cell Physiol. 208, 629–639 [DOI] [PubMed] [Google Scholar]
- 57. Dieleman L. A., Ridwan B. U., Tennyson G. S., Beagley K. W., Bucy R. P., Elson C. O. (1994) Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology 107, 1643–1652 [DOI] [PubMed] [Google Scholar]
- 58. Sutton C. E., Mielke L. A., Mills K. H. (2012) IL-17-producing γδ T cells and innate lymphoid cells. Eur. J. Immunol. 42, 2221–2231 [DOI] [PubMed] [Google Scholar]
- 59. Sutton C., Brereton C., Keogh B., Mills K. H., Lavelle E. C. (2006) A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J. Exp. Med. 203, 1685–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Koenders M. I., Lubberts E., Oppers-Walgreen B., van den Bersselaar L., Helsen M. M., Di Padova F. E., Boots A. M., Gram H., Joosten L. A., van den Berg W. B. (2005) Blocking of interleukin-17 during reactivation of experimental arthritis prevents joint inflammation and bone erosion by decreasing RANKL and interleukin-1. Am. J. Pathol. 167, 141–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Koenders M. I., Kolls J. K., Oppers-Walgreen B., van den Bersselaar L., Joosten L. A., Schurr J. R., Schwarzenberger P., van den Berg W. B., Lubberts E. (2005) Interleukin-17 receptor deficiency results in impaired synovial expression of interleukin-1 and matrix metalloproteinases 3, 9, and 13 and prevents cartilage destruction during chronic reactivated streptococcal cell wall-induced arthritis. Arthritis Rheum. 52, 3239–3247 [DOI] [PubMed] [Google Scholar]
- 62. Ando T., Langley R. R., Wang Y., Jordan P. A., Minagar A., Alexander J. S., Jennings M. H. (2007) Inflammatory cytokines induce MAdCAM-1 in murine hepatic endothelial cells and mediate α4β7 integrin dependent lymphocyte endothelial adhesion in vitro. BMC Physiol. 7, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Connor E. M., Eppihimer M. J., Morise Z., Granger D. N., Grisham M. B. (1999) Expression of mucosal addressin cell adhesion molecule-1 (MAdCAM-1) in acute and chronic inflammation. J. Leukocyte Biol. 65, 349–355 [DOI] [PubMed] [Google Scholar]
- 64. O'Sullivan B. J., Thomas H. E., Pai S., Santamaria P., Iwakura Y., Steptoe R. J., Kay T. W., Thomas R. (2006) IL-1β breaks tolerance through expansion of CD25+ effector T cells. J. Immunol. 176, 7278–7287 [DOI] [PubMed] [Google Scholar]
- 65. Peyrin-Biroulet L., Desreumaux P., Sandborn W. J., Colombel J. F. (2008) Crohn's disease: beyond antagonists of tumour necrosis factor. Lancet 372, 67–81 [DOI] [PubMed] [Google Scholar]
- 66. Cornet A., Savidge T. C., Cabarrocas J., Deng W. L., Colombel J. F., Lassmann H., Desreumaux P., Liblau R. S. (2001) Enterocolitis induced by autoimmune targeting of enteric glial cells: a possible mechanism in Crohn's disease? Proc. Natl. Acad. Sci. U.S.A. 98, 13306–13311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Iwamoto M., Koji T., Makiyama K., Kobayashi N., Nakane P. K. (1996) Apoptosis of crypt epithelial cells in ulcerative colitis. J. Pathol. 180, 152–159 [DOI] [PubMed] [Google Scholar]
- 68. Vetuschi A., Latella G., Sferra R., Caprilli R., Gaudio E. (2002) Increased proliferation and apoptosis of colonic epithelial cells in dextran sulfate sodium-induced colitis in rats. Dig Dis. Sci. 47, 1447–1457 [DOI] [PubMed] [Google Scholar]
- 69. Grimm M. C. (2009) New and emerging therapies for inflammatory bowel diseases. J. Gastroenterol. Hepatol. 24, (Suppl. 3) S69–S74 [DOI] [PubMed] [Google Scholar]
- 70. Nyska A., Waner T., Pirak M., Albeck M., Sredni B. (1989) Toxicity study in rats of a tellurium based immunomodulating drug, AS-101: a potential drug for AIDS and cancer patients. Arch. Toxicol. 63, 386–393 [DOI] [PubMed] [Google Scholar]
- 71. Sredni B., Catane R., Shani A., Gezin A., Levi E., Schlezinger M., Kalechman Y., Michlin H., Shalit F., Rosenzajn L. A., Farbstein H., Albeck M. (1990) Phase I study of AS101 (anorganotellurium compound) in patients with advanced malignancies. In Recent Advances in Chemotherapy ((Rubinstein E., Adam D., eds) p. 851, E. Lewin-Epstein, Jerusalem [Google Scholar]
- 72. D'Haens G. R. (2010) Top-down therapy for IBD: rationale and requisite evidence. Nat. Rev. Gastroenterol. Hepatol. 7, 86–92 [DOI] [PubMed] [Google Scholar]
- 73. D'Haens G., Baert F., van Assche G., Caenepeel P., Vergauwe P., Tuynman H., De Vos M., van Deventer S., Stitt L., Donner A., Vermeire S., Van de Mierop F. J., Coche J. C., van der Woude J., Ochsenkühn T., van Bodegraven A. A., Van Hootegem P. P., Lambrecht G. L., Mana F., Rutgeerts P., Feagan B. G., Hommes D., Belgian Inflammatory Bowel Disease Research Group, and North-Holland Gut Club (2008) Early combined immunosuppression or conventional management in patients with newly diagnosed Crohn's disease: an open randomised trial. Lancet 371, 660–667 [DOI] [PubMed] [Google Scholar]