

Abstract

Allylating agents were explored for the asymmetric synthesis of α-allyl-α-aryl α-amino acids by tandem N-alkylation/π-allylation. Cross-metathesis of the tandem product was developed to provide allylic diversity not afforded in the parent reaction; the synthesis of homotyrosine and homoglutamate analogues was completed. Cyclic α-amino acid derivatives could be accessed by ring-closing metathesis presenting a viable strategy to higher ring homologue of enantioenriched α-substituted proline. The eight-membered proline analogue was successfully converted to the pyrrolizidine natural product backbone.
The α,α-disubstituted α-amino acid structure, found in many natural products,1 has been employed in antibiotics2 and is important in peptidomimetics.3 Synthesis of α,α-disubstituted α-amino acids is difficult, with congeners beyond the α-Me, α-Et, and α-Bn becoming even more difficult.4 Furthermore, the construction of enantioenriched α-allyl-α-aryl α-amino acids is extremely challenging. Kinetic resolutions were first reported by Tourwe in 1997.5 Diastereoselective processes were reported by Tayama6a and Barrett.6b Maruoka and Ooi in 2000 established the use of phase-transfer catalyst to generate α,α-disubstituted α-amino acids.7a,7b Yet, in the asymmetric synthesis of α-allyl-α-aryl α-amino acids, only the parent α-allyl-α-phenylglycine α-amino acid has been made efficiently.7a−7d
Asymmetric allylic alkylation (AAA) to generate α,α-disubstituted α-amino acids was pioneered in 1997 by Trost utilizing azlactones.8 α-Aryl α-amino acids pose a challenge to AAA because of the large steric bulk of the aryl ring near the nucleophilic carbon of the α-amino acids. Recently, we developed an asymmetric method for synthesizing α-allyl-α-aryl α-amino acids,9 which relies on the formation of a chelated magnesium enolate10 that is generated in situ. This work was the first example of an asymmetric tandem N-alkylation/π-allylation of α-iminoesters (Scheme 1). The α-iminoester undergoes an umpolung N-alkylation with the Grignard reagent, generating an enolate in situ that can be used in AAA.11 This example was the first utilization of phenylglycine amino acid in AAA and enables the synthesis of previously inaccessible enantioenriched α-allyl-α-aryl α-amino acids. In this paper, the use of cross-metathesis is described to overcome challenges incurred when other allylating agents were employed. Capitalizing on the N-alkyl group introduced in the α-iminoester addition, the construction of cyclic α-amino acids by ring-closing metathesis is also reported.
Scheme 1. Tandem N-Alkylation/π-Allylation of α-Iminoesters.

Scheme 2 displays representative examples of the three-component coupling by N-alkylation/π-allylation of α-iminoesters.9 The phenyl substituent on 1 could be substituted with electron-donating or -withdrawing groups, representing the first asymmetric synthesis of α-allyl-α-aryl α-amino acids where the aryl is other than phenyl.7 Further variation of the phenyl group on 2 to the 2-naphthalene or 2-thiophene also proceeded well.9 The p-methoxyphenyl (PMP) was the best activating group for preferential N-alkylation; the methyl ester could be modified (ethyl, tert-butyl, benzyl), but enantioselectivity was reduced.9
Scheme 2. Our Previous Work on the Tandem N-Alkylation/π-Allylation of α-Iminoesters.

Table 1 describes the further exploration of the substrate scope by looking at allylating agents beyond cinnamyl acetate. Replacing the phenyl ring of cinnamyl acetate with hydrogen reduced selectivity (Table 1, 2d). This result is in line with the phenyl differentiating the termini of the allylpalladium complex (Scheme 1), which is expected to increase selectivity.12 However, modification of the phenyl group in cinnamyl acetate with either electron-donating or -withdrawing groups did not increase selectivity (Table 1, 2f and 2g). Driven by a desire to generate aspartic/glutamic acid analogues, the corresponding furyl allyl acetate was surveyed, but no product was observed (Table 1, 2h). Lastly, 1,3-disubstituted allylating agents generated no tandem product (Table 1, 2i and 2j).
Table 1. Exploration of Allylating Agent in the Tandem N-Alkylation/π-Allylation of α-Iminoesters.


Isolated yield.
Determined by chiral stationary phase (CSP) HPLC.
(R)-BINAP was substituted for (R)-DIFLUORPHOS.
α-Aryl homologues of tyrosine and glutamate have been shown to be biologically active.13 Yet, the challenges described above (Table 1) with various allylating agents prevented access to such compounds by this technology. In an alternate approach, cross-metathesis was studied to enable generation of a broad range of different allyl adducts from a single intermediate 2a (Table 2). Notably, cross-metathesis between type 1 and type 2 olefins has proven difficult.14 Cross-metathesis of α,α-disubstituted α-amino acids has been limited to α-allyl-α-methyl- and α-allylproline with the nitrogen protected as a carbamate to prevent coordination to the ruthenium catalyst.15 Indeed, amines are notoriously poor in metathesis due to deactivation of the ruthenium. However, we envisioned that the enantioenriched tandem product 2a could be an acceptable type 2 olefin for cross-metathesis because the tertiary amine does not behave as expected. For example, the hydrochloride salts of 2 could not be formed. Apparently, the N-substitution and the sterics at the α-carbon contribute to this unique profile.
Table 2. Cross-Metathesis of the Tandem Product 2a.


Isolated yield.
1,2-Dichloroethane substituted for toluene at 70 °C.
Yield based on recovered starting material.
Table 2 showcases that 2a does indeed undergo cross-metathesis with various type 1 olefins, accessing previously unknown α-amino acid analogues in good yields with retention of enantiopurity. Importantly, p-tert-butoxystyrene and methyl acrylate could be employed. Microwave conditions did not accelerate the cross-metathesis, and other catalysts were inferior to the second-generation Hoveyda–Grubbs catalyst.16 Ethylene proved recalcitrant but did afford product 2d with high enantioselectivity, in contrast to direct allylation (see Table 1, entry 1) and low conversion (with high yield based on recovered starting material).
To further our objective of generating α-amino acid homologues, hydrogenation and PMP removal was performed on 2k to provide the α-aryl tyrosine homologue 5 (Scheme 3). Notably, N-ethylamino acids can be difficult to generate due to the difficulty of reductive amination with acetaldehyde. We have shown previously that the primary amines can be generated by using but-3-enyl Grignard in the N-alkylation/π-allylation,9 where the N-butenyl is ultimately removed by an isomerization/hydrolysis using the Grotjahn catalyst.17
Scheme 3. Generation of α-Aryl Tyrosine Homologue.

There is much great synthetic and pharmaceutical interest in cyclic α-amino acid derivatives.18 Further, α-substituted prolines have been shown to be important as organocatalysts and privileged ligands. Seebach pioneered the asymmetric synthesis of α-substituted prolines in 1983,19 yet the development of asymmetric methodology for the synthesis of higher ring homologues of proline has received little attention.20 The N-substituents and the α-alkene functionality present in the α-iminoester addition products 2b and 2c described above provide an effective entry to cyclic analogues via ring-closing metathesis (Scheme 4). Microwave irradiation allowed for the formation of 6 to occur in 1 h vs 20–24 h under thermal conditions.
Scheme 4. Synthesis of Higher Ring Homologue of α-Substituted Proline.

Hydrogenation of RCM product 6 provided crystalline 7 (Scheme 5), which was triturated with dichloromethane to provide >95% ee material. The absolute configurations of products 2a–n were inferred by the X-ray crystal structure of 7.
Scheme 5. Establishing the Absolute Configuration of Products 2a–n.

The corresponding 8-membered ring analogue 8 could be formed by ring-closing metathesis of 2c (Scheme 6). Microwave irradiation not only expedited the reaction but also increased its efficiency compared to thermal conditions. Furthermore, azocine 8 presents an expeditious entry to substituted pyrrolizidines via transannular cyclization.
Scheme 6. Ring-Closing Metathesis To Generate Cyclic Pyrrolizidine Analogue.
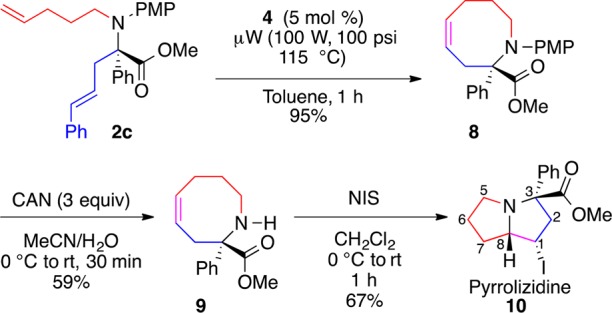
The naturally occurring pyrrolizidine structural motif exists in over 370 natural products and has been shown to have toxicity against livestock, wildlife, and humans.21 The PMP group was removed from azocine 8 to provide 9. Secondary amine 9 is poised to undergo an iodine-induced transannular cyclization.22 Treatment of 9 with NIS led exclusively to cis-iodide 10, consistent with a formal syn-addition across the olefin.23 Since most naturally occurring pyrrolizidines are substituted at C1, this route offers a labile functional group for further manipulation.21 The relative configuration of 10 was assigned by comparing the MM2* calculated bond distances and coupling constants of the four potential diastereomers with the NOESY and coupling constant data of 10.16
In summary, this paper outlines an entry to enantioenriched acylic and cyclic α,α-disubstituted amino acid derivatives by means of cross-metathesis or ring-closing metathesis of the previously disclosed products from asymmetric tandem N-alkylation/π-allylation of α-iminoesters. This approach provided rapid entry to homologues of tyrosine and glutamate as well as to higher ring homologues of proline. The eight-membered cyclic α-amino acid was further transformed to the pyrrolizidine natural product backbone in three steps.
Experimental Section
Starting material compounds 1 and 2a–c were made following our previous report.9
(S,E)-Methyl 2-(N-Ethyl-N-(4-methoxyphenyl)amino)-2-phenylpent-4-enoate (2d)
General Procedure A
Compound 1 (25 mg, 0.093 mmol) was added to a flamed dried Schlenk flask that had been charged with a stir bar and was then vacuum-purged three times under argon. THF (0.5 mL) was added, the reaction was cooled to −78 °C, and EtMgBr (3.0 M in Et2O, 50 μL, 0.15 mmol) was added. The mixture was slowly warmed to room temperature, allowed to stir for an additional 30 min and then cooled to −78 °C. A flame-dried round-bottom flask equipped with a stir bar was charged with [η3-C3H5PdCl]2 (0.85 mg, 0.0023 mmol) and (R)-DIFLUORPHOS (3.1 mg, 0.0045 mmol) and vacuum-purged three times under argon. THF (0.5 mL) and allyl acetate (11 μL, 0.10 mmol) were added. The solution was stirred for 5 min at ambient temperature under argon, cooled to −78 °C, and added to the cooled first solution via syringe. The combined mixture warmed to room temperature and stirred for 45 min. The resultant reaction mixture was quenched with satd NH4Cl (5 mL) and extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. Purification via column chromatography (prewash SiO2 with 5% NEt3/hexanes, eluent 3% EtOAc/hexanes) provided product 2d (26 mg) in 82% yield as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.43 (d, J = 8.1 Hz, 2H), 7.34 (t, J = 7.5 Hz, 2H), 7.28–7.25 (m, 1H), 7.10 (d, J = 8.9 Hz, 2H), 6.85 (d, J = 8.8 Hz, 2H), 5.43–5.35 (m, 1H), 4.72 (d, J = 10.3 Hz, 1H), 4.59 (d, J = 17.2 Hz, 1H), 3.85 (s, 3H), 3.82 (s, 3H), 3.02–2.88 (m, 2H), 2.48 (dd, J = 13.9, 6.8 Hz, 1H), 2.28 (dd, J = 13.9, 7.3 Hz, 1H), 0.79 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 173.3, 157.9, 141.5, 139.2, 134.2, 131.2, 128.0, 127.8, 127.3, 117.7, 113.8, 74.8, 55.5, 51.4, 46.6, 45.3, 14.7; IR (film) 2962, 2929, 1726, 1605, 1507, 1243 cm–1; HRMS (ESI) calcd for C21H26NO3 [M + H]+m/z = 340.1913, found 340.1900; [α]25D = +58.6 (c 0.08, 54% ee, CH2Cl2); chiral HPLC (IA, 97.5:2.5 hexanes/i-PrOH, 1 mL/min, 254 nm): tR of 2d: 4.8 min (major) and 5.2 min (minor).
(S,E)-Methyl 2-(N-Ethyl-N-(4-methoxyphenyl)amino)-2-phenyloct-4-enoate (2e)
General procedure A was followed using 1 (50 mg, 0.19 mmol) in THF (1.0 mL), 3.0 M EtMgBr (100 μL, 0.30 mmol), [η3-C3H5PdCl]2 (1.7 mg, 0.0047 mmol), R-BINAP (5.7 mg, 0.0091 mmol), and hex-2-enyl acetate (31 μL, 0.20 mmol) in THF (1.0 mL). Purification by column chromatography (8% EtOAc/hexanes) provided product 2e (49 mg) in 70% yield as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.43 (d, J = 7.5 Hz, 2H), 7.32 (t, J = 7.6 Hz, 2H), 7.27–7.22 (m, 1H), 7.10 (d, J = 8.8 Hz, 2H), 6.84 (d, J = 9 Hz, 2H), 5.03–4.88 (m, 2H), 3.84 (s, 3H), 3.81 (s, 3H), 3.03–2.86 (m, 2H), 2.42 (dd, J = 14, 6.3 Hz, 1H), 2.19 (dd, J = 14, 7.0 Hz, 1H), 1.71 (dt, J = 7.6, 6.9 Hz, 2H), 1.16–1.09 (m, 2H), 0.79 (t, J = 7.0 Hz, 3H), 0.71 (t, J = 7.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 173.2, 157.8, 141.7, 139.2, 134.1, 131.2, 128.1, 127.6, 127.0, 125.2, 113.7; IR (film) 2959, 1726, 1508, 1244 cm–1; HRMS (ESI) calcd for C24H32NO3 [M + H]+m/z = 382.2382, found 382.2388; [α]25D = +128.75 (c 0.32, 58% ee, CH2Cl2); chiral HPLC (IA, 97.5:2.5 hexanes/i-PrOH, 1 mL/min, 254 nm): tR of 2e: 4.8 min (major) and 6.8 min (minor).
(S,E)-Methyl 2-(N-Ethyl-N-(4-methoxyphenyl)amino)-5-(4-methoxyphenyl)-2-phenylpent-4-enoate (2f)
General procedure A was followed using 1 (40 mg, 0.15 mmol) in THF (1.0 mL), 3.0 M EtMgBr (80 μL, 0.24 mmol), [η3-C3H5PdCl]2 (1.36 mg, 0.0037 mmol), R-BINAP (4.6 mg, 0.0073 mmol) and (4-methoxy)cinnamyl acetate (32 mg, 0.16 mmol) in THF (1.0 mL). Purification by column chromatography (prewash SiO2 with 5% NEt3/hexanes, eluent 8% EtOAc/hexanes) provided 2f (48 mg) in 72% yield as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.46 (d, J = 8.4 Hz, 2H), 7.34 (t, J = 7.3 Hz, 2H), 7.28–7.27 (m, 1H), 7.14 (d, J = 8.8 Hz, 2H), 7.01 (m, J = 8.6 Hz, 2H), 6.86 (d, J = 8.9 Hz, 2H), 6.74 (d, J = 8.7 Hz, 2H), 5.82 (d, J = 15.8 Hz, 1H), 5.26 (dt, J = 15.8, 7.4 Hz, 1H), 3.86 (s, 3H), 3.82 (s, 3H), 3.76 (s, 3H), 3.06–2.99 (m, 1H), 2.97–2.90 (m, 1H), 2.59 (ddd, J = 13.7, 7.0, 1.3 Hz, 1H), 2.39 (ddd, J = 13.8, 7.5, 1.1 Hz, 1H), 0.81 (t, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 173.3, 158.7, 157.8, 141.7, 139.2, 132.4, 131.3, 130.9, 127.9, 127.8, 127.3, 127.1, 123.8, 113.9, 113.8, 75.2, 55.5, 55.4, 51.4, 46.6, 44.3, 14.7; IR (film) 2960, 2931, 1724, 1607, 1509, 1246 cm–1; HRMS (ESI) calcd for C28H32NO4 [M + H]+m/z = 446.2331, found 446.2322; [α]25D = +95.3 (c 0.21, 60% ee, CH2Cl2); chiral HPLC (IA, 97.5:2.5 hexanes/i-PrOH, 1 mL/min, 254 nm): tR of 2f: 9.9 min (major) and 11.8 min (minor).
(S,E)-Methyl 2-(N-Ethyl-N-(4-methoxyphenyl)amino)-5-(3-(3-trifluoromethyl)phenyl)-2-phenylpent-4-enoate (2g)
General procedure A was followed using 1 (40 mg, 0.149 mmol) in THF (1.0 mL), 3.0 M EtMgBr (80 μL, 0.240 mmol), [η3-C3H5PdCl]2 (1.36 mg, 0.0037 mmol), R-DIFLUORPHOS (4.98 mg, 0.0073 mmol), and (3-trifluoromethyl)cinnamyl acetate (38 mg, 0.156 mmol) in THF (1.0 mL). Purification by column chromatography (prewash SiO2 with 5% NEt3/hexanes, eluent 8% EtOAc/hexanes) provided 2g (55 mg) in 76% yield as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.45 (d, J = 7.9 Hz, 2H), 7.38–7.34 (m, 3H), 7.30 (t, J = 7.9 Hz, 2 H), 7.22 (d, J = 7.9, 1H), 7.14 (d, J = 8.7 Hz, 2H), 6.88 (d, J = 8.7 Hz, 2H), 5.87 (d, J = 15.8 Hz, 1H), 5.80–5.74 (dt, J = 15.6, 7.0 Hz, 1H), 3.89 (s, 3H), 3.82 (s, 3H), 3.08–3.01 (m, 1H), 2.98–2.92 (m, 1H), 2.63 (dd, J = 13.7, 6.8 Hz, 1H), 2.43 (dd, J = 13.7, 7.3 Hz, 1 H), 0.82 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 173.3, 158.0, 141.5, 139.1, 138.7, 131.6, 131.3, 130.8 (q, J = 32 Hz), 129.0, 128.8, 128.4, 128.0, 127.7, 127.5, 124.4 (q, J = 254 Hz), 123.5–123.4 (m), 122.8–122.7 (m), 113.9, 75.1, 55.5, 51.6, 46.8, 44.5, 14.7; IR (film) 3036, 2837, 1725, 1606, 1508, 1328, 1245, 1124 cm–1; HRMS (ESI) calcd for C28H29F3NO3 [M + H]+m/z = 484.2100, found 484.2099; [α]25D = +99.5 (c 0.26, 64% ee, CH2Cl2); chiral HPLC (IA, 97.5:2.5 hexanes/i-PrOH, 1 mL/min, 254 nm): tR of 2g: 6.0 min (major) and 5.6 min (minor).
(3-Trifluoromethyl)cinnamyl Acetate
Following the modified procedure previously reported.24 Reaction conditions: 3-iodobenzotrifluoride (577 μL, 4.0 mmol), allyl acetate (1.6 g, 16.0 mmol), Pd(OAc)2 (45 mg, 0.20 mmol), Ag2CO3 (662 mg, 2.4 mmol), toluene (24 mL). (3-Trifluoromethyl)cinnamyl acetate (315 mg) was obtained in 32% yield as a colorless liquid. Spectral data agreed with those reported previously.25
(S,E)-Methyl 2-(N-ethyl-N-(4-methoxyphenyl)amino)-5-(4-(tert-butoxy)phenyl)-2-phenylpent-4-enoate (2k)
General Procedure B
Compound 4 (6.0 mg, 0.0096 mmol) was added to a flame-dried 8 mL microwave vial equipped with stir bar in the glovebox. Compound 3a (85 mg, 0.48 mmol) was added followed by 2a (20 mg, 0.048 mmol) in toluene (482 μL). The vial was sealed with a Teflon cap, taken out of the glovebox, placed in a 90 °C oil bath, and stirred for 20 h. The mixture was cooled, passed through SiO2 with 30% EtOAc in hexanes, and concentrated in vacuo. Purification by column chromatography (prewash SiO2 with 5% NEt3 in hexanes, eluent 3% acetone in hexanes) provided 2k (17 mg) in 74% yield as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.47 (d, J = 7.4 Hz, 2H), 7.35 (t, J = 7.1 Hz, 2H), 7.29 (d, J = 7.0 Hz, 1H), 7.14 (d, J = 8.8 Hz, 2H), 6.97 (d, J = 8.4 Hz, 2H), 6.87 (d, J = 8.8 Hz, 2H), 6.82 (d, J = 8.4 Hz, 2H), 5.86 (d, J = 15.7 Hz, 1H), 5.58 (dt, J = 15.9, 7.3 Hz, 1H), 3.86 (s, 3H), 3.82 (s, 3H), 3.06–2.99 (m, 1H), 2.96–2.91 (m, 1H), 2.60 (dd, J = 13.9, 7.4 Hz, 1H), 2.41 (dd, J = 13.9, 7.4 Hz, 1H), 1.31 (s, 9H), 0.81 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 173.2, 157.8, 154.5, 141.6, 139.1, 133.2, 132.4, 131.3, 127.9, 127.8, 127.3, 126.5, 124.7, 124.1, 113.8, 78.6, 75.1, 55.5, 51.5, 46.6, 44.3, 29.0, 14.7; IR (film) 2975, 2855, 1725, 1604, 1507, 1241, 1162 cm–1; HRMS (ESI) calcd for C31H38NO4 [M + H]+m/z = 488.2801, found 488.2811; [α]25D = +170.1 (c 0.13, 88% ee, CH2Cl2).
(S,E)-Methyl 2-(N-Ethyl-N-(4-methoxyphenyl)amino)-5-(naphthalene-2-yl)-2-phenylpent-4-enoate (2l)
General procedure B was followed using 2a (20 mg, 0.048 mmol, 81% ee), 3b (22 mg, 0.15 mmol), and 4 (6.0 mg, 0.0096 mmol) in dichloroethane (482 μL) at 70 °C for 20 h. Purification by column chromatography (prewash SiO2 with 5% NEt3 in hexanes, eluent 7% EtOAc in hexanes) provided 2l (15 mg) in 68% yield as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.72 (dd, J = 12.0, 4.0 Hz, 2H), 7.66 (d, J = 8.6 Hz, 1H), 7.48 (d, J = 7.7 Hz, 2H), 7.42–7.33 (m, 5H), 7.29 (t, J = 8.0 Hz, 2H), 7.16 (d, J = 8.7 Hz, 2H), 6.88 (d, J = 8.7 Hz, 2H), 6.02 (d, J = 16.0 Hz, 1H), 5.84 (dt, J = 16.0, 7.1 Hz, 1H), 3.88 (s, 3H), 3.83 (s, 3H), 3.08–3.01 (m, 1H), 2.98–2.92 (m, 1H), 2.66 (dd, J = 14.0, 7.2 Hz, 1H), 2.47 (dd, J = 13.6, 7.5 Hz, 1H), 0.82 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 173.4, 157.9, 141.6, 139.2, 135.4, 133.7, 133.1, 132.7, 131.3, 128.0, 127.9 (3), 127.7, 127.4, 126.6, 126.2, 125.6, 125.5, 123.7, 113.9, 75.2, 55.5, 51.5, 46.7, 44.6, 14.7; IR (film) 2924, 2853, 1725, 1507 cm–1; HRMS (ESI) calcd for C31H32NO3 [M + H]+m/z = 466.2382, found 466.2399; [α]25D = +125.5 (c 0.21, 81% ee, CH2Cl2).
(S,E)-Dimethyl 5-(N-Ethyl-N-(4-methoxyphenyl)amino)-5-phenylhex-2-enedioate (2m)
General procedure B was followed using 2a (20 mg, 0.048 mmol), 3c (42 mg, 0.48 mmol), and 4 (6.0 mg, 0.0096 mmol) in toluene (482 μL). Purification by column chromatography (prewash SiO2 with 5% NEt3 in hexanes, eluent 3% acetone in hexanes) provided 2m (14 mg) in 72% yield as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.39 (d, J = 7.5 Hz, 2H), 7.34 (t, J = 7.4 Hz, 2H), 7.28 (t, J = 7.2 Hz, 1H), 7.10 (d, J = 8.9 Hz, 2H), 6.86 (d, J = 8.9 Hz, 2H), 6.61–6.55 (m, 1H), 5.32 (d, J = 16.5 Hz, 1H), 3.88 (s, 3H), 3.81 (s, 3H), 3.60 (s, 3H), 3.05–2.98 (m, 1H), 2.95–2.88 (m, 1H), 2.57 (ddd, J = 14.5, 7.3, 0.6 Hz, 1H), 2.43 (ddd, J = 14.4, 7.6, 0.6 Hz, 1H), 0.79 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 173.2, 166.8, 158.1, 145.1, 140.9, 138.9, 131.2, 128.2, 127.7, 127.4, 123.2, 114.0, 74.5, 55.5, 51.7, 51.4, 46.8, 43.7, 14.7; IR (film) 2951, 2855, 1724, 1655, 1508, 1245, 1035 cm–1; HRMS (ESI) calcd for C23H28NO5 [M + H]+m/z = 398.1967, found 398.1963; [α]20D = +107.2 (c 0.13, 88% ee, CH2Cl2).
2d
General procedure B was followed using 2a (20 mg, 0.0482 mmol) and 4 (8.2 mg, 0.0096 mmol) in dichloroethane (482 μL) and the following modification: ethylene gas 3d was sparged through the flask for 10 min and then subjected to microwave (100 W, 100 psi, 90 °C) conditions for 30 min. The mixture was cooled, ethyl vinyl ether (50 μL, 0.523 mmol) was added, and the mixture ws stirred for 30 min. The mixture was passed through SiO2 with 30% EtOAc in hexanes and concentrated in vacuo. Purification by column chromatography (prewash SiO2 with 5% NEt3 in hexanes, eluent 5% EtOAc in hexanes) provided 2d (3.9 mg) in 24% yield (characterization the same as 2d above) with unreacted 2a (15 mg, 0.036 mmol) recovered.
(S)-Methyl 2-(N-Ethyl-N-(4-methoxyphenyl)amino)-5-(4-tert-butoxyphenyl)-2-phenylpentanoate (2ksat)
General Procedure C
Compound 2k (17 mg, 0.035 mmol), 10% Pd/C (3.7 mg, 0.0035 mmol), and EtOAc (1.1 mL) were added to a flame-dried 10 mL round-bottom flask equipped with a stir bar. At ambient temperature, the flask was purged with one hydrogen balloon and then subjected to a hydrogen atmosphere with a new hydrogen balloon for 45 min. The flask was opened to air, passed through SiO2 with 30% EtOAc/hexanes, and concentrated in vacuo. Purification by column chromatography (10% EtOAc in hexanes) provided 2ksat (14.4 mg) in 85% yield as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.43 (d, J = 7.7 Hz, 2H), 7.34 (t, J = 7.2 Hz, 2H), 7.28 (d, J = 7.3 Hz, 1H), 7.07 (d, J = 8.6 Hz, 2H), 6.84 (d, J = 8.6 Hz, 2H), 6.77 (b, 4H), 3.83 (s, 3H), 3.01–2.83 (m, 2H), 2.25–2.19 (m, 1H), 2.15–2.09 (m, 1H), 1.72–1.66 (m, 1H), 1.56–1.50 (m, 1H), 1.29 (s, 9H), 1.28–1.25 (b, 2H); 13C NMR (125 MHz, CDCl3) δ 173.5, 157.8, 153.1, 141.9, 139.3, 137.3, 131.1, 128.6, 127.9, 127.8, 127.1, 124.0, 113.8, 78.1, 74.5, 55.5, 51.3, 46.6, 40.1, 35.5, 29.0, 26.6, 14.7; IR (film) 3337, 2974, 2930, 1724, 1507, 1241, 1162 cm–1; HRMS (ESI) calcd for C31H40NO4 [M + H]+m/z = 490.2957, found 490.2947; [α]25D = +67.9 (c 0.11, 88% ee, CH2Cl2).
(S)-Methyl 2-(Ethylamino)-5-(4-tert-butoxyphenyl)-2-phenylpentanoate (5)
General Procedure D
A solution of 2ksat (15 mg, 0.031 mmol) in MeCN (618 μL) was cooled to 0 °C and put under N2 in an 8 mL microwave vial equipped with stir bar. A solution of ceric ammonium nitrate (51 mg, 0.092 mmol) in H2O (309 μL) was added dropwise. The mixture was allowed to warm to ambient temperature, stirred for 45 min, cooled to 0 °C, and quenched with 5% NaHCO3 until a pH of 9 was obtained (∼1 mL). The mixture was diluted with 20% Na2SO3 (20 mL) and extracted with EtOAc (3 × 20 mL). The combined organic phases were washed with brine (2×), dried over Na2SO4, and concentrated in vacuo. Purification by column chromatography (5% to 30% EtOAc/hexanes) provided 5 (7.5 mg) in 64% yield as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 7.41 (d, J = 8.4 Hz, 2H), 7.31 (t, J = 7.5 Hz, 2H), 7.27–7.25 (m, 1H), 6.99 (d, J = 8.5 Hz, 2H), 6.86 (d, J = 8.4 Hz, 2H), 3.66 (s, 3H), 2.52 (t, J = 7.5 Hz, 2H), 2.32–2.24 (m, 2H), 2.18–2.12 (m, 1H), 2.06–1.97 (m, 1H), 1.54–1.39 (m, 2H), 1.31 (s, 9H), 1.09 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 175.8, 153.2, 142.0 (b), 137.3, 128.8, 128.4, 127.4, 126.2, 124.2, 78.2, 68.4, 52.4, 37.6, 35.3, 34.4, 29.0, 25.0, 15.8; IR (film) 2965, 2928, 1733, 1607, 1505, 1234 cm–1; HRMS (ESI) calcd for C24H34NO3 [M + H]+m/z = 384.2539, found 384.2547; [α]24D = −1.5 (c 0.36, 88% ee, CH2Cl2).
(S)-Methyl 1-(4-Methoxyphenyl)-2-phenyl-2,3,6,7-tetrahydro-1H-azepine-2-carboxylate (6)
General procedure B was followed using 2b (29 mg, 0.066 mmol, 81% ee) and 4 (2.8 mg, 0.003 mmol) in toluene (6 mL). The reaction was subjected to microwave (100 W, 100 psi, 115 °C) conditions for 1 h. The mixture cooled to ambient temperature, ethyl vinyl ether (75 μL, 0.78 mmol) was added, and the mixture was allowed to stir for 30 min. The mixture was passed through SiO2 with 30% EtOAc in hexanes and concentrated in vacuo. Purification by column chromatography (prewash SiO2 with 5% NEt3 in hexanes, eluent 5% EtOAc in hexanes) provided 6 (23 mg) in >98% yield as a light yellow oil. Spectral data agreed with our previous report.9
(S)-Methyl 1-(4-Methoxyphenyl)-2-phenylazepane-2-carboxylate (7)
General procedure C was followed using 6 (0.033 M ethyl acetate solution, 0.20 mmol) and 10% Pd/C (22 mg, 0.020 mmol). Purification by column chromatography (prewash SiO2 with 3% NEt3 in hexanes, eluent 6% EtOAc/hexanes) provided 7 (55 mg) in 81% yield as a white solid. Trituration of 7 in CH2Cl2 provided undissolved white solid that was close to racemic and liquid that had enhanced ee to 98%. The liquid was concentrated and recrystallized from Et2O via slow evaporation over 24 h: mp (98% ee material) = 113–110 °C; 1H NMR (500 MHz, CDCl3) δ 7.47 (d, J = 7.6 Hz, 2H), 7.29–7.26 (m, 2H), 7.22 (t, J = 7.1 Hz, 1H), 6.65–6.63 (m, 2H), 6.54–6.52 (m, 2H), 3.75–3.70 (m, 4H), 3.59 (s, 3H), 3.53 (dd, J = 15.6, 8.8 Hz, 1H), 2.61 (dd, J = 15.4, 8.9 Hz, 1H), 2.08 (dd, J = 15.1, 10 Hz, 1H), 1.96–1.93 (b, 1H), 1.88–1.84 (b, 2H), 1.72–1.69 (b, 1H), 1.63–1.60 (b, 1H), 1.50–1.41 (b, 1H); 13C NMR (125 MHz, CDCl3) δ 175.4, 151.8, 145.1, 141.1, 128.8, 127.9, 126.9, 117.0, 113.8, 72.2, 55.7, 52.4, 51.6, 44.8, 31.0, 29.7, 23.6; IR (film) 2929, 2855, 1731, 1511, 1246, 1222, 1036, 823 cm–1; HRMS (ESI) calcd for C21H26NO3 [M + H]+m/z = 340.1913, found 340.1926; [α]25D = −7.1 (c 0.34, 98% ee, CH2Cl2).
(S,Z)-Methyl 1-(4-Methoxyphenyl)-2-phenyl-1,2,3,6,7,8-hexahydroazocine-2-carboxylate (8)
General procedure B was followed using 2c (30 mg, 0.066 mmol) and 4 (2.82 mg, 0.003 mmol) in toluene (6 mL). The reaction was subjected to microwave (100 W, 100 psi, 115 °C) conditions for 1 h. The mixture was cooled to ambient temperature, and ethyl vinyl ether (75 μL, 0.784 mmol) was added and stirred for 30 min. The mixture was passed through SiO2 with 30% EtOAc in hexanes, concentrated in vacuo, and chromatographed (prewash SiO2 with 5% NEt3 in hexanes, eluent 5% EtOAc in hexanes) to provide 8 (22 mg) in 95% yield as a light yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.54 (d, J = 8.34 Hz, 2H), 7.29–7.26 (m, 2H), 7.20 (t, J = 7.0 Hz, 1H), 6.83 (d, J = 8.8 Hz, 2H), 6.70 (d, J = 8.3 Hz, 2H), 5.92–5.87 (m, 1H), 5.72–5.67 (m, 1H), 3.72 (s, 3H), 3.60–3.50 (m, 1H), 3.43 (s, 3H), 3.35–3.30 (m, 2H), 3.27–3.15 (b, 1H), 2.11–2.03 (m, 1H), 2.03–1.93 (b, 1H), 1.92–1.77 (b, 1H), 1.58–1.44 (b, 1H); 13C NMR (125 MHz, CDCl3) δ 174.2, 152.7, 142.5, 140.7, 132.9, 128.8, 128.0, 127.5, 127.4, 121.1 (b), 113.7, 75.4, 55.5, 52.4, 48.3, 33.6, 29.1, 24.1; IR (film) 3021, 2860, 1728, 1511, 1244, 1038 cm–1; HRMS (ESI) calcd for C22H26NO3 [M + H]+m/z = 352.1913, found 352.1923; [α]24D = −1.03 (c 0.33, 83% ee, CH2Cl2).
(S,Z)-Methyl 2-Phenyl-1,2,3,6,7,8-hexahydroazocine-2-carboxylate (9)
General procedure D was followed using 8 (10 mg, 0.029 mmol), CAN (50 mg, 0.086 mmol), MeCN (3.2 mL), and H2O (1.6 mL). The mixture was allowed to stir at ambient temperature for 15 min instead of 45 min then cooled to 0 °C and quenched with 10% NaHCO3 until a pH of 7 was obtained (∼0.5 mL). Purification by column chromatography (prewash SiO2 with 3% NEt3 in hexanes, eluent 5% to 30% EtOAc in hexanes) provided 9 (4 mg) in 59% yield as a yellow oil. The reaction did not scale up well so it was more effective to run multiple reactions with the scale indicated above side-by-side and then combine the material for purification: 1H NMR (500 MHz, CDCl3) δ 7.55 (d, J = 7.8 Hz, 2H), 7.34 (t, J = 7.7 Hz, 2H), 7.28–7.27 (m, 1H), 5.98–5.93 (m, 1H), 5.55–5.50 (m, 1H), 3.67 (s, 3H), 3.06–3.01 (m, 1H), 2.87–2.78 (b, 1H), 2.76–2.72 (m, 1H), 2.69–2.57 (b, 1H), 2.22–2.15 (m, 2H), 1.69–1.63 (m, 1H), 1.60–1.51 (m, 1H), 1.3 (b, 1H); 13C NMR (125 MHz, CDCl3) δ 175.0, 142.3, 134.3, 128.5, 127.6, 126.8, 126.3, 70.0, 52.6, 44.5, 35.0, 32.2, 26.0; IR (film) 3419, 2924, 1731, 1645, 1220, 1032 cm–1; HRMS (ESI) calcd for C15H20NO2 [M + H]+m/z = 246.1494, found 246.1497; [α]25D = −4.8 (c 0.09, 90% ee, CH2Cl2).
(S,S,S)-Methyl 1-Iodo-3-phenyloctahydro-1H-pyrrolizine-3-carboxylate (10)
A solution of 9 (23 mg, 0.094 mmol) in CH2Cl2 (1 mL) open to air was cooled to 0 °C in a flame-dried 5 mL round-bottom flask equipped with stirbar. NIS (freshly recrystallized from dichloromethane, 42 mg, 0.19 mmol) was added. The mixture was warmed to ambient temperature and stirred for 1.5 h. At 0 °C, the mixture was quenched with 500 μL of 10% aq Na2S2O3. The mixture was diluted with H2O (5 mL) and extracted with EtOAc (3 × 10 mL). The combined organic phases were dried over Na2SO4, filtered, and concentrated in vacuo. Purification by column chromatography (prewash SiO2 with 5% NEt3 in hexanes, gradient eluent 5% to 30% EtOAc in hexanes) provided 10 (23 mg) in 67% yield as a light yellow oil: 1H NMR (500 MHz, CDCl3) δ 7.44 (d, J = 7.0 Hz, 2H), 7.36–7.30 (m, 3H), 4.27 (ddd, J = 14, 7.5, 6.5 Hz, 1H, C1–H), 3.65 (s, 3H), 3.60–3.52 (b, 1H, C8–H), 3.16 (dd, J = 12.3, 6.1 Hz, 1H, C2–Ha), 2.88–2.80 (b, 1H, C5–Ha), 2.58 (t, J = 12.0 Hz, 1H, C2–Hb), 2.14–2.09 (m, 2H, C7–Ha, C5–Hb), 1.87–1.71 (m, 3H, C7–Hb, C6–Ha, C6–Hb); 13C NMR (125 MHz, CDCl3) δ 173.8, 137.4, 128.6, 128.2, 127.6, 77.2, 66.2, 58.1, 53.2, 52.2, 42.1, 36.7, 25.5; IR (film) 2949, 2868, 1741, 1725, 1448, 1251 cm–1; HRMS (ESI) calcd for C15H19NO2I [M + H]+m/z = 372.0461, found 372.0450; [α]25D = +8.2 (c 0.58, 90% ee, CH2Cl2).
Acknowledgments
We are grateful to the NIH (GM-087605) and the NSF (CHE0911713) for financial support of this research. Partial instrumentation support was provided by the NIH for MS (1S10RR023444) and NMR (1S10RR022442) and by the NSF for an X-ray diffractometer (CHE 0840438). Our collaboration with Dr. P. Carroll (UPenn) in obtaining the crystal structure is gratefully acknowledged, as is the help from Dr. G. Furst (UPenn) in conducting 2D NMR experiments. We also acknowledge the contributions of P. Huynh (UPenn).
Supporting Information Available
Figures giving spectroscopic data for the products as well as computational studies on 10 and the CIF for 7. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Xie W.; Zou B.; Pei D.; Ma D. Org. Lett. 2005, 7, 2775–2777. [DOI] [PubMed] [Google Scholar]
- For selected pharmaceutical examples of α,α-disubstituted α-amino acid, see:; a Savage S. A.; Waltermire R. E.; Campagna S.; Bordawekar S.; Toma J. D. R. Org. Process Res. Dev. 2009, 13, 510–518. [Google Scholar]; b Washburn W. N.; Sun C. Q.; Bisacchi G.; Wu G.; Cheng P. T.; Sher P. M.; Ryono D.; Gavai A. V.; Poss K.; Girotra R. N.; McCann P. J.; Mikkilineni A. B.; Dejneka T. C.; Wang T. C.; Merchant Z.; Morella M.; Arbeeny C. M.; Harper T. W. Bioorg. Med. Chem. Lett. 2004, 14, 3525–3529. [DOI] [PubMed] [Google Scholar]; c Ma D. W.; Tian H. Q.; Zou G. X. J. Org. Chem. 1999, 64, 120–125. [DOI] [PubMed] [Google Scholar]; d Stilz H. U.; Jablonka B.; Knolle J.; Paulus E. F.; Zoller G. J. Med. Chem. 1996, 39, 2118–2122. [DOI] [PubMed] [Google Scholar]
- For selected peptidomimetic examples with α,α-disubstituted α-amino acid, see:; a Graver A.; Konig B. Eur. J. Org. Chem. 2009, 30, 5099–5111. [Google Scholar]; b Venkatraman J.; Shankaramma S. C.; Balaram P. Chem. Rev. 2001, 101, 3131–3152. [DOI] [PubMed] [Google Scholar]
- For reviews on the asymmetric synthesis of α,α-disubstituted α-amino acid, see:; a Mosey R. A.; Fisk J. S.; Tepe J. J. Tetrahedron: Asymmetry 2008, 19, 2755–2762. [Google Scholar]; b Cativiela C.; Diaz-De-Villegas M. D. Tetrahedron: Asymmetry 2007, 18, 569–623. [Google Scholar]; c Vogt H.; Brase S. Org. Biomol. Chem. 2007, 5, 406–430. [DOI] [PubMed] [Google Scholar]; d Cativiela C.; Diaz-De-Villegas M. D. Tetrahedron: Asymmetry 1998, 9, 3517–3599. [Google Scholar]
- a Van Betsbrugge J.; Tourwe D.; Kaptein B.; Kierkels H.; Broxtermann R. Tetrahedron 1997, 53. [Google Scholar]; b Maeda K.; Miller R. A.; Szumigala R. H.; Shafiee A.; Karady S.; Armstrong J. D. Tetrahedron Lett. 2005, 46, 1545–1549. [Google Scholar]
- a Tayama E.; Orihara K.; Kimura H. Org. Biomol. Chem. 2008, 6, 3673–3680. [DOI] [PubMed] [Google Scholar]; b Jones E. P.; Jones P.; White A. J. P.; Barrett A. G. M. Beilstein J. Org. Chem. 2011, 7, 1570–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ooi T.; Takeuchi M.; Kameda M.; Maruoka K. J. Am. Chem. Soc. 2000, 122, 5228–5229. [Google Scholar]; b Ooi T.; Takeuchi M.; Ohara D.; Maruoka K. Synlett 2001, 7, 1185–1187. [Google Scholar]; c Berger R.; Duff K.; Leighton J. L. J. Am. Chem. Soc. 2004, 126, 5686–5687. [DOI] [PubMed] [Google Scholar]; Application of Maruoka’s work utilizing an analogue of phenylglycine gave 35% yield and 45% ee; see:; d Metro T.-X.; Cochi A.; Pardo D. G.; Cossy J. J. Org. Chem. 2011, 76, 2594–2602. [DOI] [PubMed] [Google Scholar]; For other enantioselective syntheses of α-allyl-α-aryl α-amino acids but with yields and ee below 50%, see:; e Belokon Y. N.; Bhave D.; D’addario D.; Groaz E.; North M.; Tagliazucca V. Tetrahedron 2004, 60, 1849–1861. [Google Scholar]; f Ito Y.; Sawamura M.; Matsuoka M.; Matsumoto Y.; Hayashi T. Tetrahedron Lett. 1987, 28, 4849–4852. [Google Scholar]
- For AAA with Pd, see:; a Trost B. M.; Ariza X. Angew. Chem., Int. Ed. 1997, 36, 2635–2637. [Google Scholar]; b Kuwano R.; Ito Y. J. Am. Chem. Soc. 1999, 121, 3236–3237. [Google Scholar]; For AAA with Mo, see:; c Trost B. M.; Dogra K. J. Am. Chem. Soc. 2002, 124, 7256–7257. [DOI] [PubMed] [Google Scholar]; For AAA with Ir, see:; d Chen W.; Hartwig J. F. J. Am. Chem. Soc. 2013, 135, 2068–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curto J. M.; Dickstein J. S.; Berritt S.; Kozlowski M. C. Org. Lett. 2014, 16, 1948–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Work on diasteroselective allylic alkylation with enantioenriched allylating agents has shown that chelation with magnesium or zinc can increase the nucleophilicity of the corresponding glycine ester enolates:Kazmaier U.; Bayer A.; Deska J. Synthesis 2013, 45, 1462–1468. [Google Scholar]
- For racemic syntheses of disubstituted α-amino acids by umpolung N-alkylation/C-alkylation, see:; a Mizota I.; Matsuda Y.; Kamimura S.; Tanaka H.; Shimizu M. Org. Lett. 2013, 15, 4206. [DOI] [PubMed] [Google Scholar]; b Shimizu M.; Takao Y.; Katsurayama H.; Mizota I. Asian J. Org. Chem. 2013, 2, 130. [Google Scholar]; c Mizota I.; Tanaka K.; Shimizu M. Tetrahedron Lett. 2012, 53, 1847. [Google Scholar]; d Dickstein J. S.; Fennie M. W.; Norman A. L.; Paulose B. J.; Kozlowski M. C. J. Am. Chem. Soc. 2008, 130, 15794. [DOI] [PubMed] [Google Scholar]; e Dickstein J. S.; Kozlowski M. C. Chem. Soc. Rev. 2008, 37, 1166. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Ariza X. J. Am. Chem. Soc. 1999, 121, 10727–10737. [Google Scholar]
- Wehbe J.; Rolland V.; Fruchier A.; Roumestant M.; Martinez J. Tetrahedron: Asymmetry 2004, 15, 851–858. [Google Scholar]
- Chatterjee A. K.; Choi T.-L.; Sanders D. P.; Grubbs R. H. J. Am. Chem. Soc. 2003, 125, 11360–11370. [DOI] [PubMed] [Google Scholar]
- For CM of α-Me-α-allyl α-amino acids, see:; Storcken R. P. M.; Panella L.; van Delft F. L.; Kaptein B.; Broxterman Q. B.; Schoemaker H. E.; Rutjes F. P. J. T. Adv. Synth. Catal. 2007, 349, 161–164. [Google Scholar]; For CM of α-allyl proline, see:; Lumini M.; Cordero F. M.; Pisaneschi F.; Brandi A. Eur. J. Org. Chem. 2008, 2817–2824. [Google Scholar]
- See the Supporting Information for further details.
- Larsen C. R.; Grotjahn D. B. J. Am. Chem. Soc. 2012, 134, 10357–10360. [DOI] [PubMed] [Google Scholar]
- Park K. H.; Kurth M. J. Tetrahedron 2002, 58, 8629–8659. [Google Scholar]
- Seebach D.; Boes M.; Naef R.; Schweizer W. B. J. Am. Chem. Soc. 1983, 105, 5390–5398. [Google Scholar]
- For Reviews on the asymmetric synthesis of cyclical α,α-disubstituted α-amino acid, see:; a Cativiela C.; Ordonez M. Tetrahedron: Asymmetry 2009, 20, 1–63. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cativiela C.; Diaz-De-Villegas M. D. Tetrahedron: Asymmetry 2000, 11, 645–732. [Google Scholar]
- Pelletier S. W.Alkaloids: Chemical & Biological Perspectives; Pergamon: New York, 1995; Vol. 9, pp 155–233. [Google Scholar]
- Wilson S. R.; Sawicki R. A. J. Org. Chem. 1979, 44, 287–291. [Google Scholar]
- Schindler C. S.; Stephenson C. R. J.; Carreira E. M. Angew. Chem., Int. Ed. 2008, 47, 8852–8855. [DOI] [PubMed] [Google Scholar]
- Pan D.; Chen A.; Su Y.; Zhou W.; Li S.; Jia W.; Xiao J.; Liu Q.; Zhang L.; Jiao N. Angew. Chem., Int. Ed. 2008, 47, 4729–4732. [DOI] [PubMed] [Google Scholar]
- Fleming S. A.; Renault L.; Grundy E. C.; Pincook J. A. Can. J. Chem. 2006, 84, 1146–1154. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.