

Abstract

Jakeman et al. recently reported the inability to distinguish the diastereomers of uridine 5′-β,γ-fluoromethylenetriphosphate (β,γ-CHF-UTP, 1) by 19F NMR under conditions we previously prescribed for the resolution of the corresponding β,γ-CHF-dGTP spectra, stating further that 1 decomposed under these basic conditions. Here we show that the 19F NMR spectra of 1 (∼1:1 diastereomer mixture prepared by coupling of UMP-morpholidate with fluoromethylenebis(phosphonic acid)) in D2O at pH 10 are indeed readily distinguishable. 1 in this solution was stable for 24 h at rt.
Ribonucleoside (NTP) and deoxyribonucleoside (dNTP) triphosphate analogues can be modified at the base, sugar, or triphosphate moiety to generate nucleotide derivatives able to function as substrate mimics or inhibitors of enzymes that utilize nucleotides.1−5 Replacing the bridging oxygen between the β- and γ-phosphates of the triphosphate moiety with a methylene carbon generates a nonhydrolyzable bisphosphonate (BP) inhibitor for enzymes that cleave or transfer the γ-phosphate, such as protein kinases.4,6−8 Conversely, these analogues can act as substrates for enzymes, such as DNA polymerases, that process nucleotides with release of pyrophosphate.7−11 Variation of carbon substitution at the CXY group when X ≠ Y generates two possible diastereomers due to the introduction of a new chiral center, in principle resulting in differing interactions with a binding enzyme.12−16 We recently synthesized the first examples of individual β,γ-CXY-stereoisomers, namely both diastereomers of β,γ-CHF- and β,γ-CHCl-dGTP.17 The configuration at the CHX carbon was found to affect both Kd and kpol with DNA polymerase β (pol β),16 an important base excision repair (BER) enzyme that typically inserts a single dNTP replacing an excised damaged or mismatched residue18,19 (in the case of these substrate analogues, the corresponding bisphosphonate is released instead of pyrophosphate).
As simple coupling20 of fluoromethylenebis(phosphonic acid) (CHF-BP, 2) with an activated dNMP (or NMP) results in the formation of both β,γ-CHF stereoisomers together, the ability to resolve their 19F NMR spectra is important, particularly because this also offers a convenient means to determine their relative reaction rates after turnover.16 We previously demonstrated that synthetic β,γ-CHF-dGTP diastereomer mixtures exhibit discrete 19F NMR (at 376 and 470 MHz) at pH 10,14,15 confirmed by preparation and analysis of the individual stereoisomers.17 Subsequently, Jakeman and co-workers studied the reaction of thymidylyltransferase Cps2L with a mixture of uridine 5′-β,γ-fluoromethylenetriphosphate (β,γ-CHF-UTP) diastereomers and found that ca. 50% of the substrate was rapidly consumed, suggesting stereospecificity for one of the two isomers present.21 However, they could not verify this intriguing result by 19F NMR at 235 MHz, due to the inability to distinguish the individual isomers, reporting21 that “...the 19F spectra of [β,γ-CHF-UTP]22 failed to show two sets of overlapping multiplets as observed by McKenna and co-workers for [β,γ-CHF-dGTP]... at the basic pH of the McKenna studies compound [β,γ-CHF-UTP] broke down.”
We found these observations to be surprising and, therefore, have sought to reproduce them. Here we report that (a) two discrete sets of 19F NMR multiplets are readily observable for an ∼1:1 mixture of 1 diastereomers in D2O under the basic conditions (pH 10) we prescribed for obtaining distinguishable 19F NMR spectra for a similar β,γ-CHF-dGTP diastereomer mixture and (b) the same β,γ-CHF-UTP diastereomer mixture is stable at rt under these solution conditions (pH ∼10) for more than 24 h.
Jakeman and Mohamady originally proposed a rapid synthesis for nucleoside triphosphate bisphosphonate analogues from nucleoside monophosphates using activation with trifluoroacetic anhydride (TFAA) and N-methylimidazole as base.22 They subsequently reported difficulties associated with the activation of UMP by this method.23 As a result, we applied standard NMP-morpholidate coupling14,15,20 to synthesize the diastereomeric mixture of 1. After dual pass HPLC purification,14 including modification of the SAX HPLC purification step to remove a persistent, minor side product with 19F NMR δ near −216.90 ppm, the product was treated with Chelex-100 to remove traces of paramagnetic metal ions. To confirm the identity of the product, its 1H, 31P, and 19F NMR (Figures S8–S10) were determined and found generally to agree with those reported by Mohamady et al. for 1,22 apart from a discrepancy in the 19F NMR spectral resolution as discussed below. The MS analysis using an ESI probe operated in negative mode gave the expected m/z of 499.1 [M – H]−.
Our 1H spectrum (500 MHz, D2O, pH 7.6, Figure S8) displays the same peaks as reported previously22 (the CHF signal is partially obscured by the large HDO peak at 4.79 ppm), and the reported J values for the H-5 and H-6 multiplets are consistent with ours (7.9 vs 8.2 Hz). Our 31P NMR spectrum (202 MHz, D2O, pH 10.4, Figure S10) displays similar J values for each phosphorus resonance, but the δ of Pβ is shifted downfield by ca. 4 ppm (a function of the pH12). We also observe additional Pα peaks, attributed to diastereomer peak resolution (Δδ 4.0 Hz), which was supported by the spectrum obtained at 243 MHz (Δδ 4.6 Hz, predicted 4.7 Hz).
Under the conditions used for resolution of the 19F NMR β,γ-CHF-dGTP diastereomers,14,15 our 19F NMR spectrum for 1 (470 MHz, D2O, pH 10.4, Figures S3 and S9) displays two sets of multiplets assigned to the diastereomers with δ1 −216.91 and δ2 −216.96 (ddd, J = 67.5, 54.6, 45.0 Hz). The largest J value (67.5 Hz) is assigned to Pβ coupling, confirmed by the 31P NMR spectrum (JPβ,F = 65.3 Hz). The second J value (54.6 Hz; 55.6 Hz for Pγ in the 31P NMR spectrum) correlates with the Mohamady et al. value at 59.8 Hz.22 The CHF proton peak is partly obscured in our 1H NMR, but the JF,H value (45.0 Hz) is similar to their JH,F (46.2 Hz).22 In the study of Mohamady et al., the 19F signal was found at δ −213.33, (reported as δ 213.33),22 with coupling constant assignments as follows: dd, JF,Pβ 85.6, JF,Pα 62.1 Hz.22 However, in their 31P NMR spectrum, they report 2JPβ,F 62.8 Hz, suggesting that the smaller coupling constant observed in the 19F spectrum is not due to splitting by Pα, but rather to splitting by Pβ, consistent with our assignment.
We next examined pH and counterion effects on the 19F NMR spectra of 1. This was of particular interest because, as obtained, the synthetic product contains both triethylammonium (TEAH+) and Na+ counterions. The expected 19F splitting pattern for a single stereoisomer of 1 is predicted to be a doublet of doublet of doublets (ddd, 8 peaks) at pH ≥ 10.14,15,17 With our experimental 1:1 diastereomer mixture, we would therefore expect up to 16 peaks depending on the degree of overlap (Figure S3). After exchange of the TEAH+ cation with Na+, K+, or NH4+ using a preparative ion-exchange resin column, 19F (470 MHz) spectra of 1 in D2O could not be resolved into contributions from discrete isomers at pH < 10 (Figure 1A). For each of these countercations, titration of the NMR sample solution to a pH ≥ 10 revealed discrete 19F NMR spectra for the two diastereomers (14 of 16 peaks, Figure 1B and Table 1), with the exception of TEAH+, which gave broad, unresolved multiplets.
Figure 1.

Effect of pH and counterion on 19F NMR spectra of β,γ-CHF-UTP 1 (∼1:1 diastereomers) purified by our previously described14 dual pass preparative HPLC method (D2O, 470 MHz, referenced to CFCl3). (A) Series of spectra for samples near neutral pH (7.4, 8.3, 7.6, and 7.9 for NH4+, K+, TEAH+, and Na+, respectively). (B) Series of spectra for samples at basic pH 10, (10.1, 12.7, 10.0, and 10.4 for NH4+, K+, TEAH+, and Na+, respectively). The minor impurity peak (1–5% by 19F NMR) that appears upfield in the resolved spectra can be completely removed by adjustment of the SAX HPLC step (see Experimental Section).
Table 1. Effect of pH and Counterions on 19F NMR of β,γ-CHF-UTP 1.
|
19F NMR |
|||||
|---|---|---|---|---|---|
| base | counteriona | pH | Δδ (Hz)b | SF (MHz)c | Δδ (ppm)b |
| dGd | Na+ | 10.5 | 22.6 | 376 | 0.060 |
| U | Na+ | 10.4 | 24.0 | 470 | 0.051 |
| U | Na+ | 10.4 | 29.6 | 564 | 0.052 |
| U | K+ | 12.7 | 23.5 | 470 | 0.050 |
| U | K+ | 12.7 | 28.4 | 564 | 0.050 |
| U | NH4+ | 10.1 | 22.5 | 470 | 0.048 |
| U | NH4+ | 10.1 | 26.8 | 564 | 0.047 |
| Ue | NH4+ | ndf | nd | 235 | nd |
The ionic radii of for Na+, K+, and NH4+ are 1.02, 1.38, and 1.50 Å, respectively.24 Ionic radii are not readily available for bulky alkylammonium cations, but instead rely on partial molar volume.25 As a representative example, the ionic radius of tetramethylammonium is 3.47 Å.24
Δδ is the δ difference between the overlapping diastereomers given in Hz and ppm, respectively.
SF is the spectrometer frequency given in MHz.
Lit. value.17
Lit. value, cation inferred.22
The pH was not reported for the spectrum given but is likely below 10.22 A simulation for SF = 235 MHz (Figure S20) assuming a line width at half height of 8 Hz and δ 12 Hz at pH 10 generated a 12-peak multiplet with substantially resolved outer peaks.
Figure 1 depicts the dramatic transition from an unresolved, broad multiplet to a sharp, distinct set of multiplets when the pH is raised from 7−8 to 10, which narrows the line width to ∼8 Hz. At 470 MHz, the spectrum at pH 10 exhibits 14 of the expected 16 peaks with the Na+, K+, and NH4+ countercations due to partial overlap (Figure 1B). To assign Δδ and J values for the spectra, they were also measured at 564 MHz (Figure 2B). To further validate the assignment, the spin systems of the individual diastereomers were simulated using MestReNova (Mnova 8.1.4). The calculated spectra display satisfactory congruence with the experimental spectra (Figure S4), yielding a Δδ of 0.05 ppm for the mixed 1 diastereomers.
Figure 2.

Effect of SF and counterion effect on 19F NMR spectra (D2O, referenced to CFCl3) of β,γ-CHF-UTP 1 (∼1:1 diastereomers). (A) Series of spectra at 470 MHz for NH4+ (J = 67.1, 56.1, and 45.4 Hz), K+ (J = 67.2, 55.3, and 45.5 Hz), Na+ (J = 67.5, 54.6, and 45.1 Hz). (B) Series of spectra at 564 MHz for NH4+ (J = 65.8, 56.0, and 45.4 Hz), K+ (J = 67.5, 54.4, and 45.6 Hz), and Na+ (J = 65.8, 55.5, and 45.7 Hz). At the higher SF, the predicted 16-peak multiplets are observed.
Although Jakeman et al. asserted that 1 decomposed under the basic conditions we previously recommended to obtain resolved 19F (and 31P) spectra of β,γ-CHF-dGTP diastereomers, no direct evidence or specific experimental conditions were given and the pH of the NMR samples was not provided.21,22 Curiously, the 19F NMR of their purified product does not show decomposition in their synthesis of 1, despite adjustment of the pH to 9.5 prior to lyophilization. Even though the actual pH was not specified, it can be inferred that the pH of their NMR sample was below pH 7.5, based on a series of 31P spectra we acquired over the range pH 7–8 (δ Pβ 2.78; pH 7.4; NH4+). Most importantly, in our hands 1 was quite stable, without detectable NMR decomposition under “basic” conditions (e.g., pH 10.1, counterion NH4+, 48 h at rt, Figure 3). Even after 72 h at rt, only a very slight decomposition to 2 (2.0%) was detected by 19F NMR (Figure 3).
Figure 3.
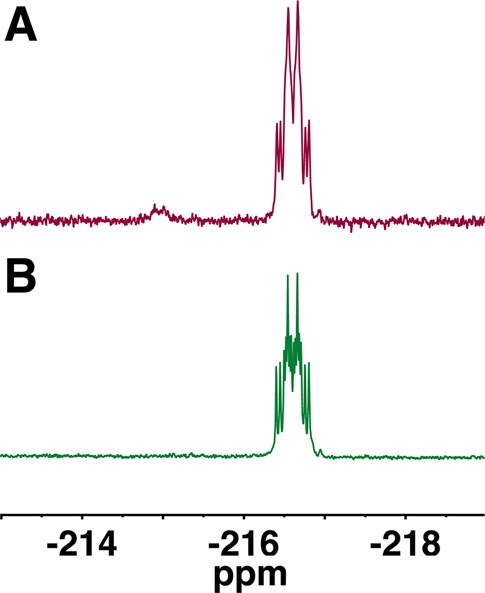
Determination of β,γ-CHF-UTP 1 (∼1:1 diastereomer mixture) stability at pH 10 by 19F NMR (D2O, 470 MHz, referenced to CFCl3). (A) Spectrum acquired prior to final preparative RP-C18 HPLC to remove 2, revealing unreacted 2 at δ −214.95, 1 at δ −216.62 in D2O solution, with the pH adjusted to 10.4. (B) Spectrum acquired after removal of 2 by the RP-C18 HPLC purification step, pH adjustment to 10.1, and then 24 h at rt. No evidence of hydrolysis to 2 can be seen.
Several monofluoromethylene (CHF) phosphonate compounds,26−30 including β,γ-CHF dNTP analogues,13,15,16 have demonstrated utility as probes in enzyme systems. The lack of fluorine in natural systems31 creates a convenient and readily available nuclear spin label for fluorinated analogues, which can use 19F NMR to readily detect stereoselective binding or consumption. 19F and 31P NMR spectroscopy have been extensively used as tools to determine pH and to explore metal cation effects, due to the sensitivity of the chemical shifts of these nuclei to the local chemical environment.32,3319F NMR is an attractive alternative to 31P NMR, due to its greater sensitivity relative to 1H (83% vs 6%), as well as the usual absence of interfering fluorine signals in natural systems. In addition to producing a stronger signal at an equivalent concentration, 19F NMR typically offers a more sensitive response to the local environment compared to 31P NMR and the detection of subtle structural changes.34
19F NMR probes have been frequently utilized as indicators of local pH.33 Between pH 7 and 9.5, the δF of 1 shows a linear downfield-shifted pH-dependence with little or no countercation effects (Figure S5A), suggesting that negative charge generation at the last ionization of the “triphosphate” moiety (pKa4 7.5)21 contributes significantly to this change in δ. When the pH significantly exceeds pKa4 (pH > 10) little or no dependence of δ on pH is observed (Figure S5B). For our present purposes of resolving β,γ-CHF-diastereomer resonances, a similar pH effect is observed. At pH 7–8, the line width is ≫10 Hz presumably due to exchange of the remaining γ-phosphate OH proton,32 in close proximity to the CHF stereocenter.
Although the individual diastereomers are not yet unequivocally assigned, it is possible that the more downfield peak in 19F NMR belongs to the (S)-CHF isomer by analogy with the corresponding dGTP analogue assignment.17
In conclusion, we have reinvestigated the 19F NMR and stability properties of 1, synthesized (for the first time) by the NMP-morpholidate route.20 Our results establish unequivocally that, under our previously reported conditions, the spectra of the individual diastereomers of 1 are easily distinguishable. Furthermore, at the “basic” pH (∼10) required to resolve the spectra by decreasing the line width, 1 was stable for 24 h at rt (and, indeed, showed little change even after 72 h).
Experimental Section
Uridine 5′-β,γ-Fluoromethylenetriphosphosphate, (R)- and (S)-β,γ-CHF-UTP, 1
The compound as an (R/S)-CHF mixture was synthesized using the standard conjugation of activated uridine 5′-monophosphate morpholidate (UMP 5′-M)20 with the tri-n-butylammonium salt of fluoromethylenebis(phosphonic acid), 2.14 Uridine 5′-monophosphate (UMP) disodium salt (265 mg, 0.72 mmol) was treated with Dowex 50WX8 (200–400 mesh, H+ form) to generate the free acid, which was evaporated to dryness. The residue was dissolved in 10 mL of t-BuOH/H2O (2:1). The pH was adjusted to 2 with 0.1 M HCl, a stir bar was added to the reaction flask, and morpholine (distilled, 311.5 μL, 3.60 mmol, 5 equiv) was added dropwise using a 500 μL gastight syringe. After stirring for 30 min, the pH of the mixture was measured and readjusted to 7.5 using 0.1 M HCl. The solution was brought to reflux. N,N′-Dicyclohexylcarbodiimide (DCC, 594 mg, 2.88 mmol, 4 equiv) was dissolved in 5 mL of t-BuOH, and the resulting solution was divided into 10 aliquots of 500 μL. An aliquot was added dropwise every 10 min to the refluxing solution. After an additional 20 min, the progress of the reaction was checked by 31P NMR (UMP 5′-M δ 7.25 (s); UMP δ 3.59 (s)). The mixture was cooled to rt, and precipitated N,N′-dicyclohexylurea was removed by vacuum filtration. The filtrate was evaporated to dryness, and the residue was taken up in 10 mL of H2O. After extraction with ether (3 × 10 mL), the aqueous layer was rotavapped and dissolved and coevaporated with dioxane (3 × 5 mL) yielding a dry white powder of the N,N′-dicyclohexyl-4-morpholinecarboxamidine salt (341 mg, 69%). It should be noted that over time degradation of the product to UMP was observed, but was accounted for using UV and 1H NMR analysis for stoichiometric determination.
2 was prepared from tetraisopropyl methylenebis(phosphonic acid) according to the literature procedure.35,362 (231 mg, 0.119 mmol, 3 equiv) was dissolved in 5 mL of EtOH/H2O (1:1). The pH was slowly adjusted to 4.5 by addition of 10% tributylamine (NBu3) in EtOH, and the solution was allowed to stir for 30 min at rt. The solvent was evaporated under vacuum, and the residue was dried by coevaporation with anhydrous DMF (3 × 3 mL). Then a solution of UMP 5′-M (28 mg, 0.04 mmol, 1 equiv) in anhydrous DMSO (2 mL) was added, and the mixture was stirred for 72 h at rt under N2. The reaction mixture was then passed through a column (25 mm x 15 cm) of a strong anion exchange (SAX) resin eluted with a gradient method (0–10 min, 0–60%; 10–15 min, 60%; 15–25 min, 60–100%) of 0.5 M triethylammonium bicarbonate (TEAB) buffer pH 7.0 with a flow rate of 8.0 mL/min (Figure S1). To eliminate minor impurity (19F NMR, δ near −216.9 ppm), a modified14,15 gradient method (0–20 min, 0%; 20–35 min, 60–100%; 35–45 min, 100%) of 0.5 M TEAB buffer pH 7.0 with a flow rate of 8.0 mL/min was used with the SAX resin column. The desired compound eluted at 30.0 min (UV detection at 259 nm; HPLC trace and 19F NMR spectra, Figure S19). The fractions containing it were evaporated to yield the product as a TEAH+ salt. 19F and 31P NMR analysis revealed traces of 2 in the purified product.
The product was next dissolved in 2 mL of 0.1 M TEAB (pH 7.0) and purified on a C18 reversed-phase (RP-C18) preparative column (5 μm, 250 mm × 21 mm) by isocratic elution with 3.75% CH3CN in 0.1 M TEAB pH 7.0 at a flow rate of 8.0 mL/min. The product 1 eluted at 18.7 min (Figure S2). Evaporation of the corresponding collected fractions gave 6.3 mg (7.8 μmol by UV, 20%) of a clear film as a TEAH+ salt. ESI-MS: m/z 499 [M – H]−. 1H NMR (500 MHz, D2O, pH 7.6, Figure S8) δ 7.99 (d, J = 8.2 Hz, 1H), 6.01 (t, J = 6.5 Hz, 2H), 4.53–4.35 (m, 2H), 4.31 (s, 1H), 4.26 (s, 2H). 19F NMR (470 MHz, D2O, pH 10.4, Figure S9) δ −216.91 and −216.96 (ddd, J = 67.5, 54.6, 45.0 Hz, Δδ 24.0 Hz). 31P NMR (202 MHz, D2O, pH 10.4, Figure S10) δ 7.46 (dd, J = 55.6, 14.3 Hz), 5.26 (ddd, J = 65.4, 28.3, 14.3 Hz), −10.55 (d, J = 28.7, ΔδPα 4.0 Hz).
Effect of Countercation and pH on 19F NMR Spectra of 1
The dual pass HPLC-purified product was further treated with Chelex-100 to remove trace metals. The 1H, 19F, and 31P NMR spectra (D2O, pH 7.6) were acquired and did not manifest distinguishable peaks for the individual diastereomers. Following treatment with Chelex-100, the compound was reisolated by evaporation and then dissolved in 2 mL of H2O, and the resulting solution was divided into four aliquots. Dowex 50WX8 (200–400 mesh, H+ form) resin was converted to alternate cation forms by treatment with 1 M HCl, rinsing with H2O until the eluate was neutral (pH paper), and subsequent treatment with the desired cation as a 10% (w/w) of its hydroxide salt solution in H2O (i.e., NaOH for Na+, KOH for K+, and NH4OH for NH4+), followed by additional washing with H2O until the eluate was again neutral. A 500 μL aliquot of 1 as described above was passed into each exchange-resin column, which was next washed with several portions of H2O. The samples were then rotavapped to yield four samples with a different countercation. NMR samples were prepared by adding 500 μL of D2O (yielding nucleotide concentrations of ca. 3.9 mM), and the pH (NMR tube electrode) was determined prior to acquiring the 1H, 19F, and 31P NMR spectra. The spectra of 1 near physiological pH did not result in resolved diastereomer-peaks, irrespective of the counterion used.
Immediately after NMR acquisition, the pH of the NMR sample was adjusted to ≥10 using a 10% (w/w) solution of the counterion hydroxide (or TEA) in D2O. Because the nucleotide concentrations in the NMR sample were low, the pH adjustment required minimal added solution. After the sample pH was determined, spectra were reacquired for each countercation sample. Overnight spectra were acquired on both 500 and 600 MHz spectrometers for each counterion sample to clarify assignment of Δδ vs J values.
Acknowledgments
This work was supported by funding from the Dana and David Dornsife College of the University of Southern California and the National Institutes of Health (U19CA177547, C.E.M.) and in part by a National Science Foundation Graduate Research Fellowship (DGE-0937362, C.S.H.). We thank Inah Kang for assistance in preparing the manuscript.
Supporting Information Available
HPLC analysis and preparative traces, counterion experimental data, 1H NMR spectrum and additional 19F, 31P NMR spectra, MS and UV–vis spectra for 1. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Engel R. Chem. Rev. 1977, 77, 349. [Google Scholar]
- Verma S.; Eckstein F. Annu. Rev. Biochem. 1998, 67, 99. [DOI] [PubMed] [Google Scholar]
- Kool E. T. Curr. Opin. Chem. Biol. 2000, 4, 602. [DOI] [PubMed] [Google Scholar]
- Elphick L. M.; Lee S. E.; Gouverneur V.; Mann D. J. ACS Chem. Biol. 2007, 2, 299. [DOI] [PubMed] [Google Scholar]
- Herdewijn P.; Marliere P. FEBS Lett. 2012, 586, 2049. [DOI] [PubMed] [Google Scholar]
- Arabshahi L.; Khan N. N.; Butler M.; Noonan T.; Brown N. C.; Wright G. E. Biochemistry 1990, 29, 6820. [DOI] [PubMed] [Google Scholar]
- Hamilton C. J.; Roberts S. M.; Shipitsin A. Chem. Commun. 1998, 1087. [Google Scholar]
- Shipitsin A.; Victorova L.; Shirokova E.; Dyatkina N.; Goryunova L.; Beabealashvilli R.; Hamilton C.; Roberts S. J. Chem. Soc., Perkin Trans. 1 1999, 1039. [DOI] [PubMed] [Google Scholar]
- Martynov B. I.; Shirokova E. A.; Jasko M. V.; Victorova L. S.; Krayevsky A. A. FEBS Lett. 1997, 410, 423. [DOI] [PubMed] [Google Scholar]
- Alexandrova L. A.; Skoblov A. Y.; Jasko M. V.; Victorova L. S.; Krayevsky A. A. Nucleic Acids Res. 1998, 26, 778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna C. E.; Kashemirov B. A.; Peterson L. W.; Goodman M. F. Biochim. Biophys. Acta 2010, 1804, 1223. [DOI] [PubMed] [Google Scholar]
- Blackburn G. M.; Kent D. E.; Kolkmann F. J. Chem. Soc., Perkin Trans. 1 1984, 1119. [Google Scholar]
- Sucato C. A.; Upton T. G.; Kashemirov B. A.; Batra V. K.; Martínek V.; Xiang Y.; Beard W. A.; Pedersen L. C.; Wilson S. H.; McKenna C. E. Biochemistry 2007, 46, 461. [DOI] [PubMed] [Google Scholar]
- McKenna C. E.; Kashemirov B. A.; Upton T. G.; Batra V. K.; Goodman M. F.; Pedersen L. C.; Beard W. A.; Wilson S. H. J. Am. Chem. Soc. 2007, 129, 15412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batra V. K.; Pedersen L. C.; Beard W. A.; Wilson S. H.; Kashemirov B. A.; Upton T. G.; Goodman M. F.; McKenna C. E. J. Am. Chem. Soc. 2010, 132, 7617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertell K.; Wu Y.; Zakharova V. M.; Kashemirov B. A.; Shock D. D.; Beard W. A.; Wilson S. H.; McKenna C. E.; Goodman M. F. Biochemistry 2012, 51, 8491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Zakharova V. M.; Kashemirov B. A.; Goodman M. F.; Batra V. K.; Wilson S. H.; McKenna C. E. J. Am. Chem. Soc. 2012, 134, 8734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes D. E.; Lindahl T. Annu. Rev. Genet. 2004, 38, 445. [DOI] [PubMed] [Google Scholar]
- Beard W. A.; Wilson S. H. Chem. Rev. 2006, 106, 361. [DOI] [PubMed] [Google Scholar]
- Roseman S.; Distler J. J.; Moffatt J. G.; Khorana H. G. J. Am. Chem. Soc. 1961, 83, 659. [Google Scholar]
- Beaton S. A.; Jiang P. M.; Melong J. C.; Loranger M. W.; Mohamady S.; Veinot T. I.; Jakeman D. L. Org. Biomol. Chem. 2013, 11, 5473. [DOI] [PubMed] [Google Scholar]
- Mohamady S.; Jakeman D. L. J. Org. Chem. 2005, 70, 10588. [DOI] [PubMed] [Google Scholar]
- Mohamady S.; Taylor S. D. J. Org. Chem. 2011, 76, 6344. [DOI] [PubMed] [Google Scholar]
- Collins K. D. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klofutar C.; Rudan-Tasic D.; Mancic-Klofutar V. J. Solution Chem. 1997, 26, 1037. [Google Scholar]
- Nieschalk J.; Batsanov A. S.; O’Hagan D.; Howard J. Tetrahedron 1996, 52, 165. [Google Scholar]
- Berkowitz D. B.; Bose M.; Pfannenstiel T. J.; Doukov T. J. Org. Chem. 2000, 65, 4498. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Qian L.; Prestwich G. D. J. Org. Chem. 2003, 68, 5320. [DOI] [PubMed] [Google Scholar]
- Cox R. J.; Gibson J. S.; Hadfield A. T. ChemBioChem. 2005, 6, 2255. [DOI] [PubMed] [Google Scholar]
- Forget S. M.; Bhattasali D.; Hart V. C.; Cameron T. S.; Syvitski R. T.; Jakeman D. L. Chem. Sci. 2012, 3, 1866. [Google Scholar]
- Berkowitz D. B.; Karukurichi K. R.; de la Salud-Bea R.; Nelson D. L.; McCune C. D. J. Fluorine Chem. 2008, 129, 731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel H. J.; Bridger W. A. Biochemistry 1982, 21, 394. [DOI] [PubMed] [Google Scholar]
- Taylor J. S.; Deutsch C. Biophys. J. 1983, 43, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerig J. T. Prog. Nucl. Magn. Reson. Spectrosc. 1994, 26Part 4293. [Google Scholar]
- Marma M. S.; Khawli L. A.; Harutunian V.; Kashemirov B. A.; McKenna C. E. J. Fluorine Chem. 2005, 126, 1467. [Google Scholar]
- McKenna C. E.; Shen P. D. J. Org. Chem. 1981, 46, 4573. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.