

Abstract
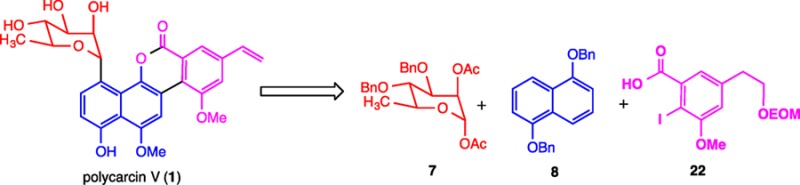
The convergent total synthesis of polycarcin V, a gilvocarcin-type natural product that shows significant cytotoxicity with selectivity for nonsmall-cell lung cancer, breast cancer, and melanoma cells, has been achieved in 13 steps from 7, 8, and 22; the sequence features a stereoselective α-C-glycosylation reaction for the union of protected carbohydrate 7 and naphthol 8. The association constant for the binding of polycarcin V to duplex DNA is similar to that previously reported for gilvocarcin V.
The gilvocarcin family of C-aryl glycoside natural products1 have been shown to exhibit high antitumor activity with low overall toxicity.2 A likely mode of action of gilvocarcin V, the most studied member of the gilvocarcins, involves intercalation of the aromatic chromophore into DNA, followed by UV-induced covalent linkage of the natural product to DNA by a [2 + 2]-cycloaddition between the vinyl moiety and a thymine residue.3 Photoactivated gilvocarcin V is also able to selectively cross-link DNA and the phosphorylated form of histone H3 and GRP78, a heat shock protein.4 Importantly, gilvocarcins M and E, both of which bear aliphatic residues instead of a vinyl group at C.8 of the chromophore, are not cytotoxic.5
Both Waring6 and McGee7 have previously demonstrated that the presence of a carbohydrate moiety on an intercalating chromophore contributes positively and significantly to the binding association with DNA. Studies on the interaction of C-aryl glycoside natural products with DNA have shown that the carbohydrate moieties typically occupy the minor groove in the bound complex, where noncovalent interactions between functional groups present on the sugar and residues in the minor groove are established.8 It has been proposed that these carbohydrate–DNA noncovalent contacts may be largely responsible for the binding-site sequence selectivity of the C-aryl glycosides.9 Furthermore, the identity of the sugar substituent of C-aryl glycosides such as the gilvocarcins may also be relevant to cell-type specificity, potency, transport, and pharmacokinetics.10
Polycarcin V (Figure 1) is a recently isolated gilvocarcin-type natural product obtained from a culture extract of Streptomyces polyformus sp. nov. (YIM 33176).11 This substance co-occurs with gilvocarcin V but possesses an α-linked l-rhamnopyranose moiety instead of a β-linked d-fucofuranose sugar; indeed, the α-C-glycosidic linkage is rare among the known C-aryl glycoside natural products.1d,1e Polycarcin V shows significant cytotoxicity with selectivity for non-small-cell lung cancer (LXF 1211 L and LXFL 529L, IC70 < 0.3 ng/mL and 0.3 ng/mL), breast cancer (MCF7, MDAMB231, MDAMB 468, IC70 from <0.3 ng/mL to 4 ng/mL), and melanoma cells (MEXF 462NL, MEXF 514 L, MEXF 520L, IC70 from <0.3 ng/mL to 0.4 ng/mL), and its antiproliferative fingerprint is virtually identical to that of actinomycin D. With the aim of evaluating the DNA binding affinity of polycarcin V for comparison with the known gilvocarcins, we sought to undertake the total synthesis of this natural product.
Figure 1.

Structures of polycarcin V (1) and gilvocarcins V (2), M (3), and E (4).
Since the Suzuki O → C glycoside rearrangement reaction, a typical strategy employed for the creation of aryl-glycosidic carbon–carbon bonds,12 favors the formation of β-glycosidic linkages, we envisioned that the carbon–carbon bond between the aromatic and carbohydrate moieties of polycarcin V could be fashioned instead by a Lewis acid mediated Friedel–Crafts-type glycosylation13 utilizing a rhamnosyl donor containing a suitable protecting group at C.2′ to direct the stereochemistry of bond formation at C.1′ (7, Figure 2). Standard oxidation and selective hydroxyl group protection would then provide a naphthol glycoside capable of carbodiimide-mediated coupling with protected iodoarene carboxylic acid 22, prepared from 3,5-dihydroxybenzoic acid. Following the precedent of Suzuki,14 palladium-catalyzed intramolecular arylation, deprotection, and hydroxyl group elimination would then provide the natural product.
Figure 2.

Retrosynthetic analysis of polycarcin V.
The synthesis commenced with assembly of the carbohydrate and aromatic fragments for coupling. For the preparation of diacetate 7,15l-rhamnose was first treated with allyl alcohol under acidic conditions (cat. H2SO4, 85 °C, 3 h; Scheme 1) to provide the corresponding allyl glycoside, which then underwent regioselective acetonide protection of the syn C.2′ and C.3′ hydroxyl groups (DMP, cat. pTsOH, DMF) to furnish 5 in 90% overall yield. Benzylation of the C.4′ hydroxyl group and acetonide hydrolysis according to the protocol of Venkateswarlu16 then gave rise to diol 6 in 80% yield, which was regioselectively benzylated utilizing Hanessian’s dibutyltin oxide method.17 Acylation of the C.2′ hydroxyl and acetolysis of the allyl glycoside under acidic conditions then provided 7 in 70% overall yield from 6.
Scheme 1. Synthesis of Diacetate 7.

Benzylation of commercially available 1,5-dihydroxynaphthalene under basic conditions provided aromatic coupling partner 8 (Scheme 2). The union of compounds 7 and 8 was accomplished by treatment of the mixture in CH2Cl2 (0.5 M) with 1.5 equiv of TMSOTf at room temperature for 30 min.13C-Glycoside 9 was obtained cleanly in 70% yield with >95:5 α:β stereoselectivity. In model studies, it was found that similar yields and stereoselectivities of glycosylated products could be obtained when rhamnosyl acetates bearing a C.2′ isobutyryl ester were employed. Anchimeric participation by the C.2′ ester group may explain the ease of this C-glycosylation reaction, both assisting the expulsion of the carbohydrate C.1′ acetate and shielding the β-face of the resulting oxocarbenium ion from attack by the naphthyl nucleophile.18
Scheme 2. C-Glycosylation of Naphthalene 8.

Elaboration of C-glycoside 9 toward the natural product commenced with formylation of the aromatic ring under Vilsmeier conditions (Scheme 3).19 After much experimentation, it was found that stirring 9 with excess POCl3 and DMF in toluene at reflux for 6 h gave aldehyde 10 in 70% yield. Baeyer–Villiger oxidation of 10 under acidic conditions (H2O2, cat. H2SO4, THF, MeOH)20 then furnishes phenol 11 cleanly in 92% yield. Sequential exposure of 11 to ceric ammonium nitrate and sodium dithionite in an oxidation/reduction sequence then provides diol 12 in 80% yield. At this point, attempts to selectively methylate the less hindered C.2 hydroxyl group gave rise to inseparable mixtures of mono- and dimethylated products. Due to the instability of 12 under basic conditions, deprotonation of the phenolic hydroxyl groups had to be performed under low-temperature conditions with NaHMDS in the presence of the electrophilic methylating agent (NaHMDS, MeOTf, THF, −78 to 0 °C). Intriguingly, careful analysis of 1H NMR data revealed that the major monomethylated product possessed the methyl group on the C.5 hydroxyl. This result, together with model studies on naphthyl systems lacking the carbohydrate moiety demonstrated that the more sterically hindered hydroxyl at C.5 is in fact more reactive toward methylation under basic conditions than the less hindered C.2 hydroxyl, likely due to resonance contributions from the C.1 benzyloxy ether.21 It was ultimately found that temporary protection of the C.5 hydroxyl with chloromethyl ethyl ether (NaHMDS, THF, −78 °C) provided the corresponding C.5 acetal 13 in 65% yield. Subsequent methylation (Me2SO4, THF, NaHMDS, −78 °C) of the C.2 hydroxyl and hydrolysis of the C.5 acetal (catalytic HCl in methanol) gives rise to the desired naphthol coupling partner 14b in 86% yield from 13.
Scheme 3. Elaboration of C-Glycoside 9 to Naphthol 14.

Next, we envisioned that the iodoarene carboxylic acid coupling partner could be fashioned from 3,5-dihydroxybenzoic acid 15 (Scheme 4). Careful methylation of 15 with dimethyl sulfate (2.2 equiv) and excess K2CO3 in DMF gave rise to a 75% yield of methyl ester 16, bearing a single phenolic hydroxyl group.22 Triflation (Tf2O, Pyr), followed by palladium-catalyzed cross coupling with allyltributyltin, furnished alkene 17 in 80% overall yield. Next, oxidative cleavage of the olefin was accomplished in two steps (OsO4, acetone, t-BuOH, NMO; KIO4, acetone, pH 6.5 buffer) to provide an intermediate aldehyde, which was immediately reduced with NaBH4 in methanol to provide primary alcohol 18 in 67% yield. Protection of the alcohol with chloromethyl ethyl ether then afforded acetal 19 in 77% yield.
Scheme 4. Preparation of 22.

Introduction of the iodine atom by directed ortho metalation23 required reduction of the methyl ester to the primary alcohol, which was accomplished by exposing 19 to LiAlH4 in ether at room temperature, giving rise to 20 in 98% yield. Treatment of 20 with excess n-BuLi in ether for 3 h at room temperature, followed by quenching with I2 in THF, furnished a 65% yield of iodide 21. Finally, sequential oxidation of 21 with PCC (PCC, DCM, KOAc) and NaClO2 (NaClO2, NaH2PO4, H2O, t-BuOH) afforded the target carboxylic acid 22 in 80% yield.
Carbodiimide-mediated coupling of naphthol 14b with carboxylic acid 22 smoothly afforded ester 23 in 98% yield (Scheme 5). Subsequent intramolecular Heck arylation with (PPh3)2PdCl2 and KOAc in DMA gave rise to lactone 24a in 64% overall ysield. Cleavage of the benzyl ether protecting groups was next accomplished by exposure of 24a to catalytic quantities of Pearlman’s catalyst in THF/EtOH under an atmosphere of hydrogen gas; acylation (Ac2O, pyr) then provided the tetraacetate 24b in 64% yield. Acetal cleavage under Lewis acidic conditions (TMSBr, CH2Cl2, −78 °C → −10 °C)14 proceeded uneventfully to furnish the corresponding primary alcohol 25. Dehydration under Grieco’s conditions (2-NO2C6H4SeCN, PBu3, THF; H2O2)24 afforded alkene 26 in 57% yield, which was identical in its NMR spectroscopic properties to an authentic sample of polycarcin V tetraacetate prepared by acylation (Ac2O, Pyr) of the natural product (see the Supporting Information for comparison of spectra). Deacylation of 26 (NaCN, MeOH) gave 1, which proved to be sensitive to light and concentration. The 1H NMR, 13C NMR, optical rotation, and HRMS of synthetic 1 were in full accord with the data reported for natural polycarcin V.11
Scheme 5. Completion of the Synthesis of 1.

The binding of 1 to duplex DNA was explored by fluorescence and UV spectroscopies. Excitation of 1 (0.5 μM in 10 mM Tris–EDTA buffer) at 380 nm in the presence of increasing concentrations of calf thymus (CT) DNA in the dark resulted in an enhancement of the fluorescence emission intensity at 470 nm; the addition of CT DNA to 1 also produced a blue shift in the fluorescence spectrum of 1, an observation consistent with what has been previously reported for both gilvocarcin V and M.5,25 Analysis of the fluorescence data by nonlinear regression based on the orthodox treatment of McGhee and von Hippel26 (see the Supporting Information) gave the association constant Ka = 1.7 (±0.1) × 106 M–1, a value which agrees with those reported for gilvocarcin V by Arce et al. (1.1 × 106 M–1)5 and Gasparro et al. (6.6 × 105 M–1).25 A similar analysis of 1 in the presence of increasing concentrations of either poly(dAdT)·poly(dAdT) or poly(dGdC)·poly(dGdC) revealed that polycarcin V binds AT-rich DNA with approximately 1 order of magnitude greater binding affinity than GC-rich DNA, a finding also consistent with the known covalent association of gilvocarcin V with thymine residues.3 Furthermore, thermal denaturation studies showed a significant (+3 °C) shift in the TM (64 °C) of salmon testes DNA in the presence of 1, even at low 1:DNA ratios (0.05).27 These data suggest that polycarcin V has a strong noncovalent association with duplex DNA in the absence of light and that the mode of association of 1 with DNA is likely similar to that of gilvocarcin V.
In conclusion, we have achieved the total synthesis of the antitumor α-C-aryl glycoside natural product polycarcin V in 3.2% overall yield from protected carbohydrate 7, naphthol 8, and arene carboxylic acid 22. This route is easily amenable to the synthesis of derivatives that incorporate alternate carbohydrate moieties and/or C.8 aryl substituents. The association constant for the binding of polycarcin V to CT DNA has been determined by fluorescence spectroscopy and was found to coincide with Ka values determined previously for gilvocarcin V. Due to the photosensitivity of 1, current efforts are directed toward the synthesis of the C.8 phenyl derivative of polycarcin, which may retain its strong DNA binding ability without the possibility of forming covalent adducts with DNA.
Acknowledgments
We thank the National Institutes of Health (SC3 GM 096899-01) for their generous support of our research program.
Supporting Information Available
Detailed experimental procedures including spectroscopic and analytical data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Hua D. H.; Saha S. Recl. Trav. Chim. Pay-B 1995, 114, 341. [Google Scholar]; b Nakano H.; Matsuda Y.; Ito K.; Ohkubo S.; Morimoto M.; Tomita F. J. Antibiot. 1981, 34, 701. [DOI] [PubMed] [Google Scholar]; c Nakano H.; Matsuda Y.; Ito K.; Ohkubo S.; Morimoto M.; Tomita F. J. Antibiot. 1981, 34, 266. [DOI] [PubMed] [Google Scholar]; d Hansen M. R.; Hurley L. H. Acc. Chem. Res. 1996, 29, 249. [Google Scholar]; e Hacksell U.; Daves G. D. Prog. Med. Chem. 1985, 22, 1. [DOI] [PubMed] [Google Scholar]
- Wei T. T.; Bryne K. M.; Warnick-Pickle D.; Greenstein M. J. Antibiot. 1982, 35, 545. [DOI] [PubMed] [Google Scholar]
- McGee L. R.; Misra R. J. Am. Chem. Soc. 1990, 112, 2386. [Google Scholar]
- Matsumoto A.; Hanawalt P. C. Cancer Res. 2000, 60, 3921. [PubMed] [Google Scholar]
- Oyola R.; Arce R.; Alegria A. E.; Garcia C. Photochem. Photobiol. 1997, 65, 802. [DOI] [PubMed] [Google Scholar]
- Bailly C.; Colson P.; Houssier C.; Rodrigues-Periera E.; Prudhomme M.; Waring M. J. Mol. Pharmacol. 1998, 53, 77. [DOI] [PubMed] [Google Scholar]
- Tse-Dinh Y. C.; McGee L. R. Biochem. Biophys. Res. Commun. 1987, 143, 808. [DOI] [PubMed] [Google Scholar]
- a Owen E. A.; Burley G. A.; Carver J. A.; Wickham G.; Keniry M. A. Biochem. Bioph. Res. Commun. 2002, 290, 1602. [DOI] [PubMed] [Google Scholar]; b Pavlopoulos S.; Bicknell W.; Wickham G.; Craik D. J. J. Mol. Recognit. 1999, 12, 346. [DOI] [PubMed] [Google Scholar]; c Pavlopoulos S.; Bicknell W.; Craik D. J.; Wickham G. Biochemistry 1996, 35, 9314. [DOI] [PubMed] [Google Scholar]; d Hansen M.; Yun S.; Hurley L. Chem. Biol. 1995, 2, 229. [DOI] [PubMed] [Google Scholar]
- a Cairns M. J.; Murray V. Biochem. Mol. Biol. Int. 1998, 46, 267. [DOI] [PubMed] [Google Scholar]; b Hardman L. C.; Murray V. Biochem. Mol. Biol. Int. 1997, 42, 349. [DOI] [PubMed] [Google Scholar]; c Murray V.; Moore A. G.; Matias C.; Wickham G. BBA-Gene Struct. Expr. 1995, 1261, 195. [DOI] [PubMed] [Google Scholar]; d Prakash A. S.; Moore A. G.; Murray V.; Matias C.; McFadyen W. D.; Wickham G. Chem.–Biol. Interact. 1995, 95, 17. [DOI] [PubMed] [Google Scholar]; e Kamei N.; Sekiguchi T.; Tsuboi M.; Ishii S.; Ohtsubo E.; Maeda Y. Nucleic Acids Res. 1994, 31, 87. [Google Scholar]; f Sun D.; Hansen M.; Hurley L. J. Am. Chem. Soc. 1995, 117, 2430. [Google Scholar]
- a Greenstein M.; Monji T.; Yeung R.; Maiese W. M.; White R. J. Antimicrob. Agents Chemother. 1986, 29, 861. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Singh K. J. Antibiot. 1984, 37, 71. [DOI] [PubMed] [Google Scholar]; c Narita T.; Matsumoto M.; Mogi K.; Kukita K.; Kawahara R.; Nakashima T. J. Antibiot. 1989, 42, 347. [DOI] [PubMed] [Google Scholar]; d Misra R.; Tritch H. R. III; Pandey R. C. J. Antibiot. 1985, 38, 1280. [DOI] [PubMed] [Google Scholar]
- Li Y. Q.; Huang X. S.; Ishida K.; Maier A.; Kelter G.; Jiang Y.; Peschel G.; Menzel K. D.; Li M. G.; Wen M. L.; Xu L. H.; Grabley S.; Fiebig H. H.; Jiang C. L.; Hertweck C.; Sattler I. Org. Biomol. Chem. 2008, 6, 3601. [DOI] [PubMed] [Google Scholar]
- a Matsumoto T.; Katsuki M.; Suzuki K. Tetrahedron Lett. 1988, 29, 6935. [Google Scholar]; b Matsumoto T.; Katsuki M.; Jona H.; Suzuki K. Tetrahedron Lett. 1989, 30, 6185. [Google Scholar]; c Matsumoto T.; Hosoya T.; Suzuki K. Tetrahedron Lett. 1990, 31, 4629. [Google Scholar]; d Matsumoto T.; Katsuki M.; Jona H.; Suzuki K. J. Am. Chem. Soc. 1991, 113, 6982. [Google Scholar]; e Yamauchi T.; Watanabe Y.; Suzuki K.; Matsumoto T. Synthesis 2006, 2818. [Google Scholar]
- For previous examples of Lewis acid promoted Friedel–Crafts-type glycosylations using anomeric acetates, see:; a Yepremyan A.; Salehani B.; Minehan T. G. Org. Lett. 2010, 12, 1580. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kalvoda L. Collect. Czech. Chem. Commun. 1973, 38, 1679. [Google Scholar]; c Vorbruggen H.; Kroliliewicz K.; Bennua B. Chem. Ber. 1981, 114, 1234. [Google Scholar]
- Hosoya T.; Takashiro E.; Matsumoto T.; Suzuki K. J. Am. Chem. Soc. 1994, 116, 1004. [Google Scholar]
- For an alternate preparation of 7, see:; a Pozsgay V.; Brison J.-R.; Jennings H. J. Can. J. Chem. 1987, 65, 2764. [Google Scholar]; See also:; b Liptak A.; Nanasi P. Carbohydr. Res. 1981, 93, 43. [Google Scholar]; c Chen Q.; Kong F.; Cao L. Carbohydr. Res. 1993, 240, 107. [DOI] [PubMed] [Google Scholar]
- Swamy N. R.; Venkateswarlu Y. Tetrahedron Lett. 2002, 43, 7549. [Google Scholar]
- David S.; Hanessian S. Tetrahedron 1985, 41, 643. [Google Scholar]
- The C.2 ester moiety not only directs the anomeric stereochemistry during the carbon–carbon bond-forming step but also preserves that stereochemistry through other synthetic steps where anomerization is possible. See ref (13a) and also:Schmidt R. R.; Castro-Palomino J. C.; Retz O. Pure Appl. Chem. 1999, 71, 729. [Google Scholar]
- Meth-Cohn O.; Stanforth S. P. Comp. Org. Synth. 1991, 2, 777. [Google Scholar]
- Roy A.; Reddy K. B.; Mohanta P. K.; Ila J.; Junjappa H.. Synth. Commun. 1999, 29, 3781.
- A similar observation was made by Suzuki: see ref (14).
- Nawrat C. C.; Palmer L. I.; Blake A. J.; Moody C. J. J. Org. Chem. 2013, 78, 5587. [DOI] [PubMed] [Google Scholar]
- Anctil E. J.-G.; Snieckus V.. The Directed Ortho-Metallation Cross-Coupling Nexus. Synthetic Methodology for the Formation of Aryl–Aryl and Aryl–Heteroatom–Aryl Bonds. In Metal-Catalyzed Cross Coupling Reactions; De Meijere A., Diederich F., Eds.; Wiley-VCH: Weinheim, 2004; Vol. 2, pp 761–813. [Google Scholar]
- Grieco P. A.; Gilman S.; Nishizawa M. J. Org. Chem. 1976, 41, 1485. [Google Scholar]
- Gasparro F.; Knobler R.; Edelson R. Chem.–Biol. Interact. 1988, 67, 255. [DOI] [PubMed] [Google Scholar]
- McGhee J.; von Hippel P. J. Mol. Biol. 1974, 86, 469. [DOI] [PubMed] [Google Scholar]; Tse W. C.; Boger D. L. Acc. Chem. Res. 2004, 37, 61. [DOI] [PubMed] [Google Scholar]
- Shi X.; Chaires J. B. Nucleic Acids Res. 2006, 34, e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.