

Abstract
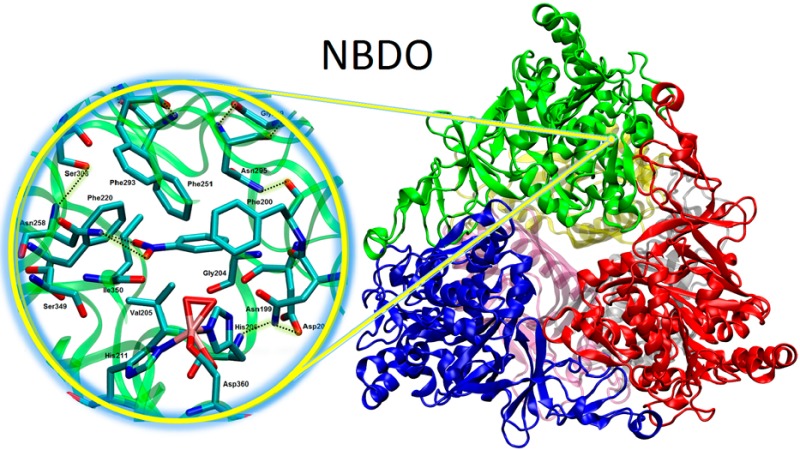
Molecular dynamics simulation of the oxygenase component of nitrobenzene dioxygenase (NBDO) system, a member of the naphthalene family of Rieske nonheme iron dioxygenases, has been carried out using the AMBER force field combined with a new set of parameters for the description of the mononuclear nonheme iron center and iron–sulfur Rieske cluster. Simulation results provide information on the structure and dynamics of nitrobenzene dioxygenase in an aqueous environment and shed light on specific interactions that occur in its catalytic center. The results suggest that the architecture of the active site is stabilized by key hydrogen bonds, and Asn258 positions the substrate for oxidation. Analysis of protein–water interactions reveal the presence of a network of solvent molecules at the entrance to the active site, which could be of potential catalytic importance.
Introduction
Nitroarene dioxygenases are members of the naphthalene family of Rieske nonheme iron dioxygenases, which are able to oxidize a variety of aromatic compounds through direct incorporation of atmospheric oxygen into the substrate, leading to the formation of a cis-dihydrodiol.1,2 Apart from cis-dihydroxylation, Rieske dioxygenases also catalyze monohydroxylation, desaturation, sulfoxidation, O- and N-dealkylation, and amine oxidation.3−8 The broad substrate specificity and versatility of Rieske dioxygenases have made them useful in the production of oxygenated compounds for biotechnology.9 Examples of their application in large-scale biosynthesis include production of indigo10 and generation of chiral precursors of drugs, such as indinavir sulfate.11−13 Being involved in bacterial metabolism, Rieske enzymes initiate degradation of many environmental contaminants, and thus remain viable targets for bioremediation platforms.14,15 Nitroarene dioxygenases are uniquely able to oxidize the aromatic ring of nitroarene compounds, resulting in the elimination of the nitro group, which facilitates further metabolization. Nitrobenzene dioxygenase (NBDO), in particular, is the only nitroarene dioxygenase that can catalyze the denitration of all mono- and dinitrotoluenes.16
NBDO, like other Rieske dioxygenases, is a three-component system that consists of an NADH-dependent flavoprotein reductase, a Rieske [2Fe-2S] ferredoxin, and an α3β3 terminal oxygenase.1,17 During the catalytic cycle two electrons are transferred one at a time from the reductase to the [2Fe-2S] cluster in ferredoxin and subsequently to the Rieske [2Fe-2S] cluster and mononuclear iron(II) center located in the terminal oxygenase component, where the reaction takes place.19−23 The oxygenase component is an α3β3 heterohexamer with a mushroom-shaped quaternary structure, in which α subunits containing the active site and Rieske domains bear catalytic function, while β subunits, which are located more than 10 Å from the active sites, are believed to have a purely structural function.16 The active site domain in each α subunit hosts a high-spin mononuclear FeII bound to two histidines and a bidentate aspartate residue, forming the 2-his-1-carboxylate facial triad motif found in many mononuclear nonheme iron(II) oxygenases.24 On the other hand, the Rieske domain contains a Rieske type [2Fe-2S] cluster coordinated by two cysteine and two histidine residues (Figure 1).
Figure 1.

Schematic representation of the interface between the mononuclear iron center located in the active site of one α subunit and the Rieske domain from a neighboring subunit connected by hydrogen bonds through Asp203 residue. Some of the possible hydrogen bond interactions involved in the contact are indicated.
Within a single α subunit, these two domains are separated by a distance of around 45 Å, which is too far for an effective electron transfer. In a tight trimer formed by three α subunits, however, the Rieske [2Fe–2S] cluster and mononuclear iron(II) center from two adjacent α subunits are only 12 Å apart and are connected through a conserved Asp residue possibly serving as a route for the transfer of the two electrons from NAD(P)H to the active site during the catalytic cycle.
The most thoroughly studied representative of Rieske dioxygenases is naphthalene dioxygenase (NDO), with which NBDO shares 80% sequence identity.16 Crystal structures of free NDO,18 as well as complexes along the reaction pathway25 have been solved and serve as the prototype for the general dioxygen activation and substrate oxidation mechanism in Rieske dioxygenases.26 As it was shown in a single turnover study of NDO, an isolated oxygenase component is capable of cis-dihydroxylation of an aromatic substrate, provided the Rieske cluster is in the reduced state.27 Reduction of the Rieske cluster and formation of the enzyme–substrate complex triggers side-on binding of O2 to the mononuclear iron(II) and its subsequent activation.16,19,26,28−30 Through extensive studies on NDO, including crystallographic data,25 single turnover27 and peroxide shunt experiments,31 and theoretical calculations,32,33 a ferric (hydro)peroxo complex formed upon dioxygen reduction has been identified as a key reaction intermediate. However, the possibility of a high-spin iron-oxo intermediate formed upon oxygen–oxygen bond cleavage being the oxidant that eventually attacks the substrate has also been indicated and found support, especially in studies of NDO reactivity using probe molecules.27,34 Ambiguities regarding the nature of the iron–oxygen intermediate oxidizing the substrate and the versatility of reactions catalyzed by Rieske dioxygenases have prompted numerous experimental and theoretical studies of their catalytic mechanism.9,30,32,35−38 An understanding of the cis-dihydroxylation mechanism of Rieske dioxygenases is particularly valuable in the development of bioremediation measures and protein engineering of dioxygenases to modify selectivity and enhance efficiency.9,26,39
Substrate specificity of Rieske dioxygenases is attributed to specific interactions between the substrate and active site residues.26 Hence, there exists an apparent need for a detailed investigation of the specific interactions taking place in the active site of these enzymes, in hopes of understanding the structural basis for their activity toward different compounds. In particular, the activity of nitroarene dioxygenases toward nitroaromatic compounds is attributed to the presence of a polar Asn residue forming a hydrogen bond with the nitro group of the substrate, positioning it for ring oxidation. Moreover, nitroarene dioxygenases catalyze both aromatic cis-dihydroxylation and monohydroxylation of alkyl side chains, with the predominant reaction dependent on the binding position of the substrate.16 It is thus important to consider the protein environment in modeling these reactions, which can be accomplished by means of biomolecular simulations of the enzymes of interest. While combined quantum mechanical/molecular mechanical (QM/MM) studies have been carried out on enzymes that also have the 2-his-1-carboxylate facial triad motif,40−42 there are currently no published force field parameters specifically developed for this moiety, and only partial atomic charges for the Rieske cluster in cytochrome bc1 complex, which also has one Fe atom bound to histidine residues and the other bound to cysteine residues, have been determined and combined with CHARMM force field parameters.43,44
In this work, we present a classical molecular dynamics simulation of the oxygenase component of the NBDO system in explicit water environment using the AMBER ff99SB force field,45 including parameters developed for the mononuclear iron center and Rieske cluster. Results of the molecular simulation provide information regarding the structure and dynamics of the enzyme in aqueous solution and specifically give insight into key hydrogen-bonding interactions within the active site region, including protein–water interactions in the substrate binding pocket. The interface between the Rieske cluster and mononuclear iron center is also analyzed and described in terms of hydrogen bonds formed between the two domains. Additionally, the results of the simulation are compared with available experimental data to estimate the performance of the new parameters developed for the metal centers of the protein. Since the residues coordinating the Rieske cluster and mononuclear iron are completely conserved within Rieske dioxygenases,16 the parameters derived herein might be applied to studying other members of this family of enzymes.
Computational Methods
System Preparation
The crystal structure of nitrobenzene dioxygenase in complex with nitrobenzene (PDB ID: 2BMQ) consisting of a single αβ dimer was used as the initial structure and transformed into an α3β3 heterohexamer by generating symmetry-related copies of monomers using Coot.46 The enzyme was assumed to be in its oxidized form, with dioxygen reduced to the form of a side-on bound hydro(peroxo) moiety. Therefore, the two water molecules coordinated to Fe in the active site (HOH2227 and HOH2401) were removed and hydro(peroxo) ligand was placed on the face of the iron center through an overlay with the O2 adduct of the NDO-indole complex (PDB ID: 1O7N). Hydrogen atoms of the protein residues were added using LeaP,47 and their standard protonation states were verified using propKa 3.1.48,49 The resulting charge of −15 was neutralized with 15 Na+ counterions. The system was then soaked in a truncated octahedral box of 26735 TIP3P water molecules extending to at least 15 Å from the protein atoms. The ff99SB force field45 was used to describe the protein, together with general Amber force field (GAFF)50 parameters for nitrobenzene. The metal centers of the protein were parametrized based on the bonded plus electrostatics model approach51 following the procedure described by Peters et al.52 Parameters were determined for the oxidized forms of the Rieske iron–sulfur cluster and mononuclear iron center, [2FeIII-2S] (S = 0) and [FeIII–OOH]2+ (S = 5/2), respectively, as this is the reactive state of the enzyme. (A description of the parametrization method and the derived partial charges and force field parameters are included in Supporting Information).
MD Simulation
The simulation was carried out using the Amber 11 Molecular Dynamics package.47 The system, prepared as described above, was first subjected to 500 steps of steepest descent and 500 steps of conjugate gradient minimization, with the protein held fixed by using position restraints with a force constant of 500 kcal mol–1 Å–2. This was followed by an additional 2500 steps of steepest descent and 2500 steps of conjugate gradient minimization. The system was then heated to the target temperature of 300 K for a period of 20 ps under constant volume periodic boundary conditions (NVT), with weak positional restraints applied to the protein atoms (force constant of 10 kcal mol–1 Å–2). Subsequently, approximately 40 ns of constant pressure and temperature simulation (NPT) was carried out to equilibrate the system, which was followed by 30 ns of production simulation performed under the same conditions. An average pressure of 1 atm was maintained by using isotropic position scaling with a relaxation time of 2 ps. Temperature was controlled via Langevin dynamics53 with collision frequency of 1 ps–1. A cutoff of 10 Å was used for nonbonded interactions and long-range electrostatic interactions were treated by means of the Particle Mesh Ewald (PME) method.54 All bonds involving hydrogen were constrained by the SHAKE method,55 and the time step for numerical integration was 2 fs. The simulation results were analyzed using the ptraj program in the Amber11 package and VMD.56 The presence of hydrogen bonds was analyzed based on a geometry criterion where a given H-bond was considered to be formed or disrupted at a cutoff distance of 3.6 Å between the donor and acceptor atom and a cutoff bond angle of 120°. Occupancy is calculated as the percent of time the hydrogen bond is formed over the trajectory. Relevant details of other structural analyses are presented in the discussion.
Results and Discussion
The stability of the overall NBDO structure throughout the simulation was investigated via root-mean-square deviations (RMSD) of backbone and all heavy atoms of the enzyme with respect to the X-ray structure (Figure 2). The RMSD values reached plateau after approximately 30 ns of the simulation, indicating that the system reached its equilibrium. The average values of 1.07 Å for the backbone and 1.51 Å for the heavy atom RMSD show that we did not observe significant deviation of the overall enzyme structure from the X-ray data.
Figure 2.

Time evolution of the root-mean-square deviation from the crystallographic structure for backbone (dotted line) and all heavy atoms (solid line) of NBDO during simulation.
The geometries of individual α and β subunits do not undergo substantial changes along the trajectory, which is reflected by small values of root-mean-square deviations between the rms-fitted average structures of the α and β subunits and their corresponding crystal structures (see Table S11 in Supporting Information). Even smaller structural changes are observed for the active site residues, with the largest heavy atom RMSD of 0.9 Å obtained for the active site located in the second α subunit. We have also investigated whether there exist any differences between single subunits in the α and β trimers, and between the three active sites. As might be expected from the overall stability of the enzyme structure, the average structures of the three α and three β subunits are very similar (Figure 3), with calculated RMSD values of less than 1 Å for the backbone atoms of the rms-fitted average structures (see Table S12 in Supporting Information).
Figure 3.
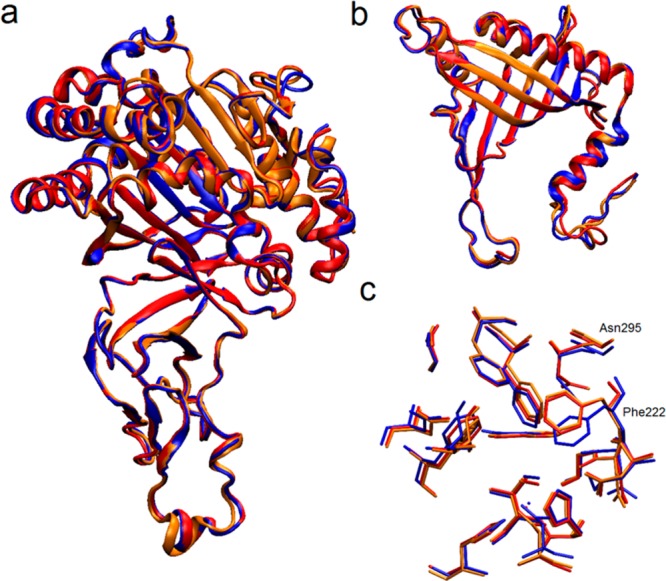
Superposition of the average structures of the three α subunits (a), three β subunits (b), and three active sites (c) calculated from the last 30 ns of the trajectory. Red, blue, and orange colored structures correspond to the subunit (or active site) 1, 2, and 3, respectively.
The average geometries of the three active sites are also similar to each other, with RMSD values ranging from 0.2 to 0.5 Å, and from 0.4 to 0.8 Å for the backbone and all heavy atoms, respectively (see Table S13 in Supporting Information). The biggest RMSD values are observed between active site 2 and the other two active sites. An overlay of the average structures presented in Figure 3c indicates that the structural difference causing the increase of the RMSD value lies in a slight change of the position of the Phe222 and Asn295 side chains, which occurs around the 47th ns of the simulation. However, since it is not accompanied by any other significant change in the active site geometry and the position of any of the surrounding residues is not altered, we believe it not to be particularly meaningful here.
The flexibility of the active site residues was also investigated in terms of B-factors calculated for heavy atoms of the active site according to the relationship:
| 1 |
where ⟨Δri2⟩ is the atomic positional variance of each heavy atom of residue i. The average (mass-weighted) B-factors computed for each residue in the three active sites indicate rather low flexibility of these residues (for details, see Figure S2 in Supporting Information), with the mean value for all active sites oscillating around 8.7 Å2 per residue. As might be expected, the largest dynamic flexibility is observed for the substrate, the B-factor of which was calculated to range from 20.8 to 33.0 Å2 in the three active sites. B-factors were also used to investigate the dynamics of a lid covering the entrance to the substrate pocket, formed by the loops between α8 and β13 (residues 211 to 238) and between β13 and α9 (residues 238 to 265) domains. The values calculated for the main chain Cα atoms range from 9.9 Å2 to 33.4 Å2 (for details, see Figure S3 in Supporting Information), with the most flexible fragment located between Val221 and Glu237. Regardless of high flexibility, the mobile part of the loop maintains a closed conformation throughout the simulation, which was confirmed by monitoring the distance between the Glu234 and Val221 Cα atoms. The average distance of 11.2 ± 0.7 Å shows that the position of the loop does not change relative to the crystal structure (dGlu234-Val221 = 11.3 Å16) during the simulation, blocking the entrance to the active site while the substrate is present.
Geometry of the Active Site and the Rieske Cluster
As already pointed out, the overall geometry of the active site observed during the simulation does not differ significantly from the crystal structure. Moreover, we observe a high degree of similarity between subunits of a particular type and between the three active sites in the simulated NBDO structure. Therefore, in the remainder of this paper, we will discuss the results obtained for active site 1 only.
The residues forming a substrate pocket do not exhibit large positional fluctuations, which are reflected by small B-factors. The position of the substrate undergoes larger fluctuations, compared to the amino acid side chains, but nevertheless, its orientation remains rather stable. Mean distances between the reacting carbon atoms of nitrobenzene and the oxygen atoms of the iron–oxygen complex are 3.27 ± 0.24 Å (C1–O) and 3.46 ± 0.27 Å (C2–O1), with the latter value slightly larger than 3.02 Å found in the starting structure for the simulation. This small change in the distance between the O1 and C2 atoms during the simulation reflects a shift in the substrate position relative to the FeIII–OOH moiety. The dihedral angle formed between the O and O1 atoms and the C1–C2 double bond of nitrobenzene changed from 39.8° in the starting structure to 0.4° in the average structure of the active site obtained from the simulation, which is the expected parallel orientation of the substrate relative to the activated dioxygen required by the cis-stereospecificity of substrate dihydroxylation. Another important aspect of substrate positioning in the active site of NBDO, namely, formation of a hydrogen bond with Asn258, will be discussed later in this paper.
The geometry of the mononuclear iron center located in the active site was monitored during the simulation and mean distances between the Fe atom and its three ligands, as well as iron–dioxygen distances, are compared with the X-ray data (Table 1).
Table 1. Binding Distances of the Ligands Coordinating Fe Ion and Key Distance Parameters for the Dioxygen Bound to Mononuclear Iron during the Catalytic Cyclea.
| distances
[Å] |
||
|---|---|---|
| MD simulation | crystal structure | |
| Fe···O | 1.83 ± 0.04 | 1.73b |
| Fe···O1 | 2.00 ± 0.10 | 1.98b |
| O···O1 | 1.47 ± 0.03 | 1.46b |
| Fe···ε-NHis206 | 2.06 ± 0.06 | 2.10 |
| Fe···ε-NHis211 | 2.08 ± 0.06 | 2.15 |
| Fe···OD1Asp360 | 2.02 ± 0.05 | 2.25 |
| Fe···OD2Asp360 | 2.02 ± 0.05 | 2.40 |
Mean values together with standard deviations for the MD simulation and values obtained from X-ray crystallography are presented.
Dioxygen position determined through an overlay with naphthalene dioxygenase crystal structure25 (see Computational Methods for details).
The values show good agreement between the simulation and the experimental data. The distances between the Fe ion and His206, His211 and Asp360 are slightly smaller than in the NBDO crystal structure, while in the average geometry of the iron–oxygen moiety, we observe slightly elongated bonds compared to the geometry of this complex adopted from the crystal structure of NDO.
The average geometry of the Rieske center located in the α subunit adjacent to the subunit containing the active site was also analyzed. The mean binding distances of the four Fe ligands and the average geometry parameters of the [2Fe-2S] cluster are presented in Table 2. Similarly to the mononuclear iron center, the geometry of the iron–sulfur cluster from the MD simulation agrees very well with the experimental X-ray structure. The binding distances between the iron and sulfur atoms, and the ligating residues are well reproduced, and the [2Fe-2S] cluster maintains the form of a flat rhombic cluster, as found in the crystal structure.
Table 2. Key Distances and Angles of the Rieske Domain Calculated from the MD Simulation and Corresponding Experimental Values Taken from the Crystal Structure of the Enzymea.
| MD simulation | crystal structure | |
|---|---|---|
| distances [Å] | ||
| Fe1···S1 | 2.29 ± 0.06 | 2.19 |
| Fe1···S2 | 2.26 ± 0.06 | 2.19 |
| Fe2···S1 | 2.19 ± 0.05 | 2.22 |
| Fe2···S2 | 2.16 ± 0.04 | 2.20 |
| Fe1···Fe2 | 2.76 ± 0.07 | 2.68 |
| Fe2···δ-NHis81 | 2.08 ± 0.06 | 2.16 |
| Fe2···δ-NHis102 | 2.06 ± 0.07 | 2.09 |
| Fe1···SCys99 | 2.25 ± 0.06 | 2.31 |
| Fe1···SCys79 | 2.26 ± 0.06 | 2.37 |
| Angles [deg] | ||
| S1–Fe1–S2 | 99.7 ± 2.4 | 105.5 |
| Fe1–S1–Fe2 | 76.1 ± 2.2 | 74.9 |
| S1–Fe2–S2 | 106.3 ± 2.5 | 104.2 |
| Fe1–S2–Fe2 | 77.3 ± 2.3 | 75.3 |
| Fe1–S1–Fe2–S2 | 3.3 ± 5.5 | 3.0 |
See Figure 1 for atom numbering.
Interactions between the Rieske cluster and the surrounding residues involve hydrogen bonds from the main chain nitrogen of Arg82 (44.8% occupancy) to the S1 sulfide ion, and from the main chain nitrogen of Trp104 (24.3% occupancy) to the S2 ion (Figure 4). Other possible hydrogen bonds between the sulfide ions and the neighboring main chain atoms of His102 and Gly103 were detected with occupancies of less than 5%. All four ligands of the iron atoms form hydrogen bonds in the second coordination shell. Main chain nitrogen atoms of Cys79 and Cys99 form hydrogen bonds with main chain oxygens of Lys84 and Trp104, respectively, with ∼100% occupancy each. Additionally, Cys99 binds through the main chain oxygen with the main chain nitrogen of Gly103 (84.7% occupancy). Both iron-coordinating histidines keep hydrogen-bond contacts with the residues located in the neighboring α subunit. His81 interacts with the carboxylic oxygens of Glu408, with 22.5 and 98.1% occupancy, and His102 forms hydrogen bonds with carboxyl group of Asp203, with 70.5 and 80.1% occupancy for the two oxygen atoms.
Figure 4.

Interactions between the Rieske cluster and the surrounding residues, together with the most highly populated hydrogen bonds formed between the four ligands of the iron atoms and their second coordination shell. Percentage occupancies of the presented hydrogen bonds are given.
Rieske Center-Active Site Interface
As already mentioned in the introduction, the catalytic center of Rieske dioxygenases, including NBDO, is formed at the interface of two neighboring α subunits by the active site containing mononuclear iron located in one subunit and the [2Fe-2S] Rieske cluster from the adjacent subunit. These two components are connected through a conserved aspartic acid residue, which is believed to be the route for electron transfer from the Rieske cluster to the nonheme mononuclear iron during the catalytic cycle. The role of this residue in the catalytic process was first suggested based on the crystal structure of NDO, where it was found to form the most direct pathway for an efficient electron transfer.18 This hypothesis was later confirmed in a site-directed mutagenesis study of NDO,57 in which it was shown that replacement at position 205 severely decreases the activity of the enzyme, and thus, Asp205 is likely the major pathway for the electron transfer in Rieske dioxygenases.
In the crystal structure of NBDO, the [2Fe-2S] Rieske cluster and mononuclear iron center are connected by Asp203, which is hydrogen bonded through the carbonyl oxygen atom to His206 and through one of the carboxyl oxygen atoms to His102.16 We have monitored these hydrogen bond contacts during the simulation, together with other possible hydrogen bonds that might be formed by Asp203 with the histidine ligands of the two connected metal centers. Figure 5 lists the analyzed hydrogen bonds together with their percentage occupancies and shows the change of the occupancy of a given bond along the trajectory. As can be seen, indeed, the major H-bond chain connecting the two centers is formed by Nδ of His206 in the active site with the carbonyl oxygen of Asp203 (96.1% occupancy) and by Nε of His102 in the Rieske cluster with either of the two carboxyl oxygen atoms of Asp203, which are involved in the bond exchangeably (80.1% and 70.5% occupancy for the OD2 and OD1 atoms, respectively). While the Asp203-His102 contact is limited to the OD2(OD1)-Nε hydrogen bond, other possible H-bonds are formed between Asp203 and His206 with significant occupancy. These include hydrogen bonds between the carboxyl oxygen of Asp203 and the main chain nitrogen of His206 or Nδ of His206, and less significant contact between the main chain atoms of the two residues. From the time evolution of the percentage occupancy shown in Figure 5, it is easily seen that Asp203(carboxyl)-His206(side chain) and Asp203(carboxyl)-His206(main chain) hydrogen contacts are formed exchangeably, in a manner obviously coupled with the Asp203(carboxyl)-His102 bonds, maintaining the hydrogen bond interaction between the carboxyl group of the aspartate and both His206 and His102 residues throughout the simulation. The possible charge transfer channels formed between the Rieske cluster and the nonheme iron center thus include the His102:Nε-Asp203:OD1(OD2) → Asp203:O-His206:Nδ and His102:Nε-Asp203:OD1(OD2) →Asp203:OD2(OD1)-His206:Nδ(N) routes.
Figure 5.
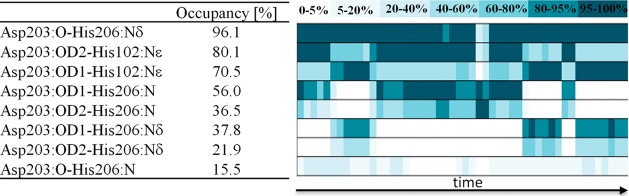
Percentage occupancy of hydrogen bond contacts formed through Asp203 between the active site mononuclear iron center and the Rieske cluster from an adjacent α subunit. Right-hand side of the picture shows time evolution of the occupancy of the listed hydrogen bonds.
Hydrogen Bonding Interactions in the Active Site
Hydrogen bonds formed by the active site residues in NBDO include intramolecular backbone–backbone, backbone–side chain, and side chain–side chain hydrogen bonds, as well as H-bonding interactions between the active site amino acids and water molecules located in their vicinity. The most highly populated bonds formed in the active site are indicated in Figure 6.
Figure 6.

Hydrogen-bond pattern found in the active site of NBDO, including hydrogen bonds formed between the active site residues and water molecules. Figure presents only those hydrogen bonds for which the calculated occupancy was greater than 10%, and the value of the occupancy of each hydrogen bond is given. The most functionally important residues and a hydrogen bond chain found at the entrance to the active site (see text for details) are presented in bold.
Majority of the 17 residues lining the substrate pocket in NBDO are hydrophobic. Thus, understandably, water molecules are not present inside the substrate cavity, but rather tend to surround it, as can be clearly seen from a volumetric map of water density around the active site of NBDO (Figure 7). The water molecules located closest to the substrate pocket form hydrogen bonds with the side chains of polar residues or backbone atoms of several active site amino acids. There is also visible water density at the entrance to the active site, in close vicinity to the iron-dioxygen complex. This density represents the presence of two water molecules forming a short hydrogen bond chain connecting dioxygen with the Asn199 residue, which will be described in detail below.
Figure 7.
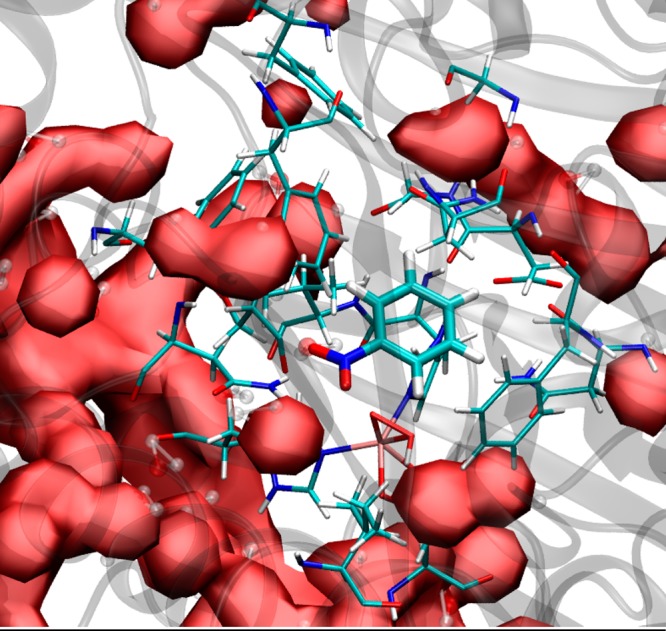
Volumetric map of water density (solid surface) around the active site of NBDO (residues in licorice representation).
Many of the hydrophobic residues located in the substrate pocket are involved in persistent backbone hydrogen-bonding interactions. The most highly occupied of these bonds (occupancy ≥95%) include backbone hydrogen bonds between Asn295 and Gly249, Phe293 and Phe251, Ala345 and Ser349. For most of the carbonyl oxygen atoms of the active site residues, backbone nitrogen atoms are the only H-bond partners. An exception to this is the Asp203 residue, which interacts with both backbone and side chain nitrogens of His206, and Ala345, which forms a hydrogen bond with the hydroxyl oxygen of Ser349 (occupancy 10.3%). On the other hand, the backbone oxygen of Phe200 is not involved in an H-bond interaction with any of the backbone amine groups but forms a hydrogen bond with the side chain of Asn295 with a high occupancy of 91.1%.
The most important hydrogen-bonding interaction occurring in the active site, from the perspective of substrate specificity of NBDO, is the hydrogen bond formed between the side chain of Asn258 and nitro group of the substrate. The formation of this bond is believed to be responsible for the proper positioning of the nitroaromatic substrate for oxidation. As it was shown in a site-directed mutagenesis study of NBDO, replacement of asparagine at this position by valine, which eliminates the said hydrogen bond, results in formation of nitrobenzyl alcohols as the major product (as much as 99%), rather than catechols, from mononitroarene substrates.58 According to the MD simulation results presented here, the amide group of Asn258 donates one of its hydrogen atoms to form an H-bond with either of the two oxygens of the substrate nitro group with an occupancy of ∼50% each, so that the hydrogen bond between Asn258 and nitrobenzene is effectively formed throughout the whole simulation. This result explains the position of the substrate observed in the simulation, with the C1–C2 double bond parallel to the attacking dioxygen and thus agrees with the role of Asn258 proposed in literature.16,58 Simultaneously to forming an H-bond with the nitro group, the amide group of Asn258 is also interacting through a second hydrogen atom with a water molecule (96%). The backbone oxygen and side chain carbonyl oxygen of Asn258 also form hydrogen bonds with water molecules and neighboring Ser349 (see Figure 6).
An interesting observation regarding other hydrogen bonds formed within the active site is that hydrogen bond interaction involving the carboxyl oxygens of Asp203, serving as a link between the mononuclear iron center and the Rieske cluster, are not limited to forming H-bonds with His206 and His102 residues, which were discussed before. Additionally, Asp203 interacts with water, and with the side chain of Asn199. As for Asn199 itself, it forms hydrogen bonds with water molecules residing at the entrance to the active site, which are at the same time interacting with the peroxide moiety of the iron-dioxygen complex. The total occupancy for the FeOOH–water and Asn199–water hydrogen bonds is 81% and 77.6% respectively, with FeOOH serving as a donor, and the carbonyl oxygen of Asn199 as acceptor in hydrogen bonding interactions with any water molecule. Closer inspection reveals, however, that there are, in fact, two specific water molecules with which FeOOH and Asn199:OD are almost exclusively interacting throughout the simulation. These two water molecules are exchangeably acting as a proton acceptor in FeOOH–water interaction (62.6% and 18.2% occupancy) and a proton donor in the Asn199:OD–water interaction (41.2% and 36.3% occupancy), being at the same time connected with each other through a water–water hydrogen bond (77.2% occupancy) (for details regarding water–water H-bond occupancies, see Table S14 in Supporting Information). This way, a short hydrogen bond chain is formed (see bottom of Figure 6), which involves the peroxide moiety bound to mononuclear iron and thus might be of potential importance to the catalytic process involving its attack on the substrate.
Conclusions
We have performed a 70 ns-long classical molecular dynamics simulation of NBDO, a representative of the naphthalene family of Rieske dioxygenases, using the AMBER force field together with a new set of force field parameters developed for the description of the active site mononuclear iron center and iron–sulfur Rieske cluster. The MD simulation was carried out in explicit water environment under NPT ensemble for a heterohexameric model of the oxidized form of the enzyme with nitrobenzene bound in the active site and (hydro)peroxide moiety attached to the mononuclear iron. Simulation results were analyzed with the focus on the comparison of the overall structure and key geometrical parameters with X-ray data, investigation of the hydrogen-bonding interactions occurring within the active site and Rieske cluster domains, as well as at the interface of the two metal centers, and role of the solvent molecules in the substrate binding pocket.
The overall structure of NBDO in aqueous solution is stable and does not undergo significant structural changes along the simulated trajectory. No structural differences are observed between the individual subunits within the α3 and β3 trimers, as well as between the three active sites formed in the heterohexamer. The average B-factors indicate rather low flexibility of the active site residues, with the highest values obtained for the substrate. A highly flexible loop covering the entrance to the active site remains in a closed conformation throughout the simulation. In general, the structure of the enzyme observed during the simulation shows very small deviation from the X-ray data. The newly developed parameters reproduce very well the geometry of the mononuclear iron center and Rieske cluster and may potentially be used in simulations of other Rieske dioxygenases.
The hydrogen bond analysis showed that the mononuclear iron center and [2Fe-2S] cluster are connected through several hydrogen bonds formed between carbonyl and carboxyl oxygen atoms of the Asp203 residue and His ligands of the two metal centers along the entire trajectory. Interaction of the substrate with the active site residues is limited to the formation of a hydrogen bond with the Asn258 residue, which is effectively formed throughout the whole simulation and positions the substrate for the reaction with the activated dioxygen. Finally, water molecules are not in general present inside the hydrophobic substrate pocket. However, there exists a short hydrogen bond chain at the entrance to the active site, which involves two water molecules, Asn199 residue and the (hydro)peroxo ligand bound to Fe ion and might thus be potentially important for the catalytic processes involving this moiety.
Acknowledgments
This work was supported by the grants: Fulbright Junior Advanced Research Grant (to A.P.), PSRP-025/2010 from the Polish-Swiss Research Program and FP7-264329 from the seventh Framework Programme, Maria Curie Action ITN “CSI:Environment” (to P.P.) and the National Institute of Health (GM62248 to D.M.Y.). This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation Grant No. OCI-1053575. The authors thank George M. Giambasu for his technical assistance in analyzing MD results.
Supporting Information Available
Computational details of the force field parametrization. Root-mean-square deviations between the average structures of the α and β subunits and their corresponding crystal structures. RMSD values for the backbone atoms of the average structures of individual α and β subunits and for the backbone atoms of the average active site structures. B-factors for the active site residues and for the residues 211 to 265, forming a lid covering the entrance to the substrate pocket. Percentage occupancy of hydrogen bonds formed between peroxide moiety, two water molecules present at the entrance to the active site and Asn199 side chain. Files containing partial charge (*.prep) and force field parameter (*.frcmod) files for catalytic site (cat-site) and Rieske cluster (rieske). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Lessner D. J.; Johnson G. R.; Parales R. E.; Spain J. C.; Gibson D. T. Molecular Characterization and Substrate Specificity of Nitrobenzene Dioxygenase from Comamonas sp. Strain JS765. Appl. Microbiol. Biotechnol. 2002, 68, 634–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson D. T.; Parales R. E. Aromatic Hydrocarbon Dioxygenases in Environmental Biotechnology. Curr. Opin. Biotechnol. 2000, 11, 236–243. [DOI] [PubMed] [Google Scholar]
- Lee J.; Zhao H. Mechanistic Studies on the Conversion of Arylamines into Arylnitro Compounds by Aminopyrrolnitrin Oxygenase: Identification of Intermediates and Kinetic Studies. Angew. Chem., Int. Ed. 2006, 45, 622–625. [DOI] [PubMed] [Google Scholar]
- Boyd D. R.; Sharma N. D.; Bowers N. I.; Dalton H.; Garrett M. D.; Harrison J. S.; Sheldrake G. N. Dioxygenase-catalysed Oxidation of Disubstituted Benzene Substrates: Benzylic Monohydroxylation vs Aryl cis-Dihydroxylation and the Meta Effect. Org. Biomol. Chem. 2006, 4, 3343–3349. [DOI] [PubMed] [Google Scholar]
- Boyd D. R.; Sharma N. D.; Byrne B. E.; Haughey S. A.; Kennedy M. A.; Allen C. C. R. Dioxygenase-catalysed Oxidation of Alkylaryl Sulfides: Sulfoxidation versus cis-Dihydrodiol Formation. Org. Biomol. Chem. 2004, 2, 2530–2537. [DOI] [PubMed] [Google Scholar]
- Resnick S. M.; Lee K.; Gibson D. T. Diverse Reactions Catalyzed by Naphthalene Dioxygenase from Pseudomonas sp strain NCIB 9816-4. J. Ind. Microbiol. Biotechnol. 1996, 17, 438–457. [Google Scholar]
- Gibson D. T.; Resnick S. M.; Lee K.; Brand J. M.; Torok D. S.; Wackett L. P.; Schocken M. J.; Haigler B. E. Desaturation, Dioxygenation, and Monooxygenation Reactions Catalyzed by Naphthalene Dioxygenase from Pseudomonas sp. strain 9816. J. Bacteriol. 1995, 177, 2615–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick S. M.; Gibson D. T. Biotransformation of Anisole and Phenetole by Aerobic Hydrocarbonoxidizing Bacteria. Biodegradation 1993, 4, 195–203. [Google Scholar]
- Wackett L. P. Mechanism and Applications of Rieske Non-Heme Iron Dioxygenases. Enzyme. Microb. Biotechnol. 2002, 31, 577–587. [Google Scholar]
- Ensley B. D.; Ratzkin B. J.; Osslund T. D.; Simon M. J.; Wackett L. P.; Gibson D. T. Expression of Naphthalene Oxidation Genes in Escherichia coli Results in the Biosynthesis of Indigo. Science 1983, 222, 167–169. [DOI] [PubMed] [Google Scholar]
- Buckland B. C.; Drew S. W.; Connors N. C.; Chartrain M. M.; Lee C.; Salmon P. M.; Gbewonyo K.; Zhou W.; Gailliot P.; Singhvi R.; Olewinski R. C. Jr.; Sun W. J.; Reddy J.; Zhang J.; Jackey B. A.; Taylor C.; Goklen K. E.; Junker B.; Greasham R. L. Microbial Conversion of Indene to Indandiol: A Key Intermediate in the Synthesis of CRIXI-VAN. Metab. Eng. 1999, 1, 63–74. [DOI] [PubMed] [Google Scholar]
- Reddy J.; Lee C.; Neeper M.; Greasham R.; Zhang J. Development of a Bioconversion Process for Production of Cis-1S,2R-indandiol from Indene by Recombinant Escherichia coli Constructs. Appl. Microbiol. Biotechnol. 1999, 51, 614–620. [DOI] [PubMed] [Google Scholar]
- Zhang N.; Stewart B. G.; Moore J. C.; Greasham R. L.; Robinson D. K.; Buckland B. C.; Lee C. Directed Evolution of Toluene Dioxygenase from Pseudomonas putida for Improved Selectivity Toward cis-indandiol During Indene Bioconversion. Metab. Eng. 2000, 2, 339–348. [DOI] [PubMed] [Google Scholar]
- Timmis K. N.; Pieper D. H. Bacteria Designed for Bioremediation. Trends Biotechnol. 1999, 17, 201–204. [DOI] [PubMed] [Google Scholar]
- Lau P. C. K.; De Lorenzo V. Genetic Engineering: The Frontier of Bioremediation. Environ. Sci. Technol. 1999, 4, 124A–128A. [DOI] [PubMed] [Google Scholar]
- Friemann R.; Ivkovic-Jensen M. M.; Lessner D. J.; Yu C.-L.; Gibson D. T.; Parales R. E.; Eklund H.; Ramaswamy S. Structural Insight into the Dioxygenation of Nitroarene Compounds: The Crystal Structure of Nitrobenzene Dioxygenase. J. Mol. Biol. 2005, 348, 1139–1151. [DOI] [PubMed] [Google Scholar]
- Parales R. E.; Huang R.; Yu C.-L.; Parales J. V.; Lee F. K. N.; Lessner D. J.; Ivkovic-Jensen M. M.; Liu W.; Friemann R.; Ramaswamy S.; Gibson D. T. Purification, Characterization, and Crystallization of the Components of the Nitrobenzene and 2-nitrotoluene Dioxygenase Enzyme Systems. Appl. Environ. Microb. 2005, 71, 3806–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppi B.; Lee K.; Carredano E.; Parales R. E.; Gibson D. T.; Eklund H.; Ramaswamy S. Structure of an Aromatic-Ring-Hydroxylating Dioxygenase–Naphthalene 1,2-Dioxygenase. Structure 1998, 6, 571–586. [DOI] [PubMed] [Google Scholar]
- Parales R. E. The Role of Active-site Residues in Naphthalene Dioxygenase. J. Ind. Microbiol. Biotechnol. 2003, 30, 271–278. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S. In Handbook of Metalloproteins; Messerschmidt A., Huber R., Poulos T., Wieghardt K., Eds.; John Wiley & Sons: Chichister, U.K., 2001; pp 613–621. [Google Scholar]
- Nordlund P. In Handbook of Metalloproteins; Bertini I., Sigel A., Sigel H., Eds.; Marcel Dekker: New York, 2001; pp 511–517. [Google Scholar]
- Karlsson A. Ph.D. thesis, Swedish University of Agricultural Science, Uppsala, 2002. [Google Scholar]
- Bugg T. D. H. Dioxygenase Enzymes: Catalytic Mechanisms and Chemical Models. Tetrahedron 2003, 59, 7075–7101. [Google Scholar]
- Hegg E. L.; Que L. Jr. The 2-His-1-Carboxylate Facial Triad—An Emerging Structural Motif in Mononuclear Non-heme Iron(II) Enzymes. Eur. J. Biochem. 1997, 250, 625–629. [DOI] [PubMed] [Google Scholar]
- Karlsson A.; Parales J. V.; Parales R. E.; Gibson D. T.; Eklund H.; Ramaswamy S. Crystal Structure of Naphthalene Dioxygenase: Side-on Binding of Dioxygen to Iron. Science 2003, 299, 1039–1042. [DOI] [PubMed] [Google Scholar]
- Ferraro D. J.; Gakhar L.; Ramaswamy S. Rieske Business: Structure-function of Rieske Non-heme Oxygenases. Biochem. Biophys. Res. Commun. 2005, 338, 175–190. [DOI] [PubMed] [Google Scholar]
- Wolfe M. D.; Parales J. V.; Gibson D. T.; Lipscomb J. D. Single Turnover Chemistry and Regulation of O2 Activation by the Oxygenase Component of Naphthalene 1,2-Dioxygenase. J. Biol. Chem. 2001, 276, 1945–1953. [DOI] [PubMed] [Google Scholar]
- Bruijnincx P. C. A.; van Koten G.; Gebbink R. J. M. K.; Mononuclear Non-Heme Iron Enzymes with the 2-His-1-carboxylate Facial Triad: Recent Developments in Enzymology and Modeling Studies. Chem. Soc. Rev. 2008, 37, 2716–2744. [DOI] [PubMed] [Google Scholar]
- Ohta T.; Chakrabarty S.; Lipscomb J. D.; Solomon E. I. Near-IR MCD of the Non-heme Ferrous Active Site in Naphthalene 1,2-Dioxygenase: Correlation to Crystallography and Structural Insight into the Mechanism of Rieske Dioxygenases. J. Am. Chem. Soc. 2008, 130, 1601–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costas M.; Mehn M. P.; Jensen M. P.; Que L. Jr. Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem. Rev. 2004, 104, 939–986. [DOI] [PubMed] [Google Scholar]
- Wolfe M. D.; Lipscomb J. D. Hydrogen Peroxide-coupled cis-Diol Formation Catalyzed by Naphthalene 1,2-Dioxygenase. J. Biol. Chem. 2003, 278, 829–835. [DOI] [PubMed] [Google Scholar]
- Bassan A.; Borowski T.; Siegbahn P. E. M. Quantum Chemical Studies of Dioxygen Activation by Mononuclear Non-heme Iron Enzymes with the 2-His-1-carboxylate Facial Triad. Dalton Trans. 2004, 3153–3162. [DOI] [PubMed] [Google Scholar]
- Bassan A.; Blomberg M. R. A.; Siegbahn P. E. M. A Theoretical Study of the cis-Dihydroxylation Mechanism in Naphthalene 1,2-Dioxygenase. J. Biol. Inorg. Chem. 2004, 9, 439–452. [DOI] [PubMed] [Google Scholar]
- Chakrabarty S.; Austin R. N.; Deng D.; Groves J. T.; Lipscomb J. D. Radical Intermediates in Monooxygenase Reactions of Rieske Dioxygenases. J. Am. Chem. Soc. 2007, 129, 3514–3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange S. J.; Que L. Oxygen Activating Non-heme Iron Enzymes. Curr. Opin. Chem. Bio. 1998, 2, 159–172. [DOI] [PubMed] [Google Scholar]
- Que L. Jr. One Motif—Many Different Reactions. Nat. Struct. Biol. 2000, 7, 182–184. [DOI] [PubMed] [Google Scholar]
- Solomon E. I.; Brunold T. C.; Davis M. I.; Kemsley J. N.; Lee S. K.; Lehnert N.; Neese F.; Skulan A. J.; Yang Y. S.; Zhou J. Geometric and Electronic Structure/Function Correlations in Non-heme Iron Enzymes. Chem. Rev. 2000, 100, 235–350. [DOI] [PubMed] [Google Scholar]
- Que L. Jr.; Ho R. Y. Dioxygen Activation by Enzymes with Mononuclear Non-heme Iron Active Sites. Chem. Rev. 1996, 96, 2607–2624. [DOI] [PubMed] [Google Scholar]
- Furukawa K. Engineering Dioxygenases for Efficient Degradation of Environmental Pollutants. Curr. Opin. Biotechnol. 2000, 11, 244–249. [DOI] [PubMed] [Google Scholar]
- Hirao H.; Morokuma K. ONIOM(DFT:MM) Study of 2-Hydroxyethylphosphonate Dioxygenase: What Determines the Destinies of Different Substrates?. J. Am. Chem. Soc. 2011, 133, 14550–14553. [DOI] [PubMed] [Google Scholar]
- Lundberg M.; Kawatsu T.; Vreven T.; Frisch M. J.; Morokuma K. Transition States in a Protein Environment—ONIOM QM:MM Modeling of Isopenicillin N Synthesis. J. Chem. Theory Comput. 2009, 5, 222–234. [DOI] [PubMed] [Google Scholar]
- Godfrey E.; Porro C. S.; de Visser S. P. Comparative Quantum Mechanics/Molecular Mechanics (QM/MM) and Density Functional Theory Calculations on the Oxo–Iron Species of Taurine/α-Ketoglutarate Dioxygenase. J. Phys. Chem. A 2008, 112, 2464–2468. [DOI] [PubMed] [Google Scholar]
- Kaszuba K.; Postila P. A.; Cramariuc O.; Sarewicz M.; Osyczka A.; Vattulainen I.; Róg T. Parameterization of the Prosthetic Redox Centers of the Bacterial Cytochrome bc1 Complex for Atomistic Molecular Dynamics Simulations. Theor. Chem. Acc. 2013, 132, 1370. [Google Scholar]
- Izrailev S.; Crofts A. R.; Berry E. A.; Schulten K. Steered Molecular Dynamics Simulation of the Rieske Subunit Motion in the Cytochrome bc1 Complex. Biophys. J. 1999, 77, 1753–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornak V.; Abel R.; Okur A.; Strockbine B.; Roitberg A.; Simmerling C. Comparison of Multiple Amber Force Fields and Development of Improved Protein Backbone Parameters. Proteins 2006, 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and Development of Coot. Acta Crystallogr. 2010, D66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.; Darden T. A.; Cheatham T. E. III; Simmerling C. L.; Wang J.; Duke R. E.; Luo R.; Walker R. C.; Zhang W.; Merz K. M.; Roberts B.; Wang B.; Hayik S.; Roitberg A.; Seabra G.; Kolossváry I.; Wong K. F.; Paesani F.; Vanicek J.; Liu J.; Wu X.; Brozell S. R.; Steinbrecher T.; Gohlke H.; Cai Q.; Ye X.; Wang J.; Hsieh M.-J.;Cui G.; Roe D. R.; Mathews D. H.; Seetin M. G.; Sagui C.; Babin V.; Luchko T.; Gusarov S.; Kovalenko A.; Kollman P. A.. AMBER 11; University of California: San Francisco, 2010. [Google Scholar]
- Olsson M. H. M.; Søndergard C. R.; Rostkowski M.; Jensen J. H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [DOI] [PubMed] [Google Scholar]
- Søndergaard C. R.; Olsson M. H. M.; Rostkowski M.; Jensen J. H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [DOI] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- Hoops S. C.; Anderson K. W.; Merz K. M. Jr. Force Field Design for Metalloproteins. J. Am. Chem. Soc. 1991, 113, 8262–8270. [Google Scholar]
- Peters M. B.; Yang Y.; Wang B.; Füsti-Molnár L.; Weaver M. N.; Merz K. M. Jr. Structural Survey of Zinc-Containing Proteins and Development of the Zinc AMBER Force Field (ZAFF). J. Chem. Theory Comput. 2010, 6, 2935–2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loncharich R. J.; Brooks B. R.; Pastor R. W. Langevin Dynamics of Peptides: The Frictional Dependence of Isomerization Rates of N-acetylalanyl-N’-methylamide. Biopolymers 1992, 32, 523–535. [DOI] [PubMed] [Google Scholar]
- Darden T.; York D.; Pederson L. Particle Mesh Ewald: An N•log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14.1, 33–38. [DOI] [PubMed] [Google Scholar]
- Parales R.; Parales J. V.; Gibson D. T. Aspartate 205 in the Catalytic Domain of Naphthalene Dioxygenase is Essential for Activity. J. Bacteriol. 1999, 181, 1831–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju K.-S.; Parales R. E. Control of Substrate Specificity by Active-Site Residues in Nitrobenzene Dioxygenase. Appl. Environ. Microbiol. 2006, 72, 1817–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.