

Abstract
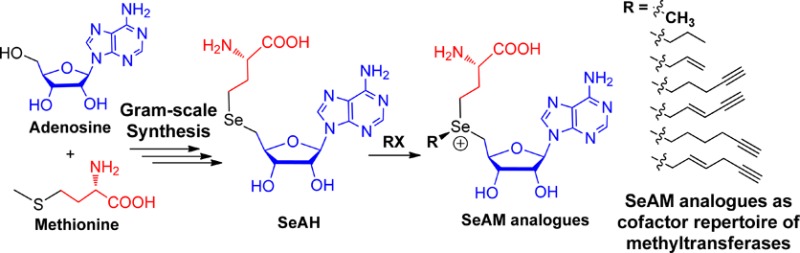
S-Adenosyl-l-methionine (SAM) analogues have previously demonstrated their utility as chemical reporters of methyltransferases. Here we describe the facile, large-scale synthesis of Se-alkyl Se-adenosyl-l-selenomethionine (SeAM) analogues and their precursor, Se-adenosyl-l-selenohomocysteine (SeAH). Comparison of SeAM analogues with their equivalent SAM analogues suggests that sulfonium-to-selenonium substitution can enhance their compatibility with certain protein methyltransferases, favoring otherwise less reactive SAM analogues. Ready access to SeAH therefore enables further application of SeAM analogues as chemical reporters of diverse methyltransferases.
S-Adenosyl-l-methionine (SAM, 1) is a ubiquitous metabolite utilized in a variety of biochemical processes, ranging from the biosynthesis of small molecules to methylation of macromolecules such as nucleic acids and proteins.1 Methylation of DNA, RNA, and protein has garnered particular attention because of their implication in epigenetic and other essential biological phenomena.2−5 In light of such importance, efforts have been made toward the development of SAM analogues to elucidate the targets and functions of various methyltransferases.6−13 Such analogues typically contain distinct chemical functionalities, such as ketones, azides, or terminal alkynes, which are compatible with bioorthogonal chemical reactions and allow the conjugation of fluorescent or affinity-based probes for target characterization.6−9,14 In conjunction with the application of these analogues, their chemical and enzymatic synthesis has also been documented in several reports.7,10,15−18
Among the reported SAM analogues, a selenium-based Se-adenosyl-l-selenomethionine (SeAM, 2) analogue, ProSeAM, has received recent interest because it stands as the SAM analogue containing the smallest bioorthogonal functionality (a propargyl group) and, more importantly, demonstrates broad compatibility toward native methyltransferases.17,19−22 ProSeAM is featured by its selenonium moiety, which is equivalent to the sulfonium of other SAM analogues.19,20 The replacement of sulfonium with selenonium for this cofactor was shown to be essential for the suppression of undesired decomposition of the propargylic handle to a ketone byproduct.7,10,19,20 Another chemical feature of Se-alkyl SeAM analogues lies in their weaker chalcogen-carbon bonds in comparison with the equivalent sulfonium analogues. Se-Alkyl SeAM analogues were thus expected to be more reactive as cofactors in methyltransferase-catalyzed transalkylation reactions,23 although this has yet to be demonstrated broadly for the various classes of methyltransferases.
Several groups have shown that bulky S-alkyl SAM analogues lacking sulfonium-β-sp1/sp2 carbons as activating groups are typically inert as methyltransferase cofactor surrogates.7,11,16 The rationale for this observation involves β-sp1- or β-sp2-carbon conjugative stabilization of the transition state during enzyme-catalyzed SN2 transalkylation reactions.11 Given the weaker carbon–chalcogen bonds of selenium-based analogues, it is of general interest to examine whether the chalcogen-β-sp1 or -sp2 functionality is dispensable for enzyme-mediated transalkylation reactions.
Despite a general interest in expanding the cofactor repertoire with Se-alkyl SeAM derivatives, their chemical synthesis and characterization have not been thoroughly examined to date. This is in part due to the challenge of readily accessing large quantities of these compounds through previously reported synthetic methods.19−22 In the present work, we present a facile, protection-free strategy to access Se-adenosyl-l-selenohomocysteine (SeAH, 3, Figure 1) in gram scale. SeAH is the last converged precursor in the synthesis of a variety of Se-alkyl SeAM analogues through a single alkylation reaction (Figure 1).19,20,24 Upon comparison of Se-alkyl SeAM analogues with their equivalent S-alkyl SAM analogues as cofactor surrogates of native and engineered protein methyltransferases (PMTs), we conclude that selenium substitution can play a role on activating otherwise less reactive sulfonium-alkyl SAM analogues. Furthermore, the presence of a selenium-β-sp2 carbon in Se-alkyl SeAM analogues is dispensable for some PMTs as cofactors. The present work therefore documents a convenient method to access a variety of SeAM analogues and facilitates further examination of their potential application as chemical reporters of methyltransferases.
Figure 1.
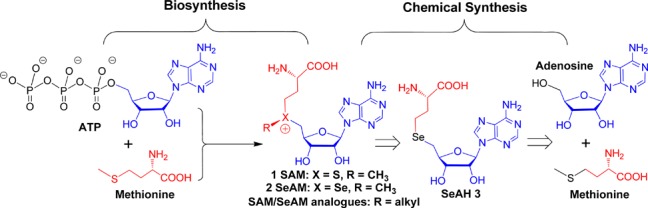
Biosynthesis of SAM and retrosynthesis of SAM, SeAM, and their chalcogen–alkyl analogues.
SAM is generated in vivo by S-adenosylmethionine synthetase, which utilizes l-methionine and ATP as substrates (Figure 1).1,17,18 Inspired by the efficient biosynthesis of SAM, we envisioned a protection-free chemical synthesis of SeAM analogues from comparable natural building blocks: l-methionine and adenosine, which can be converted into selenohomocystine and 5′-iodo-5′-deoxyadenosine building blocks, respectively (Figure 1, Scheme 1). Here, l-methionine was first treated with methyl iodide to generate S-methyl-l-methionine, which underwent intramolecular displacement to form homoserine lactone.25−27 Hydrolysis of this intermediate yielded α-amino-4-hydroxybutanoic acid 4, which was easily precipitated from solution with a yield of 74%.25,26 Treatment of 4 with hydrogen bromide in acetic acid yielded α-amino-4-bromobutanoic acid 5 with a yield of 62%.27 Compound 5 was then treated with Na2Se2, generated in situ from selenium powder and NaBH4, to give the key building block selenohomocystine 6 in 84% yield.28 Selenohomocystine was readily purified by solid-phase extraction with acid-activated Dowex 50WX4 resin. The three-step synthesis is featured by its multigram scales, good yields from readily available materials, and facile purification (precipitation or solid-phase extraction) (Supporting Information).
Scheme 1. Protection-Free Synthesis of SeAH, SeAM, and SeAM Analogues with Yields and Purification Methods Highlighted for Key Intermediates.

Another key building block in the synthesis of SeAM analogues is 5′-iodo-5′-deoxyadenosine 7, which was readily prepared from adenosine via Appel reaction at gram-scale (Scheme 1).29 SeAH 3, the last converged precursor in our synthesis of Se-alkyl SeAM analogues (Scheme 1, Figure 1), was generated through NaBH4-mediated reduction of selenohomocystine 6 to the selenide anion 8, followed by subsequent coupling with 5′-iodo-5′-deoxyadenosine 7 with a yield of 58%. Here, a convenient solid-phase extraction protocol with Amberlite XAD4 resin was used to purify both 5′-iodo-5′-deoxyadenosine 7 and SeAH 3 (Supporting Information).30 Although previous syntheses used 5′-chloro-5′-deoxyadenosine or 2′,3′-O-isopropylidene-5′-O-p-toluenesulfonyladenosine in similar coupling reactions,19,24 we found that 5′-iodo-5′-deoxyadenosine 7 can be readily prepared and gave a more robust yield under these conditions. We further noted that the efficiency of the last coupling reaction would drop significantly if not performed under inert atmosphere, likely due to oxidation of the selenide anion 8 and the resultant reformation of the diselenide 6. Collectively, this approach allowed access to SeAH through a protection-free, gram-scale synthesis with desired yields and convenient purification.
Although SeAH 3 is readily subjected to alkylation to generate SeAM 2 and Se-alkyl SeAM analogues 9a–14a, their weaker carbon–chalcogen bonds makes these compounds more susceptible to decomposition through intramolecular lactonization.31 Thus, more care must be taken during the purification and storage of these compounds than their sulfonium counterparts. We therefore freshly synthesized SeAM 2 and a set of Se-alkyl SeAM analogues 9a–14a (Figure 2a) in a milligram scale and examined their reactivity as cofactor surrogates for native and engineered methyltransferases. For comparison, the equivalent sulfonium-based SAM analogues (9b–14b, Figure 2a) were also prepared from S-adenosyl-l-homocysteine (SAH). Here, SeAH and SAH were treated with various alkyl halides in the presence of silver perchlorate (or mesolate for 12a and 12b) in a 1:1 acetic/formic acid mixture to give the desired products (Scheme 1).7,10,15 These SAM and SeAM analogues contained either β-sp2 or β-sp3 carbons for the purpose of comparing their roles on the transition-state stabilization of PMT-catalyzed SN2 transalkylation reactions.
Figure 2.
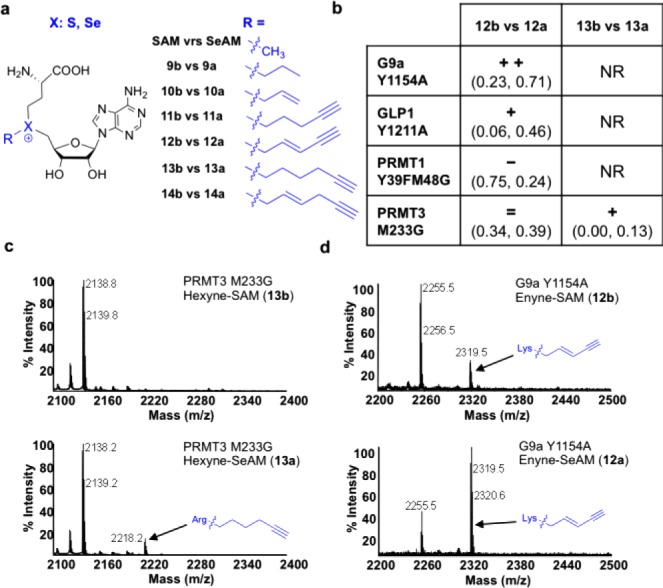
(a) SAM and SeAM analogues examined as cofactor surrogates for native and engineered PMTs. (b) Relative transalkylation efficiency (S vs Se) on known peptide substrates for selected engineered PMTs and SAM/SeAM analogues. Degrees of alkylation (equivalent to units of the consumed cofactor) per unit peptide were quantified via MALDI-MS (see the Supporting Information for more details).33 The difference between SeAM and their equivalent SAM analogues were then compared and categorized. NR, no reaction; =, no observable difference; +, an increase of 0.1–0.5 equiv alkylation/substrate; ++, an increase of >0.5 equiv alkylation/substrate; −, a decrease of 0.1–0.5 equiv alkylation/substrate from S- to Se-alkyl SAM analogues. Relative values for units of the consumed cofactors per unit peptide substrate are shown in parentheses as (S-analogue, Se-analogue) (c, d) Representative MALDI-MS analysis: PRMT3M233G-catalyzed reactions using hexyne–SAM and −SeAM cofactors (13a vs 13b); G9a Y1154A-catalyzed reactions using enyne–SAM and −SeAM cofactors (12a vs 12b).
In order to examine chalcogen–alkyl SAM or SeAM analogues as potential cofactor surrogates for PMTs, we selected a panel of native and engineered PMTs (native and Y39FM48G PRMT1, native and M233G PRMT3, native and Y1154A G9a, native and Y1211A GLP1), which have previously been shown to be active toward either native SAM or β-sp1/sp2-carbon-activated SAM analogues as cofactors.7,11,14,16,32 Enzymatic reactions with known peptide substrates were analyzed by MALDI-MS in order to assess the degrees of transalkylation with the various cofactor analogues (Figure 2b, S1–S10, Table S2, and Supporting Information).7,33 These results were then evaluated based on the difference of the efficiency of alkylation between S-alkyl SAM analogues and their Se-alkyl SeAM counterparts (Supporting Information).
Although the bulky, β-sp3-carbon-containing S-hexynyl SAM analogue 13b was completely inert as a cofactor of the examined PMTs, its equivalent SeAM analogue 13a demonstrated activity toward the PRMT3M233G mutant (Figure 2b,c; Figure S10 and Table S2, Supporting Information). Despite modest transalkylation efficiency, this reaction was enzyme-dependent, as no modification was detected in the presence of native enzyme or in a no-enzyme control (Figure 2c, S6, S9, S10, Table S2 and Supporting Information). Similarly, the S-enyne SAM analogue 12b was only modestly active toward G9a Y1154A and GLP1 Y1211A mutants,7 while the equivalent Se-enyne SeAM 12a was shown to be at least 4-fold more reactive under the same conditions (0–20% for the former versus 50–80% conversion for the latter, Figure 2b, d, S3, S5, Table S2 and Supporting Information). These observations argue that the weaker selenium–carbon bond in Se-alkyl SeAM analogues has a positive effect on accelerating transalkylation reactions for a subset of PMTs.
It was also noted that the effect of selenium substitution to facilitate PMT-catalyzed transalkylation was context-dependent. For instance, while the Se-enyne SeAM analogue (12a) is more reactive than the S-enyne SAM analogue (12b) as a cofactor for G9a Y1154A and GLP1 Y1211A mutants, such an effect was not observed for the PRMT3M233G mutant and was even reversed for the PRMT1 Y39FM48G mutant (Figure 2b; Figures S3, S5, S8, and S10 and Table S2, Supporting Information). Similarly, SeAM 2 only showed higher reactivity than SAM 1 as a cofactor for native and Y1154A G9a, native and Y1211A GLP1, and PRMT1 Y39FM48G mutant, but not for native PRMT1, native, and M233G PRMT3 (Figures S2–5 and S7–10 and Table S2, Supporting Information). In addition, the sulfonium-to-selenonium substitution in the S-allyl SAM analogue (10a versus 10b) showed no effect on their cofactor reactivity for G9a Y1154A, GLP1 Y1211A, PRMT1 Y39FM48G, and PRMT3M233G mutants (Figure S3, S5, S8, S10 and Table S2, Supporting Information). The sulfonium-to-selenonium substitution in Hey-SAM (14a versus 14b) also did not affect its cofactor activity toward G9a Y1154A and GLP1 Y1211A variants and even had a negative effect on PRMT1 Y39FM48G and PRMT3M233G variants (Figures S3, S5, S8, and S10 and Table S2, Supporting Information). Furthermore, SAM and SeAM analogues 9a,b and 11a,b were inert toward all PMTs examined (Tables S1–10, Supporting Information). According to these data, we observed the general trend that for less reactive SAM derivatives such as S-hexynyl and S-enyne SAM analogues (12b, 13b), which contain a β-sp3-carbon and a rigid bulky alkyl chain, respectively, the replacement of sulfonium with selenonium can boost their reactivity as cofactors for certain PMTs. Admittedly, this observation needs to be further explored with more PMTs and SeAM analogues.
In the present work, we described the facile synthesis of SeAH 3 as well as the converged synthesis of six SeAM analogues 9a–14a from inexpensive natural building blocks l-methionine and adenosine. The overall strategy mirrors the efficiency of the biosynthesis of SAM in vivo but allows access to a large quantity of these compounds through convenient purification steps and with decent overall yields. A key step of prior syntheses of SeAH involved the generation of a selenide anion from l-selenomethionine or Se-benzyl-selenohomocysteine through a less efficient Na/Li reduction in liquid ammonia (low-to-modest yields of <50–60%), followed by coupling to 5′-deoxy-5′-chloroadenosine or 2′,3′-O-isopropylidene-5′-O-(p-toluenesulfonyl)adenosine.19,24,34 The present synthesis avoids these less efficient steps or harsh conditions, such as the use of reactive metals.
By examining a variety of Se-alkyl SeAM analogues and comparing them with their equivalent SAM analogues, we observed context-dependent reactivity as cofactor surrogates for PMTs. In the case of protein lysine methyltransferases (PKMTs), selenium substitution can efficiently boost the reactivity of otherwise less reactive SAM analogues. Although the β-unsaturated functionality remained essential for cofactor activity of S-alkyl SAM analogues, this functionality is dispensable for certain protein arginine methyltransferases (PRMTs) when Se-alkyl SAM analogues are used. These observations illustrate a complementary role of the weaker selenium–carbon bond on activating the SN2 transition state and the potential application of Se-alkyl SeAM analogues as chemical reporters for other methyltransferases. Although the reported rates of the spontaneous cleavage of the chalcogen-carbon bonds can increase by 10-fold from SAM to SeAM,28 the efficiency of the PMT-catalyzed transalkylation reactions increases by no more than 3–5 fold from S-alkyl SAM analogues to equivalent Se-alkyl SeAM analogues. Such a discrepancy suggests that PMTs also leverage other mechanisms to control the overall catalysis, a process that can also be explored biochemically with the selenium-based SAM analogues.
Acknowledgments
We thank former and current lab members: Dr. K. Islam (University of Pittsburgh), Dr. R. Wang (Novartis), and Han Guo for enzymes and Peter Yao (Bronx H.S. of Science) for assistance. We thank NIGMS (1R01GM096056), NIH Director’s New Innovator Award Program (1DP2-OD007335), and Starr Cancer Consortium for supporting the research.
Supporting Information Available
Additional procedures, characterization of compounds, and supplementary figures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Cantoni G. L. J. Biol. Chem. 1953, 204, 403. [PubMed] [Google Scholar]
- Zhang L.; Ding X. J.; Cui J.; Xu H.; Chen J.; Gong Y. N.; Hu L. Y.; Zhou Y.; Ge J. N.; Lu Q. H.; Liu L. P.; Chen S.; Shao F. Nature 2012, 481, 204. [DOI] [PubMed] [Google Scholar]
- Van Der Werf P.; Koshland D. E. Jr. J. Biol. Chem. 1977, 252, 2793. [PubMed] [Google Scholar]
- Rea S.; Eisenhaber F.; O’Carroll N.; Strahl B. D.; Sun Z. W.; Schmid M.; Opravil S.; Mechtler K.; Ponting C. P.; Allis C. D.; Jenuwein T. Nature 2000, 406, 593. [DOI] [PubMed] [Google Scholar]
- Bernstein B. E.; Meissner A.; Lander E. S. Cell 2007, 128, 669. [DOI] [PubMed] [Google Scholar]
- Lee B. W.; Sun H. G.; Zang T.; Kim B. J.; Alfaro J. F.; Zhou Z. S. J. Am. Chem. Soc. 2010, 132, 3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam K.; Zheng W.; Yu H.; Deng H.; Luo M. ACS Chem. Biol. 2011, 6, 679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam K.; Bothwell I.; Chen Y.; Sengelaub C.; Wang R.; Deng H.; Luo M. J. Am. Chem. Soc. 2012, 134, 5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Pan Y.; Yang W.; Liu W.; Zou H.; Zhao Z. K. ChemBioChem 2013, 14, 1438. [DOI] [PubMed] [Google Scholar]
- Peters W.; Willnow S.; Duisken M.; Kleine H.; Macherey T.; Duncan K. E.; Litchfield D. W.; Luscher B.; Weinhold E. Angew. Chem., Int. Ed. 2010, 49, 5170. [DOI] [PubMed] [Google Scholar]
- Dalhoff C.; Lukinavicius G.; Klimasauskas S.; Weinhold E. Nat. Chem. Biol. 2006, 2, 31. [DOI] [PubMed] [Google Scholar]
- Comstock L. R.; Rajski S. R. J. Org. Chem. 2004, 69, 1425. [DOI] [PubMed] [Google Scholar]
- Weller R. L.; Rajski S. R. Org. Lett. 2005, 7, 2141. [DOI] [PubMed] [Google Scholar]
- Islam K.; Chen Y.; Wu H.; Bothwell I. R.; Blum G. J.; Zeng H.; Dong A.; Zheng W.; Min J.; Deng H.; Luo M. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 16778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalhoff C.; Lukinavicius G.; Klimasauakas S.; Weinhold E. Nat. Protoc. 2006, 1, 1879. [DOI] [PubMed] [Google Scholar]
- Wang R.; Zheng W.; Yu H.; Deng H.; Luo M. J. Am. Chem. Soc. 2011, 133, 7648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S.; Zhang J.; Huber T. D.; Sunkara M.; Hurley K.; Goff R. D.; Wang G.; Zhang W.; Liu C.; Rohr J.; Van Lanen S. G.; Morris A. J.; Thorson J. S. Angew. Chem., Int. Ed. 2014, 53, 3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.; Zheng W.; Luo M. Anal. Biochem. 2014, 450, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willnow S.; Martin M.; Luscher B.; Weinhold E. ChemBioChem 2012, 13, 1167. [DOI] [PubMed] [Google Scholar]
- Bothwell I. R.; Islam K.; Chen Y.; Zheng W.; Blum G.; Deng H.; Luo M. J. Am. Chem. Soc. 2012, 134, 14905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter J. M.; Chiou G.; Bothwell I. R.; Xu W.; Garg N. K.; Luo M.; Tang Y. Org. Lett. 2013, 15, 3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkuviene M.; Clouet-d’Orval B.; Cerniauskas I.; Weinhold E.; Klimasauskas S. Nucleic Acids Res. 2012, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwig D. F.; Grippe A. T.; McIntyre T. A.; Booker S. J. Biochemistry 2004, 43, 13510. [DOI] [PubMed] [Google Scholar]
- Skupin J. Rocz. Chem. 1962, 36, 631. [Google Scholar]
- Boyle P. H.; Davis A. P.; Dempsey K. J.; Hosken G. D. Tetrahedron: Asymmetry 1995, 6, 2819. [Google Scholar]
- Jamieson A. G.; Boutard N.; Beauregard K.; Bodas M. S.; Ong H.; Quiniou C.; Chemtob S.; Lubell W. D. J. Am. Chem. Soc. 2009, 131, 7917. [DOI] [PubMed] [Google Scholar]
- Koch T.; Buchardt O. Synthesis 1993, 1065. [Google Scholar]
- Klayman D. L.; Griffin T. S. J. Am. Chem. Soc. 1973, 95, 197. [Google Scholar]
- Perrone P.; Daverio F.; Valente R.; Rajyaguru S.; Martin J. A.; Leveque V.; Le Pogam S.; Najera I.; Klumpp K.; Smith D. B.; McGuigan C. J. Med. Chem. 2007, 50, 5463. [DOI] [PubMed] [Google Scholar]
- Miles R. W.; Nielsen L. P. C.; Ewing G. J.; Yin D.; Borchardt R. T.; Robins M. J. J. Org. Chem. 2002, 67, 8258. [DOI] [PubMed] [Google Scholar]
- Iwig D. F.; Booker S. J. Biochemistry 2004, 43, 13496. [DOI] [PubMed] [Google Scholar]
- Guo H.; Wang R.; Zheng W.; Chen Y.; Blum G.; Deng H.; Luo M. ACS Chem. Biol. 2014, 9, 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty D.; Islam K.; Luo M. K. Chem. Commun. 2012, 48, 1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z. S.; Smith A. E.; Matthews R. G. Bioorg. Med. Chem. Lett. 2000, 10, 2471. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.