

Abstract
Atg8 is a ubiquitin-like autophagy protein in eukaryotes that is covalently attached (lipidated) to the elongating autophagosomal membrane. Autophagy is increasingly appreciated as a target in diverse diseases from cancer to eukaryotic parasitic infections. Some of the autophagy machinery is conserved in the malaria parasite, Plasmodium. Although Atg8’s function in the parasite is not well understood, it is essential for Plasmodium growth and survival and partially localizes to the apicoplast, an indispensable organelle in apicomplexans. Here, we describe the identification of inhibitors from the Malaria Medicine Venture Malaria Box against the interaction of PfAtg8 with its E2-conjugating enzyme, PfAtg3, by surface plasmon resonance. Inhibition of this protein–protein interaction prevents PfAtg8 lipidation with phosphatidylethanolamine. These small molecule inhibitors share a common scaffold and have activity against both blood and liver stages of infection by Plasmodium falciparum. We have derivatized this scaffold into a functional platform for further optimization.
Introduction
The malaria parasite, Plasmodium, is a major public health burden in the developing world, and despite the existence of antimalarial treatment active in blood stages of infection, there is a continual need for novel drug design as the parasite develops resistance to current treatments.1 Additionally, treatment for the hypnozoite-causing species, Plasmodium vivax, requires primaquine, which has severe side effects and causes hemolysis in patients with glucose-6-phosphate dehydrogenase deficiency.2 Discovery of novel compounds in antimalarial drug development is essential for future intervention strategies.
Atg8 is the ubiquitin-like (Ubl) protein necessary for formation and maturation of autophagosomes in autophagy in Eukarya. In yeast and mammals, Atg8 is lipidated to the autophagosome membrane, but in Plasmodium, Atg8 is partially conjugated to the membrane of the apicoplast under nonstarvation conditions.3−6 The apicoplast is a nonphotosynthetic chloroplast-like organelle present in apicomplexans and is essential for isoprenoid synthesis.7 Under starvation conditions, Atg8 relocates to acidic vesicles with Rab7 in and near the food vacuole.8 Atg8 is essential to the Plasmodium parasite and has been proposed as a target for antimalarial drug design.9
In most eukaryotes, lipidation of Atg8 to phosphatidylethanolamine (PE) in membranes normally requires proteolytic processing of the C-terminus of Atg8 by Atg4 and activation via adenosine 5′-triphosphate (ATP) followed by intermediate thioester bond formation with the E1-activating enzyme Atg7. Atg8 is then transferred to its E2-like conjugating enzyme Atg3, forming a second thioester intermediate before being conjugated to the nitrogen of PE (Figure 1).10 This process also requires noncovalent interaction between Atg8 and Atg3 through a well-characterized Atg8-interacting motif (AIM) in Atg3 and two hydrophobic pockets, termed the W and L-site, in Atg8.11 Notably, in Plasmodium, Atg8 is synthesized with a C-terminal glycine and therefore does not require activation by Atg4. Recently published studies showed a drastic growth defect in Plasmodium falciparum when levels of PfAtg7 were reduced.12 This along with the essentiality of Plasmodium Atg8 suggests that targeting PfAtg8 lipidation is a good strategy for drug intervention.9
Figure 1.

Overview of Atg8 Conjugation pathway in autophagy. (A) Visual representation of the localization of Atg8 in autophagy. Atg8, represented by the rectangle, is essential to the elongation of the autophagosomal membrane. (B) Generic conjugation pathway shown for yeast system. In Plasmodium, Atg8 is synthesized with a C-terminal glycine that does not require proteolytic processing for activation. The red line indicates step of pathway targeted by our inhibitors.
We previously elucidated the protein crystal structure of P. falciparum Atg8 bound to a peptide corresponding to PfAtg3’s AIM (PDB code 4EOY).13 Regions of diversity exist between the human and Plasmodium system that may be exploitable through small molecule inhibition. Our mutational and interaction studies suggest that the Plasmodium Atg8-Atg3 interaction requires Atg8’s W/L site as well as the apicomplexan loop on Atg8 (residues 67–76), termed the A-loop.13 Here, we report the identification of a class of compounds that inhibit the Plasmodium Atg8-Atg3 interaction and that inhibit in vitro growth of P. falciparum in blood- and liver-stage assays, presumably through prevention of PfAtg8 lipidation.
Results
Screening of the MMV Malaria Box Library
Previously, we developed a surface plasmon resonance (SPR)-based competition assay to identify compounds that disrupt the PfAtg8-PfAtg3 noncovalent interaction.13PfAtg3 is immobilized onto an SPR chip, and PfAtg8 is injected in the presence of dimethyl sulfoxide (DMSO, control) or a compound (dissolved in DMSO), and binding is measured by the SPR response. The Medicines for Malaria Venture (MMV) Malaria Box of 200 druglike and 200 probelike molecules was screened at 5 μM in a primary SPR screen (Figure 2A).14 Six compounds met the cutoff for at least 25% inhibition of the PfAtg8-PfAtg3 interaction: (N-(4-methylphenyl)-4-pyridin-2-yl-1,3-thiazol-2-amine) (1), (2-methylsulfanyl-N-(4-pyridin-2-yl-1,3-thiazol-2-yl)benzamide) (2), (2-bromo-N-(4-pyridin-2-yl-1,3-thiazol-2-yl)benzamide) (3), N-[2-chloro-5-(trifluoromethyl)phenyl]-2-[2-(4-methylphenyl)pyrazolo[1,5-a]pyrazin-4-yl]sulfanylacetamide (4), 1-[4-(dimethylamino)phenyl]-6,6-dimethyl-1,3,5-triazine-2,4-diamine (5), and 2-N,3-N-bis(4-bromophenyl)quinoxaline-2,3-diamine (6) (Figure 2A,2B, Table 1). In subsequent dose-dependent studies, 4–6 demonstrated a constant level of inhibition independent of concentration of the small molecule and were not further investigated. Compounds 1–3 led to dose-dependent inhibition with an SPR inhibitory concentration (IC50 SPR) ranging from 6 to 18 μM (Figure 2C). Interestingly, these compounds shared a common scaffold: 4-pyridin-2-yl-1,3-thiazol-2-amine (PTA) incorporated as the N-substituent in various anilines or benzamides. 1–3 were tested for their effect on the stability of PfAtg8CM13 using fluorescence-based thermal shift assays (TSAs). None of the compounds significantly affected the melting temperature (Tm) indicating that they most likely did not disturb the tertiary structure of PfAtg8 (Figure 2D).
Figure 2.

Identification of a common scaffold that inhibits Atg8-Atg3 from the MMV malaria box screen. (A) Primary screen of MMV Malaria Box14 with SPR competition assay. The gray box denotes compounds meeting the threshold of greater than 25% inhibition. (B) Bar graph showing inhibition of hits in primary screen, denoted by compound number. (C) Dose-dependent inhibition of PfAtg8-PfAtg3 interaction by MMV compounds. Inhibition was measured with increasing amount of compound in SPR competition screen. Mean and standard deviation (SD) of three injections are shown. (D) Thermal stability assay of PfAtg8CM with the three MMV hits. Error bars show SD of three measurements.
Table 1. Structures and Inhibition Data for PTA Compoundsa.
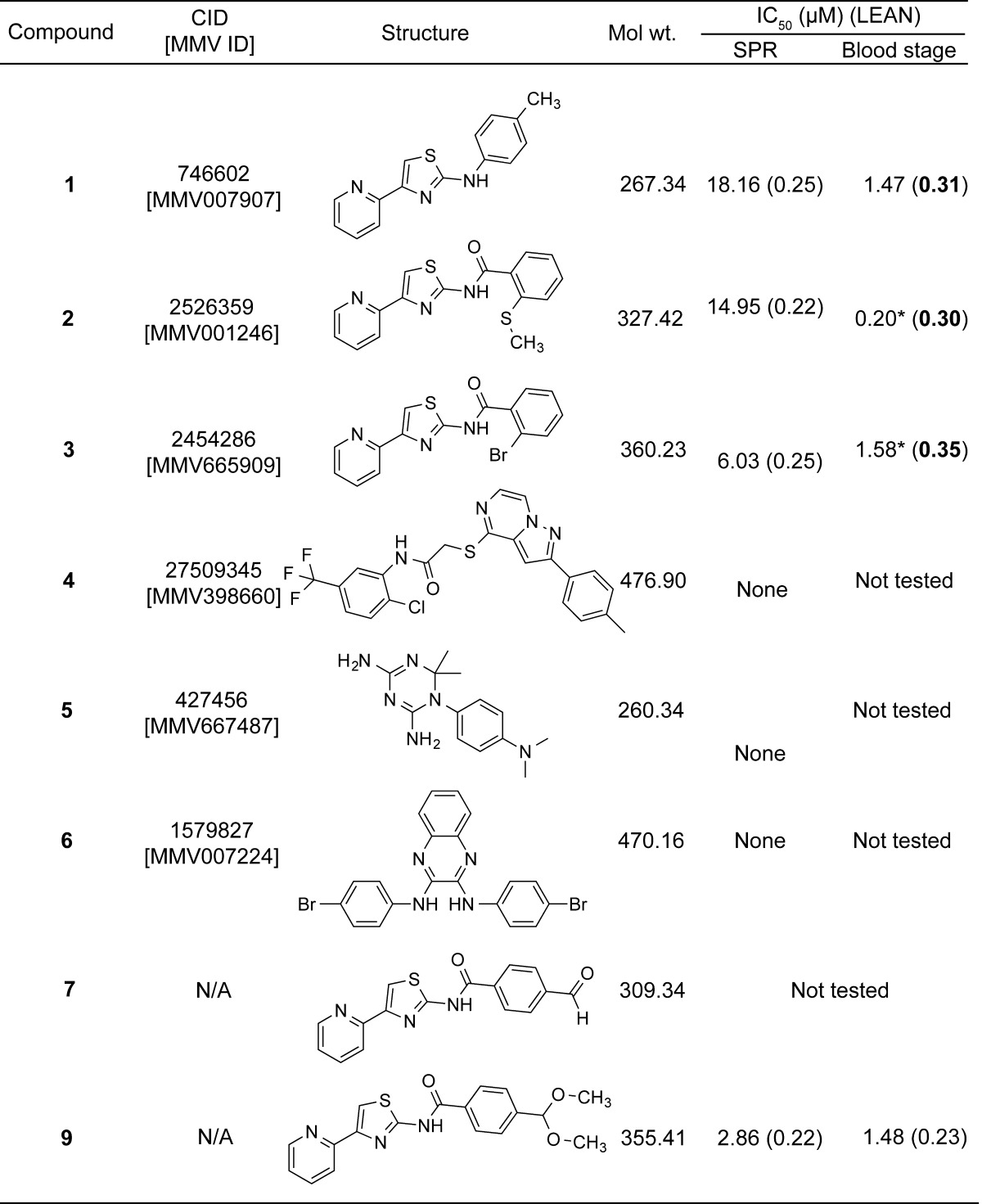
PTA-containing compounds used in studies listed with PubChem Compound Identification (CID), MMV ID, chemical structure, molecular weight (g/mol), IC50 in SPR and blood stage assays, and LEAN score, calculated as −log (IC50)/number of heavy atoms. Asterisks denote IC50 values derived from the literature.
Determination of the PTA Binding Site on PfAtg8
We tested whether these compounds bound directly to PfAtg8 using SPR. PfAtg8CM was immobilized onto an SPR Ni-nitrilotriacetic acid (NTA) chip via a 12 histidine N-terminal tag. 1, chosen for its better solubility, was injected over the chip, and binding was measured. Compound 1 led to a dose-dependent increase in SPR response, indicating binding to PfAtg8CM (Figure 3A). We next sought to determine the binding site for the PTA compounds with in silico docking.
Figure 3.
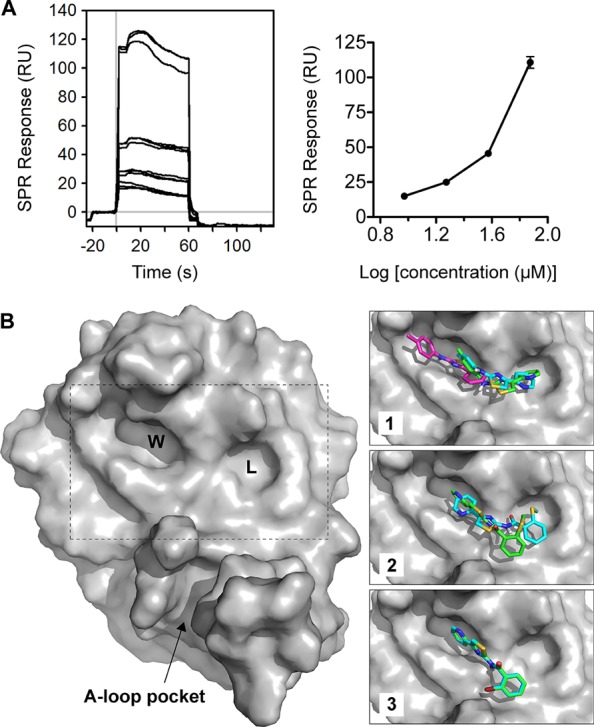
Identification of 1 binding site on PfAtg8CM. (A) Binding of 1 to PfAtg8. His12-PfAtg8CM was immobilized onto a nickel-charged NTA SPR chip. 1 was injected over immobilized PfAtg8-variants in two separate runs at four concentrations. Graph shows mean ± SD of binding from 1. (B) In silico docking of PTA compounds to PfAtg8CM (PDB code 4EOY). Overall structure of PfAtg8 with W- and L-site and A-loop demarcated. Inset shows best poses for each compound. The predicted pose for unconstrained docking is shown in cyan. Docking was also performed with a hydrogen bond constraint to the carbonyl of Lys47, located between the W- and L-site. Predicted pose for constrained docking is shown in green. For 1, an alternate pose, it is highly ranked in the unconstrained docking and is enriched in the top 10 poses as shown in magenta.
The OpenEye software package (www.eyesopen.com)15 was used to dock conformers of the compounds against the X-ray structure of PfAtg8 (PDB code 4EOY).13 All three compounds docked to the W-site of PfAtg8 (Figure 3B). 2 and 3 bound with the pyridine ring in the W-site, whereas 1 was predicted to bind with the pyridine ring in the L-site, the thiazole ring positioned between the pockets, and the methylbenzene group in the W-site. An alternate pose was enriched within the top 10 poses output by the docking study in which 1 binds solely within the W-site in a more compact conformation. 1 is missing a donor oxygen compared to 2 and 3 and, therefore, may adopt a different mode of binding to the W- and L-sites of PfAtg8. 2 docked with the pyridine ring in the W-site, and the methylsulfanylbenzene group bound to the L-site of PfAtg8. Docking was repeated using a version of the receptor for which the carbonyl of Lys47 was input as a hydrogen bond acceptor constraint (Figure 3B inset). 3 had the same docking pose in both studies. 1 had a similar binding pose to the highest ranked pose from the unconstrained docking, while 2 was slightly different with the pyridine ring rotated 90° in the W-site and the benzene ring positioned just below and to the left of the L-site pocket with the methylsulfanyl group reaching into the mostly hydrophobic L-site.
Compound 1 Shows Activity against Plasmodium Liver Stages
The half maximal inhibitory concentrations (IC50) for these compounds in P. falciparum 3D7 blood stages were previously reported and are located on the NCBI PubChem database (http://pubchem.ncbi.nlm.nih.gov). 1 has a reported IC50 of 350–400 nM (PubChem bioassay ID (AID): 660866 and 449703).16 The reported IC50 for 2 ranged from 0.20 to 6.8 μM, while 3 ranged from 1.36 to 4.52 μM (PubChem AID: 660866 and 449707).17 We focused on compound 1 for further studies because the reported cytoxicity in human cell lines is much lower than that of compounds 2 or 3 (PubChem AID: 660872, 685525, and 449705).
PfAtg8 is expressed and lipidated during the liver stage where it partially localizes to the apicoplast.4 Treatment of early liver stage parasites with the autophagy inhibitor 3-methyladenine is reported to delay conversion of the parasite into its trophozoite form.9 We therefore hypothesized that 1 would also have activity against the liver stage. 1 was previously tested in Plasmodium yoelii liver stage cultures and did not display >50% inhibition at the screening concentration of 10 μM; an IC50 was not reported (PubChem AID: 602118 and 602156).18,19P. yoelii and Plasmodium berghei are often used to test drugs for liver stage inhibition as they are easier to culture. However, these are rodent malaria models and may not be indicative of activity in P. falciparum. Analysis of the W/L-site in these three species revealed differences in the amino acid composition that could affect drugs predicted to bind in that region (Figure 4). We utilized a recently established P. falciparum liver stage in vitro model in which sporozoites isolated from infected mosquitos’ salivary glands invade HC-04 hepatocytes.20 HC-04 is a unique immortalized cell line that exhibits the expression of biochemical markers characteristic for normal hepatocytes and allows for the full development of the human malaria parasite, P. falciparum.(20−22) Using this system, we assessed the effect of 1 on the development of P. falciparum 3D7-green fluorescent protein (GFP) parasites23 in human hepatocytes in vitro. No change in the viability of HC-04 cells was detected in response to treatment with 3 μM or 30 μM of 1 for 96 h (Figure 5A). Using flow cytometry, we observed about a 50% decrease in the proportion of hepatocytes infected with P. falciparum 3D7-GFP sporozoites (GFP+/propidium iodide (PI)- cells) in response to treatment with 30 μM, but not with 3 μM of 1 (Figure 5B, C). Additionally, there was a dose-dependent reduction in the intensity of GFP fluorescence at both concentrations of 1, indicating inhibition of parasite development within hepatocytes, at least in vitro (Figure 5D). Because 1 did not affect cell survival or cell growth of HC-04 cells (Figure 5A), the compound’s effect on the parasite is unlikely to result from host cell cytotoxicity.
Figure 4.
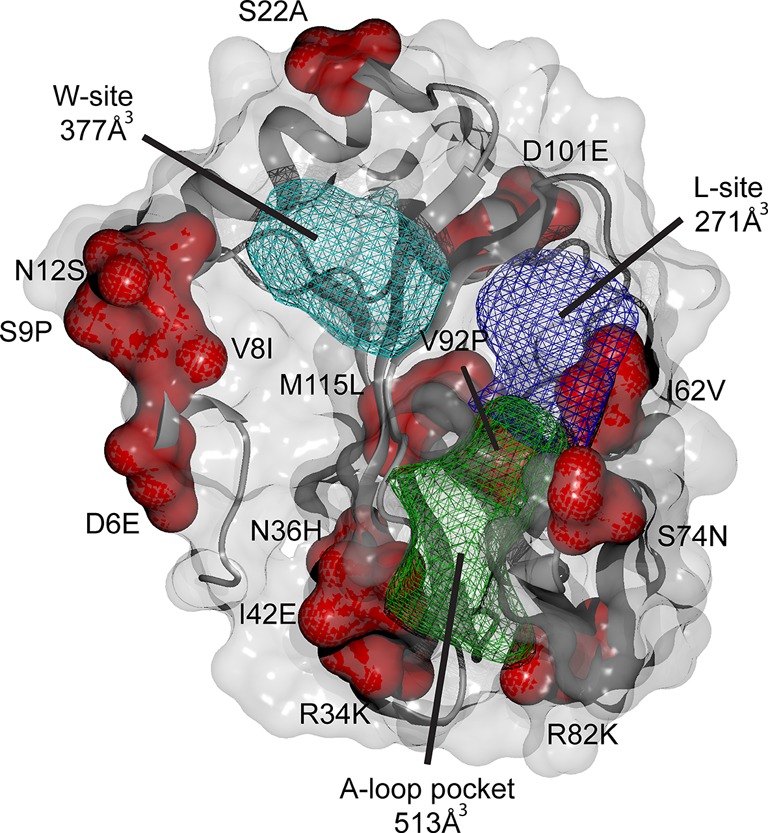
P. falciparum and P. yoelii Atg8 structural differences. PfAtg8 (PDB code 4EOY) is shown with surface and cartoon representation. Amino acid changes between the species are shown in red with P. falciparum letter and numbering followed by P. yoelii. W-site, L-site, and A-loop pockets are shown in mesh in cyan, purple, and green, respectively. P. falciparum Atg8 pocket sizes were calculated with OpenEye VIDA visualization software (www.eyesopen.com).
Figure 5.

Effect of 1 treatment on the development of P. falciparum 3D7 GFP parasite in HC-04 cells in vitro. (A) Flow cytometry based detection of Annexin-V positive and PI-positive cells in hepatocyte cultures treated with 1 (as described in Experimental section). Dot plot graphs demonstrate representative pattern of staining, and bar graphs show summary (mean ± SD) of viable cell detection obtained in three independent hepatocyte cultures. (B) Viable infected hepatocytes (GFP+/PI−) were detected by flow cytometry in HC-04 cultures 72 h post infection with P. falciparum. Dot plot graphs demonstrate representative pattern of staining, and numbers reflect percentages of GFP positive cells in total PI negative cell populations. (C) Summary (mean ± SD) of viable infected cell detection obtained in three independent hepatocyte cultures. (D) GFP-specific fluorescence was assessed in infected cultures exposed to 1 or DMSO for 72 h. Histograms demonstrate one representative staining pattern, and numbers reflect mean fluorescence intensity (MFI) in GFP-positive populations. Bar graphs reflect GFP-specific MFI (mean ± SD) in viable cell population detected in three independent hepatocyte cultures.
Validation of Drug Effect on PfAtg8 in Parasite Cultures
We next sought to determine whether 1 had an effect on PfAtg8 in P. falciparum blood stage cultures. In immunoblot assays, very low levels of endogenous PfAtg8 were detected in DMSO-treated control cells. Incubation of cells in minimal media lacking human serum for 5 h led to a very slight increase in PfAtg8. In contrast, treatment with 50 μM cytocidal levels of compound 1 led to a drastic increase in PfAtg8 protein levels as well as to a shift in mobility, likely corresponding to the unlipidated form of PfAtg8. Overall protein levels were unchanged, indicating an up-regulation or accumulation of PfAtg8 in the presence of 1 under conditions that are likely leading to cell death (Figure S1 of the Supporting Information). After treating the parasites for 6 hours, a dose-dependent increase in delipidation of PfAtg8 is already observed at 12.5 μM of 1, and at 25 μM, unlipidated PfAtg8 is the predominant species (Figure 6A, Figures S2–S4 of the Supporting Information). When investigating the soluble versus insoluble membrane fraction, PfAtg8 could only be detected in the soluble fraction with the number of parasites used per lane. We attribute this to the low amount of PfAtg8 present in the untreated control and to reaching the detection limit of our assay (data not shown). At the treatment concentration and duration used in the study, parasite morphology appeared normal (Figure 6B).
Figure 6.
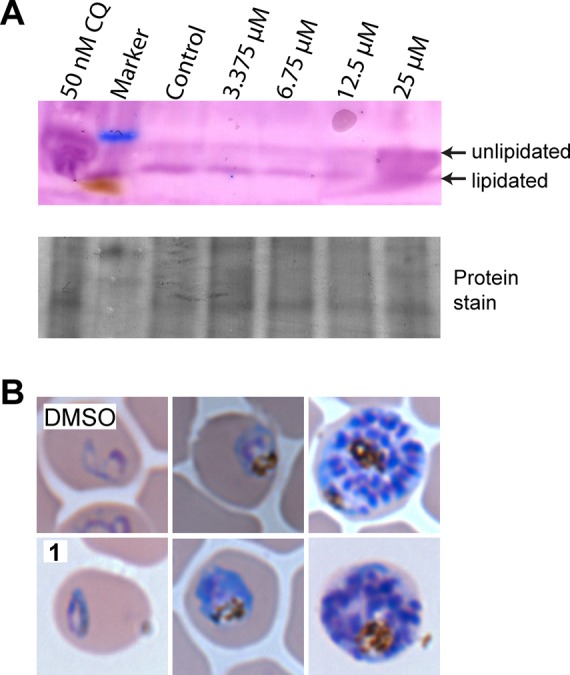
Effect of 1 on PfAtg8 protein levels. (A) Dose-dependent immunoblot analysis of P. falciparum treated with DMSO or 3.375, 6.75, 12.5, or 25 μM 1 for 6 h. Chloroquine (CQ) at 50 nM was used as a positive control of autophagy inhibition. Atg8-PE has a faster migration than unlipidated Atg8 with SDS-PAGE. Arrows indicate the migration of lipidated and unlipidated PfAtg8. The blot was probed with antibody against TgAtg8, demonstrated to be cross reactive against PfAtg8.40 (B) Blood smears of P. falciparum after treatment with DMSO or 50 μM 1 for 5 h, observed at 100× magnification. Representative images for different stages are shown, progressing from ring stage on the left to late schizont on the right.
Synthesis of a Novel PTA Derivative with a Functional Handle
Our studies indicated that the PTA scaffold is a good platform for hit-optimization. We synthesized a PTA-benzaldehyde derivative, 4-formyl-N-(4-pyridin-2-yl-1,3-thiazol-2-yl)benzamide (7), with a functional handle extending off the common hydrophobic ring system (Table 1, Scheme 1). 7 was prepared from commercially available PTA and 4-formyl benzoic acid through a dicyclohexylcarbodiimide (DCC)-promoted amide coupling. Compound 7 can be tethered through a dialkoxyamine linker to a library of aldehydes and can be screened using our primary SPR competition assay against the PfAtg8-PfAtg3 interaction.24 In docking studies, 7 bound the W- and L-site of PfAtg8 in a fashion similar to compound 2 with the functional handle positioned toward the A-loop pocket (Figure 7A). A gain in parasite selectivity for such bifunctional analogues is expected as the A-loop is missing in the human Atg8 homologues.13 To confirm binding to PfAtg8, we tethered 7 to (+)-biotinamidohexanoic acid hydrazide (BACH) through its reactive aldehyde group and tested binding with SPR. The biotinylated PTA compound 8 was immobilized onto a neutravidin coupled SPR chip. His12-PfAtg8CM injected at various concentrations led to a dose-dependent increase in SPR response indicating PfAtg8 directly binds 8 with a KD of 540 nM. In contrast, the human Atg8 homologue, microtubule-associated protein light chain 3 (hLC3) showed much lower affinity for 8 with a KD of 18 μM, indicating specificity of the PTA scaffold for P. falciparum (Figure 7B). Additionally, recombinant PfAtg3 did not bind immobilized 8 (data not shown) in agreement with our docking studies suggesting binding to the W-site of PfAtg8.
Scheme 1.

Figure 7.

Analysis of PTA-derivatives. (A) In silico docking of PTA-derivatives on PfAtg8. 7 (yellow) and 9 (green) were docked onto the constrained receptor of PfAtg8CM (PDB code 4EOY) with OpenEye docking suite.15 (B) Direct binding of the PTA scaffold to PfAtg8. PfAtg8CM and hLC3 were injected over immobilized 8, and binding was measured with SPR. (C) Superposition of small molecule hits derived from MMV Malaria Box with 9. The individual molecular surfaces with their corresponding electrostatic potential are depicted in side and top view. All four molecules share the PTA moiety and were superimposed using ROCS via shape complementarity and Tanimoto color scoring function.46 The figure was prepared with Vida and was rendered in PovRay (www.povray.org).
Compound 7 was converted to the hydrochloride salt and was subjected to acid-catalyzed acetal formation with methanol and trimethyl orthoformate under microwave irradiation to provide the dimethyl acetal compound 9, an unreactive derivative, to confirm the inhibitory activity of the starting platform (Table 1, Scheme 1). 4-(Dimethoxymethyl)-N-(4-pyridin-2-yl-1,3-thiazol-2-yl)benzamide 9 has a similar shape and distribution of the acceptor and donor pairs as the original PTA compounds (Figure 7C) and docked onto PfAtg8 in a fashion similar to 7 (Figure 7A).
SPR studies confirmed that 9 inhibited the PfAtg8-PfAtg3 interaction with an IC50 of 2.86 μM in SPR (Figure 8A). We next measured growth inhibition of P. falciparum 3D7 by 1 using the SYBR green I assay.25 This assay exploits the absence of nuclei in erythrocytes with a fluorescent dye that is unquenched upon binding to nucleic acids, preferentially double-stranded DNA. In two of three independent experiments, the IC50 of 1 was 768 nM, similar to previously published results, while in a third experiment, the IC50 was 3.3 μM, resulting in an average IC50 of 1.61 ± 1.47 μM.16 Using this assay, 9 had a potency similar to that of compound 1 against the blood stage of P. falciparum with an average IC50 of 1.48 ± 0.6 μM (Figure 8B).
Figure 8.

Validation of 7 as starting point for optimization. (A) Inhibition of PfAtg8-PfAtg3 interaction by 9. SPR response of PfAtg8 injected over immobilized PfAtg3 was measured in the presence of increasing concentration of 9. IC50 was determined as 2.86 μM. Mean ± SD of three injections are shown. (B) Inhibition of blood stage parasites 9. SYBR green I assays were used to measure inhibition by Chloroquine (CQ), 1, and 9. Growth inhibition curves are shown for one experiment with IC50 values from two to three experiments in table inset.
Discussion
We identified compound 1 through a screen for inhibitors against the Plasmodium Atg8-Atg3 protein–protein interaction (PPI). Targeting protein–protein interactions has long been overlooked in the drug development field. It was thought to be difficult because of the shallower nature of many pockets and the more extensive network of residues involved in the interaction compared to an enzymatic site. However, PPIs also offer the opportunity for more selectivity, an important concept when targeting a eukaryotic parasitic protein within a eukaryotic host. Great methodological and technological advances have been made recently using this approach with promising leads in cancer drug development.26−28 In the case of our inhibitor, in addition to its potential as a therapeutic drug, any 1-derived inhibitor could serve as a tool to elucidate the function of PfAtg8 during different stages of the malaria life cycle as its function is currently unclear.29 Treatment of P. falciparum with high levels of 1 led to a drastic increase in PfAtg8 protein levels, presumably the unlipidated form as judged by its migration in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). This increase could be due to an up-regulation of PfAtg8 synthesis to compensate for inhibition or due to a buildup of existing protein levels because of a blockade in autophagic degradation. In yeast, nitrogen starvation leads to induction of Atg8 expression while inhibition of later stages of autophagy leads to accumulation and even greater protein levels of Atg8.30,31 Further studies are necessary to determine if PfAtg8 is up-regulated at the transcriptional, translational, or degradation level in response to treatment with 1.
The IC50 in parasite cultures was much lower than against the protein–protein interaction as measured via SPR. This could be due to an accumulation of the drug inside the parasite or could be because even slight inhibition of PfAtg8 lipidation has drastic effects on parasite growth, similar to reaching the tipping point on a balance. An alternative explanation is the result of off-target effects; however, the observed delipidation of PfAtg8 could not be attributed to an off-target effect. 1 was previously reported to have low cytotoxicity with an LD50 (lethal dose) of 18.2 μM in HepG2 cells and half maximal cytotoxicity concentration (CC50) and IC50 of 32 μM in Huh7 cells (PubChem AID: 685525, 660872, 449705). In our study, we did not observe cell death in HC-04 cells at 30 μM. Together, this indicates 1 may be a good starting scaffold for antimalarial drug design.
Compound 1 had lower activity in the liver stage than in the blood stage, which could either indicate that PfAtg8 is less important in the liver stage or that less compound is delivered to the parasite in liver cells. There is precedence for this variability in the efficacy of antimalarials between the liver and blood stages.32 Compound 1 showed 50% inhibition of liver stage parasites at 30 μM, which is within the range for cytotoxicity in human cells. We suggest the use of 1 and the PTA scaffold for further probe development to increase specificity and potency in both the liver and blood stages. All PTA-containing MMV compounds had a ligand efficiency by atom number (LEAN) score greater than 0.3 in the blood stage assay, indicative of good ligand efficiency and good potential for future drug optimization.33 This combined with the higher affinity of 8 for PfAtg8 compared to human LC3 makes the PTA scaffold a promising starting point for expansion. We are currently pursuing optimization through a combinatorial oxime library approach using 7 with the goal to extend the inhibitor into the A-loop pocket and to gain selectivity toward PfAtg8.
Our SPR data confirm that 1 directly binds to PfAtg8, while our docking studies suggest that the PTA scaffolds of compounds 1–3, 7, and 9 bind the W-site. The W-sites of Atg8 homologues in mammals and yeast participate in numerous protein–protein interactions through binding to the aromatic residue of an AIM, including nonautophagic proteins.35−38 Therefore, our inhibitor and its derivatives could be used to identify novel Plasmodium Atg8 interactions as well as to confirm paralogous interactions known to occur in yeast and mammalian cells.
P. yoelii as a Liver Stage Model
Taken together, our bioinformatics analysis of the PTA-binding site and the SPR binding studies strongly suggest that the amino acid differences near the L- and W-site may have contributed to the failure of 1 in the P. yoelii liver stage drug-screening assay. Two residue differences (I8V, P9S) adjacent to the W-site may lead to a widening of the W-site in P. yoelli because of their shorter side chains, and one residue (V62I) participating directly in the L-site results in an extended side chain and therefore may decrease the volume of the L-site pocket. Additionally, L115M just below the L-site binding pocket may also contribute to a decreased volume as a secondary shell residue. Care and emphasis on choice of model system should be taken into account when studying protein–ligand interactions, as single amino acid substitutions may have drastic effects on binding.
Experimental Section
Materials
All reagents, unless specified, were purchased from Sigma–Aldrich.
Protein Expression and Purification
Proteins were cloned, expressed, and purified as described elsewhere.13 His12-PfAtg8CM variants were expressed and purified with Cobalt-NTA affinity columns similar to His6-PfAtg8CM, as previously published with the exception that proteins were eluted from cobalt-charged TALON resin (Clonetech) in buffer containing 50 mM ethylenediaminetetraacetic acid (EDTA) rather than imidazole.13
Surface Plasmon Resonance Assays
SPR runs were conducted on a Biacore 3000 instrument (GE Healthcare) at 25 °C with a flow rate of 50 μL/min, unless otherwise specified. Running buffer (RB) consisted of 1× phosphate-buffered saline (PBS) (1 mM KH2PO4, 5.6 mM Na2HPO4, 154.5 mM NaCl, pH 7.4), 0.01% v/v P20, and varying amounts of DMSO (Quality Biologicals). Binding and equilibrium constants were determined with Scrubber (BioLogic). A double referencing method was applied to correct for nonspecific binding to the chip with interspersed blank injections correcting for baseline drifts. Changes in refractive index because of DMSO were accounted for with a DMSO calibration curve.
MMV SPR Competition Assay
MBP-PfAtg3 was immobilized onto a CM5 chip (Biacore) as described previously with MBP immobilized on a reference flowcell.13 Compounds were added to 300 nM His6-PfAtg8CM in RB at a final concentration of 5 μM, and 40 μL was injected, followed by a 12.5 μL injection of 2 M MgCl2 for dissociation and regeneration of the SPR chip surface. The final DMSO concentration in SPR runs was 3%.
SPR Dose-Dependent Inhibition
Thirty microliters of PfAtg8CM at 200 nM was injected in the presence of a 2-fold dilution series of compound (highest concentration of 50 μM) or equivalent volume of DMSO (final DMSO concentration was 1%). Each injection was followed by a 10 μL injection of 2 M MgCl2. All measurements were conducted in triplicate.
Direct Binding of 1 to PfAtg8CM
His12-PfAtg8CM was injected over an NTA-chip (GE Healthcare) preconditioned with nickel, leading to capture of 3000 response units (RUs). Running buffer contained 3% DMSO. A 2-fold-dilution series of compounds, highest concentration of 75 μM, was injected over PfAtg8 variants at 40 μL/min.
Direct Binding of 8 to PfAtg8CM
8 was injected over a neutravidin-coated SPR chip (GE Healthcare) on one flowcell, with 130 RUs immobilized, while BACH was injected over a reference flowcell (150 RUs immobilized). His6-PfAtg8CM, Human LC3, or His6-PfAtg3 was injected in duplicate in running buffer containing 10 mM HEPES pH 7.5, 150 mM NaCl, 0.05% P20. Protein was dissociated from the chip after each cycle with 10 μL injection of 20 mM HEPES pH 7.4, 1% w/v SDS regeneration solution.
Synthesis of PTA Derivatives
General
All reagents were obtained from commercial suppliers and were used without further purification. Acetonitrile was distilled after drying on CaH2 and then was stored over 3 Å molecular sieves. Yields of all reactions refer to the purified products. Dynamic Adsorbents 32–63 μm silica gel was used for flash column chromatography, and 250 μm F254 plates were used for thin layer chromatography (TLC). Microwave-assisted reactions were carried out using a Biotage Initiator Microwave Synthesizer (300 W). 1H and 13C NMR spectra were acquired on a Bruker Avance III 500 spectrometer operating at 500 MHz for 1H and 125 MHz for 13C. Chemical shift values are reported as δ (ppm) relative to CHCl3 at δ 7.27 ppm and DMSO at δ 2.50 ppm for 1H NMR and CHCl3 at δ 77.0 ppm and DMSO at δ 39.51 ppm for 13C NMR. Mass spectrometry analysis was carried out at University of Illinois at Urbana-Champagne, School of Chemical Sciences, Mass Spectrometry Laboratory. The purity of synthesized compounds was ≥95% as analyzed by high-performance liquid chromatography (HPLC, Beckman Gold Nouveau System Gold) on a C18 column (Grace Alltima 3 μm C18 analytical Rocket column, 53 mm × 7 mm) using triethylammonium acetate buffer (50 mM, pH 7) and acetonitrile (ACN) as eluent, flow rate 3 mL/min, and detection at 300 nm.
4-Formyl-N-[4-(pyridin-2-yl)-1,3-thiazol-2-yl]benzamide (7)
To a solution of 4-(pyridin-2-yl)-1,3-thiazol-2-amine (0.059 g, 0.33 mmol) in acetonitrile (2.0 mL) was added sequentially dicyclohexylcarbodiimide (0.076 g, 0.37 mmol), 4-formylbenzoic acid (0.050 g, 0.33 mmol), and N,N-dimethylamino pyridine (0.012 g, 0.10 mmol). The mixture was heated at 50 °C for 17 h and then was allowed to cool to ambient temperature. Solids were removed by vacuum filtration, and the resulting filtrate was condensed under reduced pressure. The resulting yellow solid was redissolved in CHCl3 (5 mL), and 1 M HCl (5 mL) was added to give a yellow-tan emulsion at the liquid–liquid interface. This solid was collected by centrifugation at 4000 rpm for 5 min followed by manual collection of the resulting cake (this acid precipitation was necessary to remove closely eluting impurities). The solid was then purified by silica flash column chromatography (dichloromethane(DCM):MeOH:triethylamine 94:5:1) Rf =0.32. The product was obtained as a yellow powder (23 mg, 22% yield). 1H NMR (500 MHz, DMSO-d6) δ (ppm) = 12.95 (br. s., 1H), 10.13 (s, 1H), 8.63 (d, J = 3.93 Hz, 1H), 8.31 (d, J = 8.17 Hz, 2H), 8.07 (d, J = 8.33 Hz, 2H), 8.03 (d, J = 7.86 Hz, 1H), 7.92 (s, 1H) 7.91 (td, J = 2.00 Hz, 8.75 Hz, 1H), 7.35 (ddd, J = 1.10, 4.79, 7.47 Hz, 1H) 13C NMR (500 MHz, DMSO-d6) δ (ppm) = 192.90, 164.74, 158.91, 152.03, 149.54, 149.38, 138.50, 137.30, 137.15, 129.41, 128.94, 122.88, 120.06, 112.30. High-resolution mass spectrometry (HRMS, electrospray ionization, ESI) m/z: calcd 310.0650 (M – H+); found 310.0651 (M – H+).
4-[[2-[6-[5-[(3aR,4R,6aS)-2-Oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]hexanoyl]hydrazinyl]methyl]-N-(4-pyridin-2-yl-1,3-thiazol-2-yl)benzamide (8)
Twenty-five microliters 50 mM 7 dissolved in DMSO was incubated with 20 μL 50 mM BACH (5-[(3aS,4S,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]-N-(6-hydrazinyl-6-oxohexyl)pentanamide) (Sigma-Aldrich) dissolved in DMSO and 10 μL sodium acetate (pH 4.5) with 0.02% sodium azide at 37 °C overnight. Final concentration of 8 was 18 mM.
4-(Dimethoxymethyl)-N-[4-(pyridin-2-yl)-1,3-thiazol-2-yl]benzamide (9)
7 dissolved in CHCl3 was treated with 1 M HCl as described above to form the hydrochloride salt. 7 HCl (0.040 g, 0.12 mmol) was suspended in MeOH (0.5 mL), and trimethylorthoformate (0.010 mL, 0.91 mmol) was added followed by p-toulene sulfonic acid monohydrate (0.003 g, 0.012 mmol). This solution was heated by microwave irradiation at 130 °C in a sealed vial for 5 min and then was stirred at ambient temperature for 36 h at which time a precipitate formed. The solvent was removed under reduced pressure, and the residue was dissolved in DCM (10 mL) and was washed with saturated NaHCO3 (10 mL) and brine (10 mL) and was dried with Na2SO4. Condensation under reduced pressure yielded the product as a yellow powder (26 mg, 61% yield). 1H NMR (500 MHz, CDCl3) δ (ppm) = 9.81 (br s, 1H), 8.64 (d, J = 4.24 Hz, 1H), 7.96 (d, J = 8.17 Hz, 2H), 7.91 (d, J = 7.86 Hz, 1H), 7.74 (s, 1H), 7.74 (td, J = 1.73, 7.70 Hz, 1H), 7.62 (d, J = 8.17 Hz, 2H), 7.22 (dd, J = 4.95, 6.84 Hz, 1H), 5.47 (s, 1H), 3.35 (s, 6H) 13C NMR (500 MHz, CDCl3) δ (ppm) = 164.24, 158.12, 152.24, 149.85, 149.62, 143.32, 136.84, 131.80, 127.51, 127.29, 122.69, 120.50, 112.21, 102.08, 52.71. HRMS (ESI) m/z: calcd 356.1069 (M – H+); found 356.1070 (M – H+).
Thermal Shift Assays
Assays were conducted in 1× PBS with 1:1800 final dilution of SYPRO orange dye (Invitrogen). Measurements were made in triplicate for each condition. The concentration of His12-PfAtg8CM or His6-PfAtg8CM in assay was 65 μM. Fluorescence was measured from 20 to 80 °C in a Biorad C1000 thermal cycler. One hundred microliters of PTA compounds or equivalent volume of DMSO was added to His6-PfAtg8CM.
In Vitro Infection of Human Hepatocytes with P. falciparum 3D7-GFP
The HC-04 cell line (ATCC, Manassas, VA, U.S.) was maintained in complete medium (IMDM containing 2.5% FCS, 100 units/mL penicillin, 100 μg/mL streptomycin, and 2 mM l-glutamine, all from GIBCO, Life Technologies, Grand Island, NY). P. falciparum 3D7-GFP parasite strain23 was propagated in the Parasitology Core facility, the Johns Hopkins Malaria Research Institute. In vitro infection of human hepatocytes was done as described previously.20 Briefly, salivary glands were sequestered from infected Anopheles gambiae mosquitoes at day 17 after exposure to infective blood meal, and homogenates were separated on an OptiPrep Density Gradient (Sigma-Aldrich, St. Louis, MO) at 12 000g for 10 min. Sporozoites were collected from the gradient interface, were washed in complete medium, were counted using a hemocytometer, and were incubated with HC-04 cells at 3:1 sporozoite to hepatocyte ratio for 2 h at 37 °C. Infected cultures were further propagated in complete medium alone or in medium supplemented with 3 μM or 30 μM of 1. Flow cytometry based detection of infected cells was done 72 h post infection using FACSCalibur flow cytometer (BD Biosciences) and was analyzed using FlowJo software (Tree Star, Inc., Ashland, OR).
Effect of 1 on the viability of in vitro propagated HC-04 cells was monitored as follows: 0.3 × 106 cells per well were seeded into the 24-well plate and were treated with 3 μM or 30 μM of 1 in complete medium for 96 h, and a relevant amount of DMSO was used as a vehicle control. Detection of Annexin-V positive and PI-positive cells in hepatocyte cultures was done by flow cytometry according to the manufacturer’s instruction (Invitrogen, Life Technologies, Grand Island, NY, U.S.).
P. falciparum Blood Stage Culturing
P. falciparum 3D7 and FCR3 cultures were maintained using modified, previously published methods at 37 °C, 2% hematocrit of human red blood cells.39 Complete culture media consisted of sterile RPMI 1640 media (Life Technologies) supplemented with 10% human serum and 0.005% hypoxanthine and buffered with final concentrations of 0.6% HEPES and 0.26% NaHCO3. The FCR3 strain was maintained at 3% CO2 and 5% O2, 92% N2 atmosphere, while the 3D7 strain was maintained at 5% CO2, 5% O2, and 90% N2 atmosphere.
Immunoblot Analysis of P. falciparum Blood Stage
P. falciparum FCR3 (generously provided by Dr. J. Smith, Seattle BioMed) asynchronous culture, 25% parasitemia, was washed in starvation media lacking human serum and was resuspended in complete media with 50 μM compound 1 or equivalent DMSO or in starvation media with equivalent DMSO for 5 h. RBCs were harvested with centrifugation and were lysed with 0.2% saponin, and RBC lysate was removed through three 1× PBS washes. Parasites were harvested by centrifugation and were washed in 1× PBS with complete EDTA-free protease inhibitors (Roche) and were lysed by repeated vortexing and boiling in SDS reducing sample buffer. Lysates were separated with SDS-PAGE on a 4−20% polyacrylamide gel and were subjected to Western blotting with 1:400 α-TgAtg8, demonstrated to be cross-reactive with PfAtg840 (generously provided by Dr. P. Roepe, Georgetown University). HRP-conjugated secondary antibodies (Southern Biotech) were detected by SuperSignal West Femto (Thermo Scientific) or Amersham ECL Prime (GE Healthcare) chemiluminescent substrate. AP-conjugated secondary antibodies (EMD Millipore) were detected using NBT/BCIP (Promega) colorimetric stain. Total protein levels were visualized with ProAct Membrane Stain (Amresco) and were quantified with ImageJ.41 Parasite morphology at time of harvesting was visualized with light microscopy at 100× magnification on Olympus BX53 system microscope (Olympus America, Inc.).
SYBR Green I Growth Inhibition Assay
Ten microliters of 10× compound diluted in RPMI 1640 media (Gibco) with a constant concentration of 1% DMSO was added to a 96 well plate (Costar), 90 μL of 1.5% ring stage, synchronized with 5% w/v sorbitol P. falciparum 3D7 parasites, 1% hematocrit, in culture media with 10% v/v human serum with 10 μg/mL gentamycin. Each compound concentration and 1% v/v DMSO controls were run in triplicate. Plates were incubated at 37 °C in 5% O2, 5% CO2, and 90% N2 for 72 h. Plates were frozen, thawed, and incubated with 100 μL 2× SYBR green in lysis buffer (20 mM Tris pH 7.5, 5 mM EDTA, 0.008% Saponin, 0.08% TritonX-100) in the dark for at least 1 h. Fluorescence was measured with a plate reader (HTS 7000, PerkinElmer) at excitation/emission wavelengths of 485/535 nm.
In Silico Docking
Docking was conducted using the OpenEye software package using standard parameters if not specified otherwise (www.eyesopen.com).15 A receptor for PfAtg8 was made with make_receptor from OEDocking toolkit without constraints covering the whole molecule to detect potential binding pockets on the surface. Three main pockets (W-site, L-site, A-site) were detected with 377, 271, and 513 Å3 volumes, used in first docking studies. A second receptor was generated with specific constraints to the carbonyl of Lys47 as hydrogen bond donor. A maximum of 2000 conformers for each compound from the MMV Malaria Box was prepared with Omega2.42,43 FRED was used to dock these conformers onto both the constrained and unconstrained receptor.15 Docking results were visualized using the OpenEye visualization software, VIDA.
Generation of Homology Models for P. yoelii and P. berghei Atg8
Using iTasser, models of P. yoelii and P. berghei Atg8 were generated with PDB code 4EOY as the parent molecule.13,44 Default values as suggested by the Web server were used to generate these models. Sequences for P. yoelii (PYYM_0504500) and P. berghei (PBANKA_050410) were obtained from PlasmoDB.45
Human Subjects and Hazards
P. falciparum, a human pathogen, is cultivated in human erythrocytes. The organisms are maintained in licensed BSL2 facilities, and approvals are obtained annually for all consortia laboratories. Human erythrocytes are obtained either commercially or from healthy volunteers under Johns Hopkins IRB-approved protocols. Because these cells are provided to the lab without identifiers, their use does not constitute human investigation.
Acknowledgments
We thank the five anonymous reviewers, who have helped us to improve our manuscript. We thank Stephanie Trop, Peter Dumoulin, and Jinxia Ma for dissection of P. falciparum-infected mosquitoes for our assays. We thank Dr. Krista Matthews for technical help. We thank the Johns Hopkins Malaria Research Institute parasite and insectary facility for assistance with our experiments. This work was partially funded through The Bloomberg Family Foundation (J.B.), a grant (NIH AI099704 for C.F. M. and D.B.), and a Predoctoral fellowship from the Johns Hopkins Malaria Institute for A.U.P.H. We thank Dr. P. Roepe (Georgetown University), Dr. Anthony Sinai (University of Kentucky), and Dr. N. Kumar (Tulane University) for antibodies and Dr. J. Smith (Seattle BioMed) for the FCR3 strain of P. falciparum. We thank The Medicines for Malaria Venture for supply of the malaria box.
Glossary
Abbreviations
- Pf
P. falciparum
- SPR
surface plasmon resonance
- TSA
thermal shift assay
- CM
cysteine mutant
- AIM
Atg8 family interacting motif
- RBC
red blood cell
- RU
response units
- PTA
4-pyridin-2-yl-1,3-thiazol-2-amine
Supporting Information Available
A detailed analysis of the PfAtg8 Western blot with quantification. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. D.B. and C.F.M. synthesized compounds. A.U.H., J.L., N.S., A.S.M., D.S., and J.B. designed research. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Phyo A. P.; Nkhoma S.; Stepniewska K.; Ashley E. A.; Nair S.; McGready R.; ler Moo C.; Al-Saai S.; Dondorp A. M.; Lwin K. M.; Singhasivanon P.; Day N. P.; White N. J.; Anderson T. J.; Nosten F. Emergence of artemisinin-resistant malaria on the western border of Thailand: a longitudinal study. Lancet 2012, 379, 1960–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarlov A. R.; Brewer G. J.; Carson P. E.; Alving A. S. Primaquine sensitivity. Glucose-6-phosphate dehydrogenase deficiency: an inborn error of metabolism of medical and biological significance. Arch. Int. Med. 1962, 109, 209–234. [DOI] [PubMed] [Google Scholar]
- Kitamura K.; Kishi-Itakura C.; Tsuboi T.; Sato S.; Kita K.; Ohta N.; Mizushima N. Autophagy-related Atg8 localizes to the apicoplast of the human malaria parasite Plasmodium falciparum. PLoS One 2012, 7, e42977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eickel N.; Kaiser G.; Prado M.; Burda P. C.; Roelli M.; Stanway R. R.; Heussler V. T. Features of autophagic cell death in Plasmodium liver-stage parasites. Autophagy 2013, 9, 568–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayabalasingham B.; Voss C.; Ehrenman K.; Romano J. D.; Smith M. E.; Fidock D. A.; Bosch J.; Coppens I. Characterization of the ATG8-conjugation system in 2 Plasmodium species with special focus on the liver stage: Possible linkage between the apicoplastic and autophagic systems?. Autophagy 2014, 10, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes S.; Bunnik E. M.; Saraf A.; Conner C. M.; Escalante A.; Sardiu M. E.; Ponts N.; Prudhomme J.; Florens L.; Le Roch K. G. The multifunctional autophagy pathway in the human malaria parasite, Plasmodium falciparum. Autophagy 2014, 10, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh E.; DeRisi J. L. Chemical rescue of malaria parasites lacking an apicoplast defines organelle function in blood-stage Plasmodium falciparum. PLoS Biol. 2011, 9, e1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlins A. M.; Ben-Rached F.; Willams R. A. M.; Proto W. R.; Coppens I.; Ruch U.; Gilberger T. W.; Coombs G. H.; Mottram J. C.; Müller S.; Langsley G. Plasmodium falciparum Atg8 implicated in both autophagy and apicoplast formation. Autophagy 2013, 9, 1540–1552. [DOI] [PubMed] [Google Scholar]
- Duszenko M.; Ginger M. L.; Brennand A.; Gualdron-Lopez M.; Colombo M. I.; Coombs G. H.; Coppens I.; Jayabalasingham B.; Langsley G.; de Castro S. L.; Menna-Barreto R.; Mottram J. C.; Navarro M.; Rigden D. J.; Romano P. S.; Stoka V.; Turk B.; Michels P. A. Autophagy in protists. Autophagy 2011, 7, 127–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura Y.; Kirisako T.; Takao T.; Satomi Y.; Shimonishi Y.; Ishihara N.; Mizushima N.; Tanida I.; Kominami E.; Ohsumi M.; Noda T.; Ohsumi Y. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [DOI] [PubMed] [Google Scholar]
- Yamaguchi M.; Noda N. N.; Nakatogawa H.; Kumeta H.; Ohsumi Y.; Inagaki F. Autophagy-related protein 8 (Atg8) family interacting motif in Atg3 mediates the Atg3-Atg8 interaction and is crucial for the cytoplasm-to-vacuole targeting pathway. J. Biol. Chem. 2010, 285, 29599–29607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker D. M.; Mahfooz N.; Kemme K. A.; Patel V. C.; Spangler M.; Drew M. E. Plasmodium falciparum erythrocytic stage parasites require the putative autophagy protein PfAtg7 for normal growth. PLoS One 2013, 8, e67047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hain A. U.; Weltzer R. R.; Hammond H.; Jayabalasingham B.; Dinglasan R. R.; Graham D. R.; Colquhoun D. R.; Coppens I.; Bosch J. Structural characterization and inhibition of the Plasmodium Atg8-Atg3 interaction. J. Struct. Biol. 2012, 180, 551–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenberg T.; Burrows J. N.; Kowalczyk P.; McDonald S.; Wells T. N.; Willis P. The open access malaria box: a drug discovery catalyst for neglected diseases. PLoS One 2013, 8, e62906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGann M. FRED and HYBRID docking performance on standardized datasets. J. Comput.-Aided Mol. Des. 2012, 26, 897–906. [DOI] [PubMed] [Google Scholar]
- Plouffe D.; Brinker A.; McNamara C.; Henson K.; Kato N.; Kuhen K.; Nagle A.; Adrian F.; Matzen J. T.; Anderson P.; Nam T. G.; Gray N. S.; Chatterjee A.; Janes J.; Yan S. F.; Trager R.; Caldwell J. S.; Schultz P. G.; Zhou Y.; Winzeler E. A. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 9059–9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiguemde W. A.; Shelat A. A.; Bouck D.; Duffy S.; Crowther G. J.; Davis P. H.; Smithson D. C.; Connelly M.; Clark J.; Zhu F.; Jimenez-Diaz M. B.; Martinez M. S.; Wilson E. B.; Tripathi A. K.; Gut J.; Sharlow E. R.; Bathurst I.; El Mazouni F.; Fowble J. W.; Forquer I.; McGinley P. L.; Castro S.; Angulo-Barturen I.; Ferrer S.; Rosenthal P. J.; Derisi J. L.; Sullivan D. J.; Lazo J. S.; Roos D. S.; Riscoe M. K.; Phillips M. A.; Rathod P. K.; Van Voorhis W. C.; Avery V. M.; Guy R. K. Chemical genetics of Plasmodium falciparum. Nature 2010, 465, 311–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbyshire E. R.; Prudencio M.; Mota M. M.; Clardy J. Liver-stage malaria parasites vulnerable to diverse chemical scaffolds. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 8511–8516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister S.; Plouffe D. M.; Kuhen K. L.; Bonamy G. M.; Wu T.; Barnes S. W.; Bopp S. E.; Borboa R.; Bright A. T.; Che J.; Cohen S.; Dharia N. V.; Gagaring K.; Gettayacamin M.; Gordon P.; Groessl T.; Kato N.; Lee M. C.; McNamara C. W.; Fidock D. A.; Nagle A.; Nam T. G.; Richmond W.; Roland J.; Rottmann M.; Zhou B.; Froissard P.; Glynne R. J.; Mazier D.; Sattabongkot J.; Schultz P. G.; Tuntland T.; Walker J. R.; Zhou Y.; Chatterjee A.; Diagana T. T.; Winzeler E. A. Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science 2011, 334, 1372–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J.; Trop S.; Baer S.; Rakhmanaliev E.; Arany Z.; Dumoulin P.; Zhang H.; Romano J.; Coppens I.; Levitsky V.; Levitskaya J. Dynamics of the major histocompatibility complex class I processing and presentation pathway in the course of malaria parasite development in human hepatocytes: implications for vaccine development. PLoS One 2013, 8, e75321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattabongkot J.; Yimamnuaychoke N.; Leelaudomlipi S.; Rasameesoraj M.; Jenwithisuk R.; Coleman R. E.; Udomsangpetch R.; Cui L.; Brewer T. G. Establishment of a human hepatocyte line that supports in vitro development of the exo-erythrocytic stages of the malaria parasites Plasmodium falciparum and P. vivax. Am. J. Trop. Med. Hyg. 2006, 74, 708–715. [PubMed] [Google Scholar]
- Lim P. L.; Tan W.; Latchoumycandane C.; Mok W. C.; Khoo Y. M.; Lee H. S.; Sattabongkot J.; Beerheide W.; Lim S. G.; Tan T. M.; Boelsterli U. A. Molecular and functional characterization of drug-metabolizing enzymes and transporter expression in the novel spontaneously immortalized human hepatocyte line HC-04. Toxicol. In Vitro 2007, 21, 1390–1401. [DOI] [PubMed] [Google Scholar]
- Talman A. M.; Blagborough A. M.; Sinden R. E. A Plasmodium falciparum strain expressing GFP throughout the parasite’s life-cycle. PLoS One 2010, 5, e9156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y. L.; Krosky D. J.; Seiple L.; Stivers J. T. Uracil-directed ligand tethering: an efficient strategy for uracil DNA glycosylase (UNG) inhibitor development. J. Am. Chem. Soc. 2005, 127, 17412–17420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett T. N.; Paguio M.; Gligorijevic B.; Seudieu C.; Kosar A. D.; Davidson E.; Roepe P. Novel, rapid, and inexpensive cell-based quantification of antimalarial drug efficacy. Antimicrob. Agents Chemother. 2004, 48, 1807–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullard A. Protein-protein interaction inhibitors get into the groove. Nat. Rev. Drug Discovery 2012, 11, 173–175. [DOI] [PubMed] [Google Scholar]
- Zimmermann G.; Papke B.; Ismail S.; Vartak N.; Chandra A.; Hoffmann M.; Hahn S. A.; Triola G.; Wittinghofer A.; Bastiaens P. I.; Waldmann H. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature 2013, 497, 638–642. [DOI] [PubMed] [Google Scholar]
- Semba C. P.; Torkildsen G. L.; Lonsdale J. D.; McLaurin E. B.; Geffin J. A.; Mundorf T. K.; Kennedy K. S.; Ousler G. W. A phase 2 randomized, double-masked, placebo-controlled study of a novel integrin antagonist (SAR 1118) for the treatment of dry eye. Am. J. Ophthalmol. 2012, 153, 1050–1060. [DOI] [PubMed] [Google Scholar]
- Hain A. U. P.; Bosch J. Autophagy in Plasmodium, a multifunctional pathway?. Comput. Struct. Biotechnol. J. 2013, 8, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W. P.; Scott S. V.; Kim J.; Klionsky D. J. The itinerary of a vesicle component, Aut7p/Cvt5p, terminates in the yeast vacuole via the autophagy/Cvt pathways. J. Biol. Chem. 2000, 275, 5845–5851. [DOI] [PubMed] [Google Scholar]
- Kirisako T.; Baba M.; Ishihara N.; Miyazawa K.; Ohsumi M.; Yoshimori T.; Noda T.; Ohsumi Y. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J. Cell. Biol. 1999, 147, 435–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara C. W.; Lee M. C.; Lim C. S.; Lim S. H.; Roland J.; Nagle A.; Simon O.; Yeung B. K.; Chatterjee A. K.; McCormack S. L.; Manary M. J.; Zeeman A. M.; Dechering K. J.; Kumar T. R.; Henrich P. P.; Gagaring K.; Ibanez M.; Kato N.; Kuhen K. L.; Fischli C.; Rottmann M.; Plouffe D. M.; Bursulaya B.; Meister S.; Rameh L.; Trappe J.; Haasen D.; Timmerman M.; Sauerwein R. W.; Suwanarusk R.; Russell B.; Renia L.; Nosten F.; Tully D. C.; Kocken C. H.; Glynne R. J.; Bodenreider C.; Fidock D. A.; Diagana T. T.; Winzeler E. A. Targeting Plasmodium PI(4)K to eliminate malaria. Nature 2013, 504, 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May P. C.; Dean R. A.; Lowe S. L.; Martenyi F.; Sheehan S. M.; Boggs L. N.; Monk S. A.; Mathes B. M.; Mergott D. J.; Watson B. M.; Stout S. L.; Timm D. E.; Smith Labell E.; Gonzales C. R.; Nakano M.; Jhee S. S.; Yen M.; Ereshefsky L.; Lindstrom T. D.; Calligaro D. O.; Cocke P. J.; Greg Hall D.; Friedrich S.; Citron M.; Audia J. E. Robust central reduction of amyloid-β in humans with an orally available, non-peptidic β-secretase inhibitor. J. Neurosci. 2011, 31, 16507–16516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend K. N.; Hughson L. R.; Schlie K.; Poon V. I.; Westerback A.; Lum J. J. Autophagy inhibition in cancer therapy: metabolic considerations for antitumor immunity. Immunol. Rev. 2012, 249, 176–194. [DOI] [PubMed] [Google Scholar]
- Ho K. H.; Chang H. E.; Huang W. P. Mutation at the cargo-receptor binding site of Atg8 also affects its general autophagy regulation function. Autophagy 2009, 5, 461–471. [DOI] [PubMed] [Google Scholar]
- Noda N. N.; Ohsumi Y.; Inagaki F. Atg8-family interacting motif crucial for selective autophagy. FEBS Lett. 2010, 584, 1379–1385. [DOI] [PubMed] [Google Scholar]
- Tung Y. T.; Hsu W. M.; Lee H.; Huang W. P.; Liao Y. F. The evolutionarily conserved interaction between LC3 and p62 selectively mediates autophagy-dependent degradation of mutant huntingtin. Cell. Mol. Neurobiol. 2010, 30, 795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thielmann Y.; Weiergräber O. H.; Mohrlüder J.; Willbold D. Structural framework of the GABARAP-calreticulin interface--implications for substrate binding to endoplasmic reticulum chaperones. FEBS J. 2009, 276, 1140–1152. [DOI] [PubMed] [Google Scholar]
- Trager W.; Jensen J. B. Human malaria parasites in continuous culture. Science 1976, 193, 673–675. [DOI] [PubMed] [Google Scholar]
- Gaviria D.; Paguio M. F.; Turnbull L. B.; Tan A.; Siriwardana A.; Ghosh D.; Ferdig M. T.; Sinai A. P.; Roepe P. D. A Process Similar to Autophagy Is Associated with Cytocidal Chloroquine Resistance in Plasmodium falciparum. PLoS One 2013, 8, e79059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramoff M. D.; Magalhaes P. J.; Ram S. J. Image Processing with ImageJ. Biophotonics Int. 2004, 11, 36–42. [Google Scholar]
- Hawkins P. C.; Skillman A. G.; Warren G. L.; Ellingson B. A.; Stahl M. T. Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins P. C.; Nicholls A. Conformer generation with OMEGA: learning from the data set and the analysis of failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [DOI] [PubMed] [Google Scholar]
- Roy A.; Kucukural A.; Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurrecoechea C.; Brestelli J.; Brunk B. P.; Dommer J.; Fischer S.; Gajria B.; Gao X.; Gingle A.; Grant G.; Harb O. S.; Heiges M.; Innamorato F.; Iodice J.; Kissinger J. C.; Kraemer E.; Li W.; Miller J. A.; Nayak V.; Pennington C.; Pinney D. F.; Roos D. S.; Ross C.; Stoeckert C. J. Jr.; Treatman C.; Wang H. PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res. 2009, 37, D539–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins P. C.; Skillman A. G.; Nicholls A. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 2007, 50, 74–82. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.