

Abstract
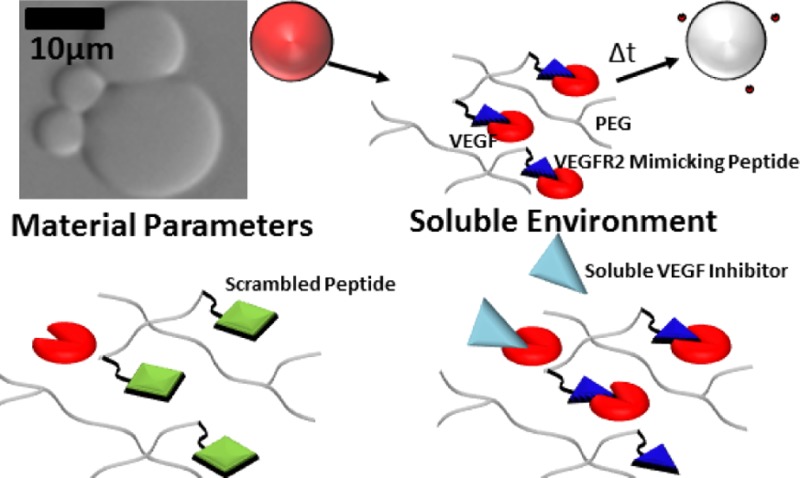
Vascular endothelial growth factor (VEGF) activity is highly regulated via sequestering within the ECM and cell-demanded proteolysis to release the sequestered VEGF. Numerous studies have demonstrated that VEGF activity mediates cellular events leading to angiogenesis and capillary formation in vivo. This has motivated the study of biomaterials to sustain VEGF release, and in many cases, the materials are inspired by the structure and function of the native ECM. However, there remains a need for materials that can bind to VEGF with high specificity, as the in vivo environment is rich in a variety of growth factors (GFs) and GF-binding moieties. Here we describe a strategy to control VEGF release using hydrogel microspheres with tethered peptides derived from VEGF receptor 2 (VEGFR2). Using biomaterials covalently modified with varying concentrations of two distinct VEGFR2-derived peptides with varying serum stability, we analyzed both biomaterial and environmental variables that influence VEGF release and activity. The presence of tethered VEGF-binding peptides (VBPs) resulted in significantly extended VEGF release relative to control conditions, and the resulting released VEGF significantly increased the expansion of human umbilical vein endothelial cells in culture. VEGF release rates were also strongly influenced by the concentration of serum. The presence of Feline McDonough Sarcoma-like tyrosine kinase 1 (sFlt-1), a serum-borne receptor fragment derived from VEGF receptor 1, increased VEGF release rates, although sFlt-1 was not sufficient to recapitulate the release profile of VEGF in serum. Further, the influence of serum on VEGF release was not due to protease activity or nonspecific VEGF interactions in the presence of serum-borne heparin. VEGF release kinetics correlated well with a generalizable mathematical model describing affinity-mediated release of VEGF from hydrogel microspheres in defined conditions. Modeling results suggest a potential mechanism whereby competition between VEGF and multiple VEGF-binding serum proteins including sFlt-1, soluble kinase insert domain receptor (sKDR), and α2-macroglobulin (α2-M) likely influenced VEGF release from microspheres. The materials and mathematical model described in this approach may be useful in a range of applications in which sustained, biologically active GF release of a specific GF is desirable.
Introduction
Growth factor regulation is a key function of the extracellular matrix (ECM) and is particularly important for proper blood vessel growth and maturation during wound healing.1 Blood vessel sprouting associated with angiogenesis is required for effective healing,2 and it is highly dependent on the ECM to regulate growth factor (GF) activity via sequestering, spatial patterning, and cell-demanded release.3 One particularly well-characterized example involves regulation of vascular endothelial growth factor (VEGF) activity. VEGF is an important factor during angiogenesis,4,5 and previous investigations have demonstrated blood vessel sprouting within a limited VEGF concentration range in vivo.6 In the native ECM, VEGF activity can be regulated via binding to ECM components, such as heparan sulfate proteoglycans (HSPGs)7,8 and collagens.9,10 In addition, cell-demanded proteolytic degradation (via matrix metalloproteinases) of ECM components11 can increase unbound VEGF and consequently increase local VEGF activity.12 The need to maintain VEGF activity in a particular concentration range during angiogenesis has motivated the use of therapeutic interventions to regulate VEGF activity when natural regulation is dysfunctional, such as during diabetic wound healing13 and tumor growth.14,15
Various synthetic biomaterials have been designed to include ECM-mimicking moieties and thereby control GF release. Biomaterials functionalized with ECM-mimicking moieties such as heparin,16−19 fibrin,20,21 or collagen9,22 have been used to deliver pro-angiogenic GFs in vitro and in vivo. However, these moieties bind GFs promiscuously,21,23−25 and are therefore not ideal for regulating the activity of a specific GF such as VEGF. To achieve more specific VEGF regulation with a chemically defined biomaterial, we have recently developed biomaterials functionalized with VEGF receptor-mimicking peptides that specifically bind to VEGF. These biomaterials have been shown to regulate VEGF availability and VEGF-dependent endothelial cell behavior in vitro,26−28 though there has been only limited characterization of the role of complex biological environments (e.g., serum) on GF binding and release. In view of the number of factors in biological environments that may influence growth factor binding and release (e.g., albumins, globulins, antibodies, GFs, GF receptor fragments),29,30 there is a need to understand the effect of the soluble environment on GF binding and release.
Here we focused on understanding the role of complex biological environments on VEGF release from hydrogels functionalized with specific VEGF-binding peptides (VBPs). This information is of critical importance to cell culture and in vivo applications involving these and similar materials, as the soluble environment is a latent variable in many biomaterial studies. We specifically examined VEGF release from poly(ethylene glycol) (PEG) microspheres functionalized with peptides derived from VEGF receptor type 2 (VEGFR2), which are known to bind specifically to VEGF.31 We explored two distinct peptides: (1) a wild-type VEGF-binding peptide derived from the VEGFR2 sequence,32 termed “VBPWT”; and (2) a D-amino acid substituted VEGF-binding peptide derivative termed “VBP”.31 The VBP included four D-amino acids that increased stability against protease-mediated degradation.31 Further, we applied a kinetic mathematical model to understand how microsphere characteristics, including peptide identity and content, and the protein composition of the soluble microenvironment influenced GF release from microspheres. Kinetic equations were established based on previous models of controlled drug release from hydrogels33,34 with additional terms describing affinity-mediated VEGF binding and release. Finally, we probed the ability of released VEGF to modulate VEGF-dependent human umbilical vein endothelial cell (HUVEC) expansion in serum-containing cell culture solutions. Results demonstrated that both biomaterial characteristics and the identity of proteins in the microenvironment influence VEGF release. Specifically, biomaterial characteristics such as peptide identity and concentration influenced release rates. In the soluble microenvironment, soluble Feline McDonough Sarcoma-like tyrosine kinase 1 (sFlt-1), a soluble receptor fragment derived from VEGF receptor 1, increased VEGF release rates, though neither heparin nor proteases influenced VEGF release. This prompted modeling analysis that demonstrated VEGF-binding proteins in serum, including sFlt-1, soluble kinase insert domain receptor (sKDR), and α2-macroglobulin likely influenced VEGF release rates.
Materials and Methods
Peptide Synthesis and Characterization
Two peptides identified from a previous study, VEGF binding peptide (VBP) sequence CEFdAdYdLdIDFNWEYPASK and wild type (VBPWT) sequence CELNVGIDFNWEYPASK,31,32 and a peptide with the same amino acids but in a scrambled sequence (Scramble), CDAdPYNFdEFAWEYdVISLdK were synthesized using standard Fmoc solid phase peptide synthesis. The peptides were amidated at the carboxy terminus by synthesizing on MBHA Rink Amide resin (EMD Novabiochem) as previously described.28 Peptide identity was determined by matrix-assisted laser desorption/ioniziation time-of-flight mass spectrometry (Bruker). Peptide purity of >85% was determined with C18 reverse-phase high performance liquid chromatography (Shimadzu, Supelco silica column). The dry weight percent (wt %) peptide content was determined by measuring free thiol groups with Ellman’s Assay (Thermo Fisher).
PEG-Norbornene Synthesis
Four-arm poly(ethylene-glycol) (PEG; Mn = 20,000; Jenkem) was functionalized with norbornene moieties at each arm in order to utilize thiolene photopolymerization as introduced by Anseth and co-workers35 and described previously26−28 to generate PEG-norbornene (PEG-NB). Briefly, 4-Arm PEG, terminated at each arm with hydroxyl functional group, was reacted under constant stirring in a flask, which was purged with argon during dissolution and reaction, with 10 molar equivalents (with respect to the number of PEG arms) of 5-norbornene-2-carboxylic acid (Sigma-Aldrich) in dichloromethane (Fisher), five molar equivalents of N,N′-dicyclohexylcarbodiimide (Sigma), half molar equivalent of 4-dimethylaminopyridine (Sigma-Aldrich), and five molar equivalents of pyridine (Sigma-Aldrich). Derivatization was determined as >90% using 1H nuclear magnetic resonance by comparing the chemical shift expected for ether bonds associated with PEG (∼3.4 ppm) with the chemical shift expected for the norbornene group (∼5.8–6.2 ppm).
Microsphere Synthesis
PEG microspheres were synthesized using a water-in-water emulsion,36 as described previously for generating microspheres of a controlled size.26−28 Microspheres containing covalently immobilized VEGF-binding peptides (VBP, VBPWT, or Scramble) at various concentrations and microspheres containing no peptide (Blank) were synthesized using an aqueous emulsion of two phases, a PEG-rich discontinuous phase and a dextran-rich continuous phase. In the PEG-rich phase, PEG-NB was mixed with a half molar equivalent of PEG3400 dithiol (Laysan Bio) along with a peptide solution for VBP, VBPWT, and Scramble conditions and Irgacure 2959 (Ciba) photoinitiator at a final concentration of 0.05 wt %. The peptide solutions were prepared at 3.1%, 1.6%, 0.8%, 0.4%, or 0% concentrations, which refer to the percentile molar equivalent with respect to norbornene groups. These peptide solutions were mixed into the PEG-rich phase, which contained 10 wt % of PEG-NB and PEG3400 Dithiol. This PEG-rich phase was diluted 6-fold into a nitrogen-purged dextran-rich phase composed of 40 wt % dextran T40 (Mn = 40 000; Alfa Aesar) in pH 8 buffer containing 0.22 M KCl and 10 mM NaPO4. To form the microspheres, the PEG-dextran mixture was vortexed for 1 min and photopolymerized under ultraviolet light at an intensity of 1.1 J cm–2. Unreacted microsphere components and dextran were removed by diluting the microspheres 25 fold in deionized (DI) water, mixing, and centrifuging at 1600g. Microspheres were washed 3-fold as above and were lyophilized for storage until use. Peptide concentration was determined using a micro bicinchoninic acid assay (Micro BCA Assay; Thermo Scientific). Peptide concentration is hereafter reported as the percent molar equivalent of PEG-NB arms occupied by peptide in the coupling reaction.
Assays for VEGF Release from Biomimetic Microspheres
For all studies, microspheres were incubated in 10 ng mL–1 VEGF, as this concentration has previously been shown to result in maximal endothelial cell proliferation in vitro8 and has been observed during in vivo wound healing.37 A schematic demonstrating the VEGF loading, washing, and release is shown in Figure 1A. Before incubation, 1.5 mL microcentrifuge tubes were blocked overnight with a 0.1 wt % solution of bovine serum albumin (BSA; Fisher Scientific) in phosphate-buffered saline (PBS; Fisher Scientific) at pH 7.4 followed by two washing steps in deionized (DI) water and subsequent lyophilization for storage. Blank microspheres were studied throughout experiments to control for the influence of biomaterial characteristics on VEGF release. Lyophilized microspheres were loaded with VEGF by incubating at 37 °C and 95% relative humidity for 4 h at 1 mg mL–1 microspheres in 9.9 ng mL–1 recombinant human Vascular Endothelial Growth Factor–165 (rhVEGF165 hereafter referred to as VEGF; R&D Systems) and 0.1 ng mL–1 of Iodine-125 tagged rhVEGF165 (hereafter referred to as [125I]VEGF; PerkinElmer). Previous studies demonstrated rapid VEGF sequestering in buffered solutions within the first 30 min.26 Microspheres were therefore incubated in VEGF-containing solution for 4 h as previously described26−28 to allow for equilibrium sequestering in all solutions examined. Radiolabeled VEGF was used for release studies for the required sensitivity of detecting VEGF levels below 1 pg mL–1, a range typically not detectable via enzyme-linked immunosorbent assays utilizing fluorescent antibodies. Microspheres were loaded with VEGF in a buffered serum-containing or protein-containing solution, termed release solution, whose composition is defined in Figures 1–6 and in the section “Analysis of Serum-Dependence of VEGF Release from Microspheres”. After incubation in VEGF, microspheres were washed three times by centrifuging at 10 800g, removing supernatant, adding 1 mL of release solution, and mixing. After washing, microspheres were incubated in release solution, and at various time points, supernatants were collected on the days denoted in Figures 1–6, and fresh release solution was added. Supernatant counts per minute were measured using a γ-counter (PerkinElmer, Cobra II Auto-Gamma), and then directly correlated to the VEGF concentration released at each time point via a [125I]VEGF standard curve.
Figure 1.

Influence of peptide concentration on VEGF release from microspheres. (A) Schematic of VEGF release from PEG microspheres in albumin-only (Serum-Free) solution. Top panel: Blank (no peptide) PEG microspheres imaged with phase contrast, under 20X objective with an Olympus IX51 inverted epifluorescence microscope. Schematic shows VEGF-bound state of a microsphere (in red), followed by a change in time and subsequent release of VEGF from the PEG microsphere. (B–E) Fractional cumulative VEGF release from PEG microspheres that were incubated in 9.9 ng mL–1 VEGF, 0.1 ng mL–1 [125I]VEGF in 0.1 wt % BSA in PBS at various peptide concentrations in % of norbornene groups functionalized with peptide. Graphs represent PEG microspheres containing 0.4% peptide (B), 0.8% peptide (C), 1.6% peptide (D) and 3.2% peptide (E). Fractional release was calculated by dividing the release at each time point by the cumulative amount of VEGF released at the final time point.
Figure 6.

Influence of serum-borne heparin and sFlt-1 on VEGF release. (A) Time for 50% release, t50, calculated for 1.6% VBP and VBPWT microspheres releasing bound VEGF into medium containing albumin-only solution with or without supplemented heparin. Briefly, microspheres were incubated in an albumin-only solution containing VEGF with or without heparin. Subsequent release was measured in albumin-only solution with or without heparin. t50 values were calculated as described in the Materials and Methods section. Data is shown for VBP (black bars) and VBPWT microspheres (white bars) for both treatment groups described on the x-axis. Scramble and Blank microspheres were omitted because data could not be modeled using logarithmic regression analysis. Statistical significance was only observed between VBP and VBPWT in albumin-only solution at p-value <0.05 and denoted by asterisk (*). No significant differences were observed between t50 values for VEGF release into albumin-only medium with or without heparin. (B) Normalized t50 values for VEGF release from VBP and VBPWT microspheres in albumin-only solution with 1 or 10 ng mL–1 sFlt-1. Values were normalized to t50 in 1 ng mL–1 sFlt-1 for comparison. Statistical significance was observed for VEGF release from VBP microspheres (p-value <0.05), denoted by an asterisk (*).
Analysis of Serum-Dependence of VEGF Release from Microspheres
In order to elucidate the serum-dependence of VEGF sequestering, VEGF release was measured in solutions containing different protein identity and content and serum concentrations. Experiments varying microsphere peptide concentrations were performed in 0.1 wt % BSA in PBS at pH 7.4. Experiments varying serum concentration were performed in fetal bovine serum (serum; Gibco) at 25 vol %, 10 vol %, and 2 vol % in pH 7.4 PBS. Above 25 vol % serum, microspheres were unable to be segregated by centrifugation, most likely due to viscosity effects at high protein content; microspheres in 5 wt % BSA in PBS (50 mg mL–1, similar to total protein concentration of pure serum29) were similarly unable to be segregated following centrifugation. In order to recapitulate the total protein concentration in 25 vol % serum, a BSA solution was prepared at 1.25 wt % in pH 7.4 PBS (denoted Albumin-only solution). Experiments determining the protease-dependence of VEGF release were performed using 1.6% and 0.4% peptide microspheres (representing high and low peptide concentration, respectively) in the highest serum concentration tested, 25 vol % serum in pH 7.4 PBS, with 1X concentration of HALT Protease Inhibitor Cocktail (Thermo). Experiments specifically examining the influence of heparin on VEGF release were carried out using 1.6% peptide microspheres incubated in a buffered solution containing 0.1 wt % BSA in pH 7.4 PBS with a physiologic level of supplemented heparin (unfractionated porcine heparin; Sigma), 10 μg mL–1 (refs (38 and 39)). Experiments specifically examining the influence of sFlt-1 on VEGF release were carried out using 1.6% peptide microspheres incubated in a buffered solution containing 0.1 wt % BSA in pH 7.4 PBS with physiologic concentrations of supplemented human sFlt-1 (Sino Biological), 1 and 10 ng mL–1 (refs (40−42)).
Mathematical Model for Determining Effect of Affinity and Diffusion on VEGF Release from Microspheres
In order to understand the influence of intrinsic and extrinsic variables on VEGF sequestering and release, we developed a mathematical model incorporating microsphere variables including the concentration and identity of peptide. The model describes the kinetics of the interaction between VEGF and peptide (VBP, VBPWT, Scramble), resulting in sequestered VEGF (VEGF–peptide; eq 1).
| 1 |
A mass balance on all possible species resulted in one partial differential equation (PDE) and two ordinary differential equations (ODE), written as the change in concentration Cx over time, t, where ‘x’ represents all three species in our model (eqs 1S–3S)
The solution to the coupled PDE–ODE system utilized spatial discretization of one-dimensional parabolic equations43 via the MATLAB 2012 PDEPE function. The simultaneous solution of three nonlinear equations (NLE) describing mass balances on all observed species at equilibrium (eqs 4S–6S) resulted in initial value conditions for the model via Levenberg–Marquardt algorithm in MATLAB. The solution to the PDE–ODE system was obtained with initial conditions (eq 7S) and boundary conditions (eqs 8S–9S), which implicated a Dirichlet condition at the outermost boundary to assume a perfect “sink” for VEGF and a Neumann condition at the innermost boundary to describe symmetry at the source. At each time point, released VEGF was removed. Considering that the volume of microspheres at 1 mg mL–1 is at least 20-fold lower than the solution (data not shown), and that the only source of VEGF during release studies was the microspheres, it is reasonable to assume that the concentration of VEGF in the microspheres was at least 20-fold higher than in the release solution at each time point. Thus, the bulk solution would constitute a “perfect sink” for VEGF, and released VEGF in the bulk solution would not be expected to influence equilibrium VEGF-peptide interactions during release. Assuming symmetry about the θ and φ axes, the flux of VEGF out of a given microsphere (eq 10S) was normalized to the final time point and graphed with experimental data for analysis.
Equilibrium dissociation data for the VEGF–VBP interaction were determined via a Klotz plot analysis of published VBP data,31 and the association kinetic rate constant for VEGF–VBP was assumed to take the value of a similar study demonstrating association of a 100-amino acid portion of the extracellular domain of VEGFR2 to VEGF.44 In addition, our previous analysis of the affinity of VEGF-binding affinity to Scramble, VBP, and VBPWT microspheres (data not shown) supported an approximately 10-fold increase in the dissociation constant for the Scramble–VEGF interaction versus the VBP–VEGF interaction. The equilibrium dissociation parameter for Scramble peptide was therefore ascribed a value 10-fold higher than the same for VBP. A scaling factor for the VEGF diffusion coefficient, DVEGF, was required to fit experimental release results to model predictions of VEGF release from Scramble and VBP microspheres, and we posited that the scaling factor was proportional to peptide–VEGF rebinding during release. An effective diffusion coefficient, DVEGF,eff, was calculated to take into account the probability of VEGF–peptide rebinding during release. Protein–ligand rebinding has been well-established for diffusible species interacting with an immobilized binding partner on a surface.45 The probability of VEGF–peptide rebinding was calculated based on known or assumed values for the peptide–VEGF affinity, VEGF diffusion coefficient, and both VEGF and peptide concentrations.45 This rebinding probability was used to scale the value of DVEGF, an established protein diffusion coefficient describing diffusion through PEG hydrogels,46,47 to DVEGF,eff, an effective VEGF diffusion coefficient describing diffusion through peptide-containing PEG hydrogels.
Modeling the Influence of Protein Identity and Content on VEGF Release
Similarly to the aforementioned method, we developed a second mathematical model to understand the impact of solution variables, including the identity and concentration of specific proteins, on VEGF sequestering and release from microspheres. We posited that competition between serum-borne VEGF-binding proteins including sFlt-1 would interfere with VEGF sequestering and release in serum, and therefore we proposed a model (eq 2) describing the kinetics of the interaction between VEGF and VEGF-binding serum proteins including sFlt-1, sKDR, and α2-M. The competition model is based on the competitive interaction between Competitori
 |
where ‘i’ is defined as sFlt-1, sKDR, or α2-M, and the parameters ki,r and ki,f are defined as the dissociation and association rate constants respectively for the interaction between VEGF and Competitori (Table 1). The analysis was performed as previously described with revised partial and ordinary differential eqs (eqs 2S–3S and 11S–13S), nonlinear eqs (eqs 4S–6S and 14S–16S) for deriving initial conditions, and boundary conditions (eqs 8S–10S and 17S). The solution of VEGF flux (eq 10S) was normalized as previously described and plotted versus time.
Table 1. Constants Used in Numerical Approximation of the VEGF Release Model.
| constanta | valueb | descriptionc |
|---|---|---|
| KD,VBP-VEGF | 79 nM | affinity of VBP–VEGF interaction, derived from31 |
| kf, VBP,Scramble-VEGF | 4.2 × 10–6 nM–1 d–1 | association rate constant,VEGF–VEGFR2(ED3)44 |
| KD,Scramble-VEGF | 10*KD,VBP-VEGF | assumed affinity of Scramble–VEGF interaction |
| kf,sFlt-1-VEGF | 3.456 × 102 nM–1 d–1 | association rate constant, sFlt-1–VEGF77 |
| KD,sFlt-1-VEGF | 1 × 10–3 nM | affinity of sFlt-1–VEGF interaction78 |
| kf,sKDR-VEGF | 4.51 × 102 nM–1 d–1 | association rate constant, sKDR–VEGF79 |
| KD,sKDR-VEGF | nM | affinity of sKDR–VEGF interaction52,67 |
| kf,α2-M-VEGF | 2.16 × 10–2 nM–1 d–1 | association rate constant, α2-M–VEGF80 |
| KD,α2-M-VEGF | 420 nM | affinity of α2-M–VEGF interaction65 |
| DVEGF | 2.4 × 105 μm2/d | diffusion of ∼40 kDa protein in PEG hydrogel51 |
| DVEGF, eff | DP*(p2 – 2p + 1) μm2/d | effective VEGF diffusion coefficient, p = rebind prob.45 |
| Di-VEGF | 1.6 × 105 μm2/d | where ‘i’ is sFlt-1, sKDR, or α2-M |
| diffusion of ∼100 kDa protein in PEG hydrogel51 |
Representative constants used in VEGF release model equations.
Value of constants in first column with associated units.
Description of derivation of constant values with citations provided.
Assays of VEGF Biological Activity
The biological activity of released VEGF was determined by measuring endothelial cell expansion in culture (Figure 8A). HUVECs (Lonza) were cultured as described previously.26,28 Cells were expanded in “growth medium”, consisting of EGM2 SingleQuots with 2 vol % serum (Lonza), medium 199 (M199; CellGro) with Earle’s salts and l-glutamine, 2.2 g L–1 sodium bicarbonate (Acros), and a penicillin/streptomycin solution (Hyclone) giving a final concentration of 100 units mL–1 penicillin and 100 μg mL–1 streptomycin. For experiments, HUVECs were used between passages 2 and 4 (corresponding to population doublings between 5 and 10). On Day 0 of experiments, HUVECs were dissociated with 0.05 wt % buffered Trypsin (Lonza), diluted in 10 vol % serum in basal medium (M199 with Earle’s salts, l-glutamine, sodium bicarbonate, P/S) and centrifuged at 200g for 5 min. Cells were counted on a hemacytometer and suspended at 40 000 cells mL–1 in basal medium with 2 vol % serum, hereafter referred to as “serum starvation medium”. Assay plates were coated with 0.1 wt % gelatin (Sigma) in DI water for 1 h prior to experiments. Cells were added at 100 μL per well in serum starvation medium into a 96 well plate and incubated overnight at 37 °C, 95% relative humidity, and 5% CO2. This serum-starvation step was employed to synchronize the HUVECs in the G0 phase of the cell cycle before beginning cell expansion experiments.48,49
Figure 8.

HUVEC number upon VEGF release from Blank and 1.6% VBP, VBPWT, and Scramble microspheres. (A) Schematic demonstrating the difference between cumulative VEGF release from VBP/VBPWT microspheres versus Scramble and Blank controls and the effect on HUVEC number in culture. (B) Graphical representation of the number of HUVECs in 4 × 4 images at10× magnification in various conditions. The concentration of VEGF in the initial microsphere incubation are listed on the x-axis, and the bars represent the different microsphere conditions (legend in B). Graphs represent HUVEC number upon VEGF release in different serum concentrations - microspheres and cells were cultured in 2 vol % serum (B), 10 vol % serum (C), or 25 vol % serum (D). Significant differences between conditions in brackets are shown with an asterisk denoting p-value < 0.05.
On Day 1 of VEGF bioactivity experiments, microspheres were prepared by incubating Blank and 1.6% VBP, VBPWT, and Scramble microspheres at 1 mg mL–1 in 10 ng mL–1 VEGF and 0.1 wt % BSA in pH 7.4 PBS for 4 h and subsequently washed with 2, 10, or 25 vol % serum in basal medium as appropriate for each condition. Cell culture media was diluted 1:1 into basal medium with 4, 20, or 50 vol % serum also containing 2 mg mL–1 microspheres (with 1.6% peptide or 0% peptide in the case of Blank) for a total of 2, 10, and 25 vol % serum respectively and 1 mg mL–1 microspheres in culture. Following 2 days of culture in serum- and microsphere-supplemented media, cells were fixed using 10% buffered formalin, washed in PBS, and stained with 1 μg mL–1 DAPI (Invitrogen) in PBS. Imaging was performed using a Nikon Ti Eclipse inverted epifluorescence microscope equipped with 10X objective and filter cubes for DAPI, FITC, and TexasRed channels (Nikon). 4 × 4 images of each well were taken and stitched together using NIS Elements v3.2 software. The images were thresholded uniformly across all wells and counted using built-in object counting in NIS Elements software.
Statistical Analysis
For cumulative VEGF release comparisons, error was propagated using the additive property of variance at each time point. A Student’s t test at p-value <0.05 was used to compare the cumulative VEGF released at the final time point. For all VEGF release curves, error bars represent one propagated standard deviation about the mean cumulative release at each time point. For all fractional VEGF release data, statistical analyses were performed on logarithmic regressions of each release sample, normalized to cumulative VEGF released. The cumulative VEGF release was divided by the cumulative release at the final time point to arrive at a normalized cumulative VEGF release for each condition and time point. Data for each sample fit to the general form y = a*ln(x) + b, and the regression coefficients for each sample were used to calculate an average t50 value, which we defined as the time necessary to reach 50% normalized release for all conditions. This analysis was performed on all microspheres except Blank and Scramble microspheres in serum-containing medium because the burst release in these conditions could not be modeled using logarithmic regression. Data is presented as t50 ± standard deviation. Comparisons of t50 values were performed using the Student’s t test for each condition tested at p-value <0.05. Release experiments were performed at n = 3. For all bar graphs, error bars represent one standard deviation about the mean.
For model comparisons, each model prediction for VEGF flux was normalized and compared to normalized VEGF release data for Blank, 1.6% Scramble, and 1.6% VBP microspheres. The goodness-of-fit of the model was determined using the coefficient of determination, R2, where an R2 value of 0.9 or higher was defined as a good model fit. Finally, conditions were performed at n = 5 for VEGF bioactivity experiments, consisting of five independent wells per condition, where each sample represented one well wherein 4 × 4 stitched images were acquired for quantifying DAPI-stained cells. Averaged cell counts for each condition were compared using Student’s t test, where significance was determined at p-value <0.05.
Results
VEGF Release from VEGF-Binding Microspheres
Microspheres with VEGF binding peptides exhibited sustained VEGF release, with release kinetics dependent on peptide concentration. Both VBP and VBPWT microspheres exhibited significantly sustained release (Figure 1B–E) and higher t50 values (t50 = average time for 50% VEGF release) when compared to Scramble and Blank microspheres at all peptide concentrations tested (Figure 2). Thus, the VEGF binding affinity of VBP and VBPWT microspheres extended the time frame of VEGF release, as expected. The peptide concentration in the microspheres did not influence the total amount of VEGF released from VBP and VBPWT microspheres (Figure 1S,A–D). This phenomenon may be attributed to the large excess of peptide in the microspheres relative to VEGF that would not be expected to increase VEGF-binding capacity or the cumulative VEGF release in the range of peptide concentrations tested. However, the t50 values increased with increasing peptide concentration. Specifically, increasing peptide concentration increased the t50 values of VEGF released from 0.8% and 1.6% relative to 0.4% VBP microspheres, from 1.6% and 3.2% relative to 0.8% VBPWT microspheres, and from 1.6% and 3.2% relative to 0.8% Scramble microspheres (Figure 2). We also observed increased t50 values for VEGF release from Scramble microspheres relative to Blank microspheres at 1.6 and 3.2% peptide (Figure 2). Additionally, cumulative VEGF release amounts from Scramble microspheres increased significantly at 3.2% peptide concentration relative to Scramble microspheres at 0.4–1.6% concentrations (Figure 1S D), which may be attributed to nonspecific VEGF binding to the Scrambled peptides at 3.2% peptide concentration. Therefore, in subsequent analysis we focused on 0.4 (“low”) and 1.6% (“high”) peptide concentrations for specific VEGF binding and release, and we did not explore 3.2% peptide concentrations further.
Figure 2.

Time for 50% release presented for 0.4%, 0.8%, 1.6%, and 3.2% microspheres and Blank microspheres in albumin-only (Serum-Free) solution. Significance is reported by asterisks comparing each microsphere condition to the same condition at 0.4% (*) and at 0.8% (#) peptide concentrations (p < 0.05). Statistical significance comparing different peptides at a given peptide concentration is denoted by % (p < 0.05). In all cases, VBP and VBPWT microspheres exhibited significantly higher t50 values than Scramble at each peptide concentration and Blank microspheres.
The rate of VEGF release was strongly dependent on the presence and concentration of serum in the release medium. Increasing serum concentration significantly increased VEGF release rates, as release profiles were shifted toward burst release (Figure 3E–H), and t50 values were significantly decreased (Figure 4B) for the 1.6% VBP and VBPWT microsphere conditions. No significant differences in VEGF release profile were observed from 0.4% VBP and VBPWT microspheres relative to Scramble and Blank controls in any of the serum concentrations tested (Figure 3A–D). However, the t50 values for VEGF release from 0.4% VBP and VBPWT microspheres were significantly higher in 2 vol % serum and serum-free medium relative to 25 vol % serum (Figure 4A). Taken together, these results suggest that increasing serum concentration increased the rate of VEGF release from low peptide concentration (0.4%) and high peptide concentration (1.6%) VBP and VBPwt microspheres. Since the total soluble protein concentration in the serum-free (albumin-only) solution was equivalent to the total protein concentration in 25 vol % serum,29,30 we could conclude that increased VEGF release rates in serum were not caused by higher total protein concentration in solution, but instead were likely influenced by the identity and concentration of particular serum molecules other than albumin.
Figure 3.

(A–H) Fractional cumulative release of VEGF measured from binding in 9.9 ng mL–1 VEGF, 0.1 ng mL–1 [125I]VEGF in various loading solutions containing albumin-only or serum. Release of bound VEGF from 0.4% microspheres (A–D) and 1.6% microspheres (E–H) was measured in albumin-only, 1.25 wt % BSA in PBS solution (A,E), 2 vol % serum in PBS (B,F), 10 vol % serum in PBS (C,G), and 25 vol % serum in PBS (D,H).
Figure 4.

Time for 50% release, t50, calculated for microspheres releasing VEGF into medium containing 25 vol % serum, 10 vol % serum, 2 vol % serum, and albumin-only (serum-free) solution. (A) t50 values were calculated for 0.4% peptide microspheres. (B) t50 values were calculated for 1.6% peptide microspheres. Data is shown for VBP (black diamonds) and VBPWT microspheres (gray squares). Scramble and Blank microspheres were omitted because data could not be adequately modeled using logarithmic regression analysis. Statistical significance is reported at p-value <0.05 compared to VBP microspheres in 25 vol % serum (*) and 10 vol % serum (**),compared to VBPWT microspheres in 25 vol % serum (&) and 10 vol % serum (%), and between VBP and VBPWT where indicated (#).
To understand the influence of serum proteases on VEGF release, we measured VEGF release in the presence of a protease inhibitor cocktail. Interestingly, protease inhibition did not significantly influence VEGF release rates in serum. Specifically, VEGF-bound microspheres incubated in the presence of a protease inhibitor cocktail exhibited no differences in VEGF release profiles compared to no protease inhibition at both low and high peptide concentrations (Figure 4S). Protease inhibition also did not significantly influence t50 values for VEGF release from VBP or VBPWT microspheres (Figure 5). The protease inhibitor cocktail can inhibit serine proteases, amino-peptidases, cysteine proteases, metalloproteases, and aspartic acid proteases, so our results indicate that protease activity related to these protease families did not significantly influence the amount or rate of VEGF release.
Figure 5.

Time for 50% release, t50, calculated for microspheres supplemented with 9.9 ng mL–1 VEGF, 0.1 ng mL–1 [125I]VEGF in 25 vol % serum with or without protease inhibitor (PI). Release of bound VEGF was measured in 25 vol % serum with or without PI. For each sample, logarithmic regression analysis was calculated as described in Materials and Methods, and t50 values were calculated for 0.4% and 1.6% VBP and VBPWT microspheres. Scramble and Blank microspheres were omitted from this analysis because the data could not adequately be modeled using logarithmic regression analysis. No significant differences were observed between conditions (α = 0.05).
To further understand the influence of particular serum components on VEGF release, we examined the effects of soluble heparin and soluble sFlt-1 on cumulative VEGF release and release rate. We hypothesized that heparin and sFlt-1 in serum could interfere with VEGF during release and increase release rates by “competing” with VEGF–peptide interactions. The release profile for VEGF release from VBP and VBPWT microspheres were not influenced by the presence of a physiologic concentration of heparin38,39 in the release medium (Figure 5S). Additionally, heparin did not influence the t50 values for VEGF release from VBP and VBPWT microspheres (Figure 6A). In contrast, cumulative release from Scramble and Blank microspheres was significantly reduced in heparin-containing solution when compared to the serum-free (albumin-only) solution (Figure 5S), which reflects the lower amount of bound VEGF in these conditions. Importantly, the t50 values for VEGF release from VBP microspheres was significantly reduced in the presence of a physiologic concentration of sFlt-140−42 (Figure 6B). Taken together, these data indicate that serum significantly influenced VEGF release from VBP and VBPWT microspheres, although this effect was independent of serum-borne heparin (Figure 6A) or proteases (Figure 5). Results suggest that competition between serum-borne components including sFlt-1 (Figure 6B) may have accelerated VEGF release rates in the presence of serum.
Mathematical Model of VEGF Release
We established a mathematical model to determine the relative influence of peptide-VEGF affinity, VEGF diffusion, and serum-borne protein-VEGF interactions on VEGF binding and release. We defined a kinetic rate equation to describe the VEGF-peptide interaction (eq 1) with kinetic and equilibrium parameters derived from literature values (Table 1). We transformed eq 1 into partial differential equations defining the rate of change for each species in our model—VEGF, peptide, and VEGF–peptide complex—while including terms to describe Fickian diffusion of VEGF. We derived an effective diffusion coefficient, DVEGF,eff, for VEGF based on an established diffusion coefficient, DVEGF, of a ∼40 kDa protein with similar Stokes’ radius to VEGF diffusing through PEG hydrogels.47,50,51 The value of DVEGF,eff was calculated by multiplying DVEGF by a scaling factor approximating the influence of VEGF–peptide rebinding during release. We posited that this scaling factor was needed to describe VEGF–peptide rebinding during the release phase, a phenomenon that has been modeled previously on surfaces presenting an antigen-binding component.45
The predicted normalized VEGF flux from microspheres correlated well with experimental VEGF release data from Blank, Scramble, and VBP microspheres. Model VEGF release results using a nonscaled diffusion coefficient, DVEGF, was consistent with VEGF release data from Blank microspheres containing no peptide, as exhibited by R2 of 0.916 (Figure 7A). A scaled diffusion coefficient, DVEGF,eff, was required to adequately fit the release profile of Scramble and VBP microspheres. Diffusion coefficients, DVEGF,eff (Table 1), were scaled using Scramble–VEGF and VBP–VEGF equilibrium dissociation constants, KD (Table 1). Model VEGF release prediction correlated with experimental VEGF release from Scramble and VBP microspheres, with R2 equal to 0.954 and 0.952, respectively (Figure 7A). Our analysis demonstrated that the scaled diffusion-affinity model based on kinetic and equilibrium phenomena correlated well with experimentally observed release of VEGF from both blank microspheres and peptide-containing microspheres.
Figure 7.
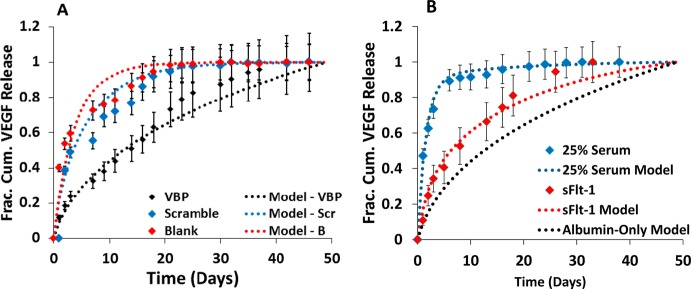
Modeling correlations to experimental release from microspheres. (A) Replot of VEGF released from Blank (red diamonds), 1.6% Scramble (blue diamonds), and 1.6% VBP (black diamonds) microspheres preincubated in 0.1 wt % BSA in PBS supplemented with 9.9 ng mL–1 VEGF, 0.1 ng mL–1 [125I]VEGF. Subsequent release was measured in 0.1 wt % BSA in PBS without VEGF supplementation. Data is fit to model of normalized VEGF flux from Blank microspheres exhibiting passive diffusion of VEGF from Blank microspheres (red dotted line; R2 = 0.916), Scramble microspheres (blue dotted line; R2 = 0.954) and from VBP microspheres (black dotted line; R2 = 0.952). (B) Plot of normalized VEGF release from 1.6% VBP microspheres in albumin-only solution supplemented with 10 ng mL–1 sFlt-1 (red diamonds). VEGF release data in sFlt-1 is fit to model of normalized VEGF flux from 1.6% VBP microspheres releasing into solution containing no protein, sFlt-1 (red dotted line; R2 = 0.98). Graph also contains a replot of normalized VEGF release data from 1.6% VBP microspheres in 25 vol % serum (blue diamonds) and modeling results for VEGF release into albumin-only solution (black dotted line). Experimental VEGF release data in 25 vol % serum is fit to model of normalized flux from 1.6% VBP microspheres releasing into solution containing physiologic concentrations of three serum proteins, sFlt-1, sKDR, and α2 M (blue dotted line; R2 = 0.99).
Further, we developed a mathematical model to address the hypothesis that a specific, serum-borne VEGF inhibitor could increase the rate of VEGF release from microspheres in serum. We based the model on three VEGF-binding molecules known to be present in serum (i.e., sFlt-1, sKDR, and α2-M), and defined a kinetic rate equation to describe the VEGF-Competitori interaction, where i = sFlt1, sKDR, or α2-M (eq 2). Together with the VEGF-peptide interaction modeled previously (eq 1), we derived differential equations to define the rate of change of each of the five species in our competition model: VEGF, peptide, Competitori, VEGF–peptide, and VEGF–Competitori.
Modeling predictions suggest that sFlt-1 present in the release medium could increase VEGF release rates. Model VEGF release predictions were calculated based on physiologic serum concentrations of sFlt-1.40 VEGF release from VBP microspheres in the presence of a physiologic concentration of sFlt-1,40 10 ng mL–1, correlated with the competition model incorporating sFlt-1 (Figure 7B; R2 = 0.98), though model prediction of VEGF release in the presence of sFlt-1 yielded poor fit to experimental release in 25% serum (R2 = 0.134; Figure 7B). These results suggest that serum-borne sFlt-1 could increase VEGF release rates (Figure 6B) but alone would not be sufficient to recapitulate the increased release rates observed in serum (Figure 7B). Thus, we posited that other serum-borne proteins in addition to sFlt-1 increased VEGF release rate in serum by binding with VEGF. We developed a model incorporating physiologic concentrations of multiple VEGF-binding serum proteins, sFlt-1, sKDR,52 and α2-M.53 Interestingly, increased VEGF release rates predicted in the presence of all three proteins were consistent with our experimentally determined VEGF release in 25 vol % serum, with R2 equal to 0.99 (Figure 7B). These data suggest that modeling approaches may be useful to understand the simultaneous influence of intrinsic variables, such as peptide identity and concentration, as well as soluble environmental conditions, on VEGF release from microspheres.
Biological Activity of Released VEGF
VEGF released from VBP and VBPWT microspheres increased HUVEC expansion in culture, which is consistent with the known mitogenic activity of VEGF.8,54 Microspheres were first pre-incubated in a solution containing varied VEGF concentrations and serum concentrations, and subsequently added to cell culture media to examine the effect of released VEGF on HUVEC expansion (Figure 8A). 1.6% VBP and VBPWT microspheres preincubated in 10 ng mL–1 VEGF significantly increased HUVEC expansion relative to Scramble and Blank microspheres after 3 days in culture (Figure 8B–D; representative DAPI images in Figure 6S), and this effect was independent of serum concentration in culture. Conversely, no significant differences in HUVEC expansion were observed between microspheres preincubated in medium with 0 or 1 ng mL–1 VEGF (Figure 8B–D) regardless of peptide identity or serum concentration. Thus, the VEGF sequestered to VBP and VBPWT microspheres was biologically active, as measured by enhanced VEGF-dependent HUVEC expansion.
Discussion
Here we examined in detail the release of VEGF from PEG microspheres functionalized with either VBPWT, which was derived from VEGFR2 and contained only natural amino acids,32 or VBP, which was a partially D-substituted version of VBPWT with higher stability against protease-mediated degradation.31 Previously, VBP and VBPWT were each shown to bind VEGF with high affinity in soluble form,31 motivating the current approach to sustain VEGF release over time using tethered versions of each of these peptides in PEG microspheres. We found that release of VEGF from both VBP and VBPWT microspheres was sustained 4-fold longer compared to Scramble and nearly 5-fold longer compared to Blank microspheres at high peptide concentration. VEGF has been previously shown to rapidly release from hydrogel microspheres, exhibiting t50 values of 1 and 2.5 days from alginate microspheres in the absence and presence of heparin respectively,55 ∼6 days from alginate microspheres within a chitosan scaffold,56 and 2.5 and 4 days from collagen microspheres incubated in collagenase and culture media, respectively.9 However, these previously used materials were loaded in solutions containing supraphysiologic concentrations of VEGF in order to elicit a biological effect upon release, as the loading efficiency for these materials was 10–30%9,55 and only as high as 55% if the VEGF was incorporated during material cross-linking.56 In contrast, VBP and VBPWT microspheres were previously shown to sequester 40–60% of VEGF28 in a solution containing a physiologic concentration of VEGF, 10 ng mL–1 (ref (37)), which demonstrates that these chemically defined materials can regulate VEGF within a physiologic concentration range.37 VBP and VBPWT microspheres sustained VEGF release substantially, as they exhibited t50 values of 5.5 and 8 days at low and high concentrations of VEGF-binding peptides respectively (Figure 2). Additionally, previous studies have not examined the influence of the soluble environment on VEGF release rates. We found that release of bound VEGF was substantially influenced by serum concentration. However, the decreased cumulative release (Figure 2S-3S) and increased release rates observed (Figure 4) were not dependent on protease activity (Figure 5) or the presence of a physiologic concentration of soluble heparin (Figure 6A), but VEGF release rates were significantly increased in the presence of a physiologic concentration of sFlt-1 (Figure 6B). We additionally demonstrated that released VEGF from VBP and VBPWT microspheres stimulated endothelial cell expansion, a standard measure of VEGF biological activity (Figure 8). Finally, we correlated VEGF release data (Figure 7) with a generalizable model describing affinity-mediated release of a growth factor from peptide-containing hydrogels.
Serum substantially accelerated VEGF release kinetics, and a comparison of VEGF release in the 2% serum condition versus the albumin-only condition provides an illustration of the potential mechanism (Figure 4). Although the total protein concentration in 2 vol % serum (approximately 1 mg mL–1 total protein29,30) was substantially less than the albumin-only solution (12.5 mg mL–1 total protein), the release rate from VBP and VBPWT microspheres was similar in 2 vol % serum and serum-free (albumin-only) conditions (Figure 4A,B). Thus, we concluded that total protein content in the release solution did not influence VEGF release rates. Furthermore, previous experiments demonstrated that buffered solution did not influence Blank microsphere diameter over a 20 day time frame (Figure 7S), and thus we concluded that biomaterial degradation was not an operative mechanism governing VEGF release. Given the similar ionic strength57 and pH58 of PBS and serum-containing medium, we hypothesized that specific biological molecules in the serum likely increased the VEGF release rates independent of total protein concentration and biomaterial degradation.
Serum contains numerous protein components, including proteases, proteoglycans, glycosaminoglycans, growth factors59−61 and various carrier proteins.29,30 Surprisingly, our experiments indicated that neither protease activity (Figures 5 and 4S) nor the presence of soluble heparin (Figures 6A and 5S) were responsible for the effect of serum on VEGF release. Therefore, we hypothesized that biological molecules other than proteases and heparin could directly compete with VEGF–peptide binding and increase the VEGF release rate. The rate of VEGF release from VBP microspheres in the presence of sFlt-1 (a receptor fragment from VEGFR1) was increased approximately 2-fold (Figure 6B), suggesting that sFlt-1 in serum could have increased VEGF release rate in serum. However, serum increased the release rate of VEGF by almost 8-fold compared to albumin-only solution (Figure 4B), which together with modeling results (Figure 7B) suggests that sFlt-1 was not sufficient to recapitulate the effect of serum on VEGF release. We utilized a mathematical model to predict the influence of physiological concentrations of multiple VEGF-binding proteins in serum: sFlt-1, sKDR (a receptor fragment from VEGFR2), and α2-macroglobulin (α2-M). Consistent with our VEGF release model in the presence of sFlt-1 (Figure 7B), modeling of VEGF release in the presence of sKDR and α2-M alone could not have increased VEGF release rates in serum (data not shown). Conversely, model VEGF release prediction in the presence of sFlt-1, sKDR, and α2-M increased VEGF release rates relative to no protein. Remarkably, the model taking into account all three putative VEGF binding proteins correlated well with experimental release results in 25 vol % serum (Figure 7B). These experimental results and model predictions suggest that specific interactions, such as those between VEGF and serum-borne receptor fragments, may have influenced VEGF release. This concept is consistent with previous studies describing sFlt-1,62,63 sKDR,52,64 and α2-M65 binding to VEGF and acting as “sinks” for free VEGF. It is reasonable to posit that given the intermediate affinity of VEGF for VBP (KD ∼80 nM), a combination of high-affinity competitors like sFlt-1 and sKDR and high abundance promiscuous GF-binding competitors like α2-M may collectively interfere with VEGF-peptide interactions. Our modeling results are supported by literature describing compartmentalization of VEGF to both cell surface receptors in vivo66 and soluble receptor fragments in silico.67,68 These results suggest that VEGF sequestering in solution and to cell surface receptors may be the operative mechanism for reducing growth factor half-life,66,69,70 and further that protease activity may have a negligible effect on growth factor pharmacokinetics. The results of this study demonstrate that soluble competitors present in the biological environments intended for affinity-based GF delivery should be considered before therapeutic application.
The observed biological activity of VEGF released from VBP and VBPWT microspheres agrees with previous observations that released VEGF acts as an endothelial cell mitogen.26 Importantly, serum concentration alone did not influence HUVEC proliferation (Figure 8) in agreement with previous literature,71,72 which enabled us to specifically study the impact of serum concentration on VEGF activity in vitro. The biological activity of released VEGF in serum-containing medium (Figure 8) is consistent with our observation that VEGF release is not influenced strongly by protease activity in a high concentration of serum (Figure 5). In particular, if protease activity was an operative mechanism dictating VEGF release, then one could expect considerable levels of proteolytic VEGF degradation and poor VEGF biological activity, neither of which was observed. In addition, we observed that only VBP and VBPWT microspheres preincubated in 10 ng mL–1 VEGF increased HUVEC expansion relative to controls, while microspheres preincubated in 1 ng mL–1 VEGF did not significantly increase HUVEC expansion. This difference is likely due to the total amounts of VEGF released in these experimental conditions. The t50 for VEGF release from 1.6% VBP and VBPWT microspheres preincubated in 10 ng mL–1 VEGF was 5 days and 0.5 days in 2 vol % serum and 25 vol % serum, respectively (Figure 4B). Based on the VEGF release data, these conditions would have resulted in approximately 1 and 4 ng mL–1 of total released VEGF into the cell culture solutions with 2 vol % and 25 vol % serum, respectively (Figure 3S,A) during the 60 h of HUVEC culture. Microspheres preincubated in 1 ng mL–1 VEGF would have released substantially lower amounts of VEGF, approximately 0.1 and 0.4 ng mL–1 in 2 vol % and 25 vol % serum, respectively (Figure 3S,A). A released VEGF amount less than 1 ng mL–1 would be unlikely to act as an effective endothelial cell mitogen, as a previous study showed that 1–1.2 ng mL–1 VEGF was required in culture to elicit increased endothelial cell proliferation8 and a plateau of HUVEC proliferation was observed at greater than 5 ng mL–1 VEGF in culture.54 These conclusions are consistent with our observed results and similar studies,16,26 wherein released VEGF concentrations greater than 1 ng mL–1 were used to enhance HUVEC expansion.
Here we established a model to correlate observed VEGF release profiles with a generalizable mathematical description of affinity–GF interactions. Experimental results were directly correlated to model VEGF predictions, which used a coupled diffusion–affinity coefficient, DVEGF,eff, that was weighted to account for the probability of VEGF–peptide rebinding during VEGF release. The resulting DVEGF,eff from 1.6% VBP microspheres was approximately 3–4 orders of magnitude lower than established DVEGF of VEGF release from PEG hydrogels without peptide. This result is consistent with a previous study implicating protein–receptor rebinding for slowing diffusion of a target molecule from a surface with tethered receptor.73 The modeling in this study is also consistent with similar modeling of affinity-mediated growth GF from peptide-containing fibrin hydrogels, in which authors used a system of equations similar to those derived here to describe the contribution of affinity parameters and diffusion coefficients on GF release.74 However, previous models have lacked direct correlation to experimental release data. Here, we have correlated modeled VEGF release with experimental sustained release data from microspheres containing VEGF-binding ligands, as well as from control microspheres with no inherent affinity for VEGF. Furthermore, the VEGF release profile in the presence of sFlt-1 correlated well with a mathematical model incorporating sFlt-1 interactions with VEGF, though results suggested that sFlt-1 was insufficient to fully recapitulate the influence of serum on VEGF release. Our competition model suggested that competitive interactions between multiple VEGF-binding serum proteins (sFlt-1, sKDR, and α2-M) could increase VEGF release rates.
Conclusion
Here, we have analyzed the contribution of both intrinsic biomaterial parameters and extrinsic solution parameters on VEGF release from biomimetic hydrogel microspheres. Collectively, the results of our in vitro and in silico analysis of a biological environment suggest that affinity-based platforms should reflect understanding of both intrinsic material properties and extrinsic soluble microenvironment properties before translating these materials to a biological environment. The increased use of affinity-based materials for GF delivery suggest that the approach and mathematical model used in the current study may be applicable to similar emerging approaches in biomaterials design and controlled release.75,76
Acknowledgments
The authors acknowledge support from the National Institutes of Health (T32 HL007936-12, RO1HL093282, R21 EB016381, and 5T32GM08349).
Supporting Information Available
Cumulative VEGF release from microspheres with different material parameters (peptide identity and density) and from microspheres releasing into serum-containing solution; comparison of fractional VEGF release curves in the presence of serum with protease inhibitor for microspheres containing a low and high concentration of peptide; cumulative VEGF release from microsphere releasing into heparin-containing solution; description of mathematical model describing affinity-mediated VEGF release from microspheres containing a low (Scramble), high (VBP), or no affinity for VEGF (Blank); description of mathematical model describing competition-mediated VEGF release in solution containing one or multiple soluble VEGF inhibitors; representative fluorescent micrographs of DAPI-stained HUVECs in VEGF bioactivity assay; experimental data and representative phase contrast images of blank PEG-norbornene microspheres in buffered solution over time. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Midwood K. S.; Williams L. V.; Schwarzbauer J. E. Int. J. Biochem. Cell Biol. 2004, 36, 1031–1037. [DOI] [PubMed] [Google Scholar]
- Phelps E. A.; Garcia A. J. Regen. Med. 2009, 4, 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo A. G.; Iruela-Arispe M. L. Cardiovasc. Res. 2010, 86, 226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunewald M.; Avraham I.; Dor Y.; Bachar-Lustig E.; Itin A.; Jung S.; Yung S.; Chimenti S.; Landsman L.; Abramovitch R.; Keshet E. Cell 2006, 124, 175–189. [DOI] [PubMed] [Google Scholar]
- Maharaj A. S. R.; D’Amore P. A. Microvasc. Res. 2007, 74, 100–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee R. J.; Springer M. L.; Blanco-Bose W. E.; Shaw R.; Ursell P. C.; Blau H. M. Circulation 2000, 102, 898–901. [DOI] [PubMed] [Google Scholar]
- Vempati P.; Popel A. S.; Mac Gabhann F. BMC Syst. Biol. 2011, 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N.; Henzel W. J. Biochem. Biophys. Res. Commun. 1989, 161, 851–858. [DOI] [PubMed] [Google Scholar]
- Nagai N.; Kumasaka N.; Kawashima T.; Kaji H.; Nishizawa M.; Abe T. J. Mater. Sci. 2010, 21, 1891–1898. [DOI] [PubMed] [Google Scholar]
- Wang A. Y.; Leong S.; Liang Y.-C.; Huang R. C. C.; Chen C. S.; Yu S. M. Biomacromolecules 2008, 9, 2929–2936. [DOI] [PubMed] [Google Scholar]
- Belotti D.; Paganoni P.; Manenti L.; Garofalo A.; Marchini S.; Taraboletti G.; Giavazzi R. Cancer Res. 2003, 5224–5229. [PubMed] [Google Scholar]
- Ferrara N. Mol. Biol. Cell 2010, 21, 687–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem H.; Tomic-Canic M. J. Clin. Invest. 2007, 117, 1219–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulapaditharom B.; Boonkitticharoen V.; Sritara C. J. Oncol. 2012, 2012, 687934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P. Nat. Med. 2003, 9, 653–660. [DOI] [PubMed] [Google Scholar]
- Tae G.; Scatena M.; Stayton P. S.; Hoffman A. S.; Allan S. J. Biomater. Sci. 2006, 17, 187–197. [DOI] [PubMed] [Google Scholar]
- Chung Y.-I.; Kim S.-K.; Lee Y.-K.; Park S.-J.; Cho K.-O.; Yuk S. H.; Tae G.; Kim Y. H. J. Controlled Release 2010, 143, 282–289. [DOI] [PubMed] [Google Scholar]
- Pike D. B.; Cai S.; Pomraning K. R.; Firpo M. A.; Fisher R. J.; Shu X. Z.; Prestwich G. D.; Peattie R. A. Biomaterials 2006, 27, 5242–5251. [DOI] [PubMed] [Google Scholar]
- Oliviero O.; Ventre M.; Netti P. A. Acta Biomater. 2012, 8, 3294–3301. [DOI] [PubMed] [Google Scholar]
- Nakatsu M. N.; Sainson R. C. A.; Aoto J. N.; Taylor K. L.; Aitkenhead M.; Pérez-del-Pulgar S.; Carpenter P. M.; Hughes C. C. W. Microvasc. Res. 2003, 66, 102–112. [DOI] [PubMed] [Google Scholar]
- Wong C.; Inman E.; Spaethe R.; Helgerson S. Thromb. Haemost. 2003, 89, 573–582. [PubMed] [Google Scholar]
- Borselli C.; Ungaro F.; Oliviero O.; D’Angelo I.; Quaglia F.; La Rotonda M. I.; Netti P. A. J. Biomed. Mater. Res. Part A 2010, 92, 94–102. [DOI] [PubMed] [Google Scholar]
- Mosesson M. W. J. Thromb. Haemost. 2005, 3, 1894–1904. [DOI] [PubMed] [Google Scholar]
- Capila I.; Linhardt R. J. Angew. Chem. 2002, 41, 391–412. [DOI] [PubMed] [Google Scholar]
- Rider C. C. Biochem. Soc. Trans. 2006, 34, 458–460. [DOI] [PubMed] [Google Scholar]
- Impellitteri N. A.; Toepke M. W.; Lan Levengood S. K.; Murphy W. L. Biomaterials 2012, 33, 3475–3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toepke M. W.; Impellitteri N. A.; Lan Levengood S. K.; Boeldt D. S.; Bird I. M.; Murphy W. L. Adv. Healthcare Mater. 2012, 1, 457–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belair D. G.; Murphy W. L. Acta Biomater. 2013, 9, 8823–8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X.; Baker H.; Hancock W. S.; Fawaz F.; McCaman M.; Pungor E. Biotechnol. Prog. 2006, 22, 1294–1300. [DOI] [PubMed] [Google Scholar]
- Issaq H. J.; Xiao Z.; Veenstra T. D. Chem. Rev. 2007, 107, 3601–3620. [DOI] [PubMed] [Google Scholar]
- Germeroth L.; Piossek C.; Thierauch K.-H.; Schneider-Mergener J.; Volkmer-Engert R.; Bachmann M. F.; Korff T.; Augustin H. G. Thromb. Haemostasis 2003, 501–510. [DOI] [PubMed] [Google Scholar]
- Piossek C.; Schneider-Mergener J.; Schirner M.; Vakalopoulou E.; Germeroth L.; Thierauch K. H. J. Biol. Chem. 1999, 274, 5612–5619. [DOI] [PubMed] [Google Scholar]
- Wu N.; Wang L.-S.; Tan D. C.-W.; Moochhala S. M.; Yang Y.-Y. J. Controlled Release 2005, 102, 569–581. [DOI] [PubMed] [Google Scholar]
- Lin C.-C.; Metters A. T. Adv. Drug Delivery Rev. 2006, 58, 1379–1408. [DOI] [PubMed] [Google Scholar]
- Fairbanks B. D.; Schwartz M. P.; Halevi A. E.; Nuttelman C. R.; Bowman C. N.; Anseth K. S. Adv. Mater. 2009, 21, 5005–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franssen O.; Hennink W. E. Int. J. Pharm. 1998, 168, 1–7. [DOI] [PubMed] [Google Scholar]
- Howdieshell T. R.; Riegner C.; Gupta V.; Callaway D.; Grembowicz K.; Sathyanarayana; McNeil P. L. Ann. Surg. 1998, 228, 707–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama H.; Sato K.; Okudaira M.; Morita C.; Takahashi C.; Suzuki D.; Sakai H.; Iwamoto Y. Kidney Int. 1999, 56, 650–658. [DOI] [PubMed] [Google Scholar]
- Hofmann-Kiefer K.; Kemming G.; Chappell D.; Flondor M.; Kisch-Wedel H.; Hauser A.; Pallivathukal S.; Conzen P.; Rehm M. Eur. J. Med. Res. 2009, 14, 526–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgore F. M.; Blann A. D.; Lip G. Y. Clin. Sci. 2001, 100, 567–575. [PubMed] [Google Scholar]
- Lip P. L.; Belgore F.; Blann A. D.; Hope-Ross M. W.; Gibson J. M.; Lip G. Y. Invest. Ophthalmol. Vis. Sci. 2000, 41, 2115–2119. [PubMed] [Google Scholar]
- Kaitu’u-Lino T. J.; Whitehead C. L.; Ngian G.-L.; Permezel M.; Tong S. PLoS One 2012, 7, e32509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeel R. D.; Berzins M. SIAM J. Sci. Stat. Comput. 1990, 11, 1–32. [Google Scholar]
- Zhang J.; Li H.; Chen W.; Cao P.; Wang M. Biotechnol. Prog. 2009, 25, 1703–1708. [DOI] [PubMed] [Google Scholar]
- Lagerholm B. C.; Thompson N. L. Biophys. J. 1998, 74, 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zustiak S. P.; Durbal R.; Leach J. B. Acta Biomater. 2010, 6, 3404–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zustiak S. P.; Boukari H.; Leach J. B. Soft Matter 2010, 6, 3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper S. FASEB J. 2003, 17, 333–340. [DOI] [PubMed] [Google Scholar]
- Welsh C. F.; Roovers K.; Villanueva J.; Liu Y.; Schwartz M. A.; Assoian R. K. Nat. Cell Biol. 2001, 3, 950–957. [DOI] [PubMed] [Google Scholar]
- Zustiak S. P. Y.; Leach J. B. Biotechnol. Bioeng. 2011, 108, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber L. M.; Lopez C. G.; Anseth K. S. J. Biomed. Mater. Res. Part A 2010, 90, 720–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebos J. M. L.; Bocci G.; Man S.; Thorpe P. E.; Hicklin D. J.; Zhou D.; Jia X.; Kerbel R. S. Mol. Cancer Res. 2004, 2, 315–326. [PubMed] [Google Scholar]
- Tunstall A. M.; Merriman J. M. L.; Milne I.; James K. J. Clin. Pathol. 1975, 133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soker S.; Gollamudi-Payne S.; Fidder H.; Charmahelli H.; Klagsbrun M. J. Biol. Chem. 1997, 272, 31582–31588. [DOI] [PubMed] [Google Scholar]
- Lee K. W.; Yoon J. J.; Lee J. H.; Kim S. Y.; Jung H. J.; Kim S. J.; Joh J. W.; Lee H. H.; Lee D. S.; Lee S. K. Transplant. Proc. 2004, 36, 2464–2465. [DOI] [PubMed] [Google Scholar]
- De la Riva B.; Sánchez E.; Hernández A.; Reyes R.; Tamimi F.; López-Cabarcos E.; Delgado A.; Evora C. J. Controlled Release 2010, 143, 45–52. [DOI] [PubMed] [Google Scholar]
- Honn K. V.; Singley J. A.; Chavin W. Proc. Soc. Exp. Biol. Med. 1975, 344–347. [DOI] [PubMed] [Google Scholar]
- Dill B.; Daly C. J. Biol. Chem. 1937, 569–579. [Google Scholar]
- Banks R. E.; Forbes M. A.; Kinsey S. E.; Stanley A.; Ingham E.; Walters C.; Selby P. J. Br. J. Cancer 1998, 77, 956–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor-McCourt M. D.; Wakefield L. M. J. Biol. Chem. 1987, 262, 14090–14099. [PubMed] [Google Scholar]
- Takano S.; Yoshii Y.; Kondo S.; Suzuki H.; Maruno T.; Shirai S.; Nose T. Cancer Res. 1996, 56, 2185–2190. [PubMed] [Google Scholar]
- Hornig C.; Barleon B.; Ahmad S.; Vuorela P.; Ahmed A.; Weich H. A. Lab. Invest. 2000, 80, 443–454. [DOI] [PubMed] [Google Scholar]
- Barleon B.; Totzke F.; Blanke S.; Kremmer E.; Siemeister G.; Marmé D.; Herzog C.; Marme D.; Martiny-Baron G. J. Biol. Chem. 1997, 10382–10388. [DOI] [PubMed] [Google Scholar]
- Pavlakovic H.; Becker J.; Albuquerque R.; Wilting J.; Ambati J. Ann. N.Y. Acad. Sci. 2010, 1207, E7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee G.; Asplin I. R.; Wu S. M.; Gawdi G.; Pizzo S. V. J. Biol. Chem. 2000, 275, 26806–26811. [DOI] [PubMed] [Google Scholar]
- Eppler S. M.; Combs D. L.; Henry T. D.; Lopez J. J.; Ellis S. G.; Yi J.-H.; Annex B. H.; McCluskey E. R.; Zioncheck T. F. Clin. Pharmacol. Ther. 2002, 72, 20–32. [DOI] [PubMed] [Google Scholar]
- Stefanini M. O.; Wu F. T. H.; Mac Gabhann F.; Popel A. S. BMC Syst. Biol. 2008, 2, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen P.; Finley S. D.; Engel-Stefanini M. O.; Popel A. S. PLoS One 2011, 6, e27514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Wang G.-J.; Sun J.-G. Acta Pharmacol. Sin. 2004, 25, 991–995. [PubMed] [Google Scholar]
- Nguyen C. B.; Harris L.; Onyi S. Z.; Baughman S. A.; Hale V. G.; Dybdal N. O.; Sadick M. D.; Escandon E. Drug Metab. Dispos. 2000, 28, 598–607. [PubMed] [Google Scholar]
- Bala K.; Ambwani K.; Gohil N. K. Tissue Cell 2011, 43, 216–222. [DOI] [PubMed] [Google Scholar]
- Walker C.; Mates G.; Pumford D.; Daniel M. J. Cell Sci. 1987, 87Pt 5739–747. [DOI] [PubMed] [Google Scholar]
- Oh D.; Ogiue-ikeda M.; Jadwin J. A.; Machida K.; Mayer B. J.; Yu J. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 14024–14029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakiyama-Elbert S. E.; Hubbell J. A. J. Controlled Release 2000, 65, 389–402. [DOI] [PubMed] [Google Scholar]
- Zhu J. Biomaterials 2010, 31, 4639–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudalla G. A.; Murphy W. L. Adv. Funct. Mater. 2011, 21, 1754–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Tiedemann B.; Bilitewski U. Biosens. Bioelectron. 2002, 17, 983–991. [DOI] [PubMed] [Google Scholar]
- Tanaka K.; Yamaguchi S.; Sawano A.; Shibuya M. Jpn. J. Cancer Res. 1997, 867–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham S. A.; Tran T. M.; Arrate M. P.; Brock T. A. J. Biol. Chem. 1999, 274, 18421–18427. [DOI] [PubMed] [Google Scholar]
- Finley S. D.; Dhar M.; Popel A. S. Front. Oncol. 2013, 3, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.