

Abstract
Mass spectrometry (MS)-based shotgun proteomics is an effective technology for global proteome profiling. The ultimate goal is to assign tandem MS spectra to peptides and subsequently infer proteins and their abundance. In addition to database searching and protein assembly algorithms, computational approaches have been developed to integrate genomic, transcriptomic, and interactome information to improve peptide and protein identification. Earlier efforts focus primarily on making databases more comprehensive using publicly available genomic and transcriptomic data. More recently, with the increasing affordability of the Next Generation Sequencing (NGS) technologies, personalized protein databases derived from sample-specific genomic and transcriptomic data have emerged as an attractive strategy. In addition, incorporating interactome data not only improves protein identification but also puts identified proteins into their functional context and thus facilitates data interpretation. In this paper, we survey the major integrative bioinformatics approaches that have been developed during the past decade and discuss their merits and demerits.
Keywords: shotgun proteomics, proteogenomics, personalized proteomics, data integration, peptide identification, protein identification, Next Generation Sequencing, RNA-Seq
1. Introduction
Proteins are key functional molecules in cells and serve as a link between genotype and phenotype. Global proteomic analysis allows direct measurements of proteins, and when integrated with genomic and transcriptomic studies, provides a great opportunity to understand the information flow from DNA to protein to phenotype.
Among different high-throughput proteomic technologies, mass spectrometry (MS)-based shotgun proteomics has had the greatest impact in biological and biomedical research. Recent technology advances have made this approach increasingly applicable for global profiling of cell and tissue proteomes, with the capacity to detect more than 10000 proteins from a single biological sample.1 Figure 1 illustrates the typical workflow of a shotgun proteomics study. In the experimental phase, proteins are enzymatically digested into peptides, which are fractionated and analyzed by liquid chromatography–tandem mass spectrometry (LC–MS/MS). In the data analysis phase, tandem mass spectra are interpreted to peptides by computational algorithms and then assembled into proteins. The most widely used method for peptide identification is database searching by computational tools such as SEQUEST, Mascot,2 X!Tandem,3 or MyriMatch.4 These tools first perform an in silico digestion of all proteins in a reference protein database to enumerate all candidate peptide sequences and then construct a theoretical spectrum for each candidate peptide sequence. Experimentally observed fragment ion spectra are compared to the theoretical spectra and then linked to corresponding peptides if a comparison produces a statistically significant peptide-spectrum match (PSM) score. Finally, identified peptides are transformed into a list of identified proteins through protein assembly tools such as IDPicker,5 MassSieve,6 or ProteinProphet,7 among others.
Figure 1.
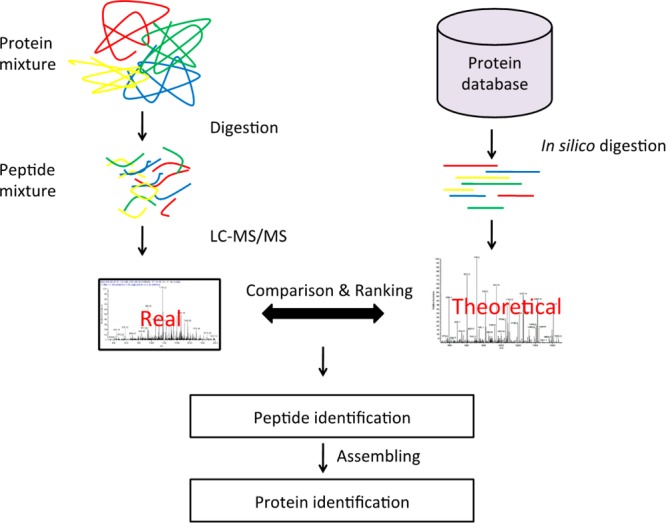
A typical workflow of shotgun proteomics.
Although this strategy has been successful, there are several critical challenges that cannot be fully addressed by simply improving database searching and protein assembly algorithms. On one hand, all database searching algorithms rely on a reference protein database, which is typically incomplete. First, novel protein coding genes are still being continuously identified.8 Second, a single gene locus can produce multiple transcript and protein isoforms through alternative splicing, and it remains difficult to completely catalogue all protein coding transcripts that can be generated from known gene loci.9 Moreover, sequence variants including single nucleotide polymorphisms (SNPs), somatic mutations, insertions, deletions, and gene fusions are often neglected in commonly used reference protein databases. On the other hand, despite substantial improvements, reliable identification of low-abundant proteins remains challenging.
During the past decade, various computational methods have been developed to integrate orthogonal data sources to improve peptide and protein identification in shotgun proteomics studies. These approaches take advantage of the rapidly growing volumes of genomic, transcriptomic, and interactome data. Here we review different integrative bioinformatics strategies that have been used to address the above-mentioned challenges. Relevant studies are summarized in Table 1 and the list continues to grow. This review is limited to human and mouse studies, and studies focusing on microbes or plants are not included.
Table 1. List of Published Orthogonal Data Assisted Proteomics Studies.
| Genomic Information | ||
|---|---|---|
| Choudhary et al. | six-frame translation using the draft of human genome | (13) |
| Fermin et al. | six-frame translation of whole human genome | (19) |
| Sevinsky et al. | six-frame translation of whole human genome | (18) |
| peptide isoelectric point (pI) | ||
| Bitton et al. | prescreening searches on databases translated from individual chromosomes; matched entries were then combined with the Celera database entries and used for a second time search | (12) |
| Mo et al. | exon–exon junction database | (29) |
| Power et al. | noncontiguous junction peptides in a “full length transcript” | (30) |
| Gatlin et al. | generating dynamically all possible SNPs | (40) |
| Roth et al. | creating a highly annotated database, including splicing, PTMs, and SNPs | (47) |
| Bunger et al. | reference protein database | (41) |
| tryptic peptide database created from dbSNP | ||
| peptide pI | ||
| Schandorff et al. | elongating IPI sequences with theoretical N-terminal peptides, variant peptides from cSNP, variant peptides from conflict annotation in Swiss-Prot, and proteolytic enzyme and keratin sequences | (37) |
| Xi et al. | human disease-related variants from OMIM, PMD, and Swiss-Prot | (44) |
| Nijveen et al. | 20-mer variant peptides generated by three-frame translation from mRNA sequences including SNPs in dbSNP | (35) |
| Li et al. | combined database of normal proteins and variant peptides | (46) |
| modified FDR estimation | ||
| Su et al. | a pipeline of nontargeted proteomics for identifying SAP peptides in human plasma and quantifying them using targeted proteomics | (43) |
| Khatun et al. | whole genome proteogenomic mapping to identify novel protein coding regions for ENCODE cell line proteomics data | (8) |
| Transcriptomic Information | ||
|---|---|---|
| Tanner et al. | using genomic data and EST data to construct the exon graph, which is a compact representation of all putative exons, splice variants and polymorphisms | (31) |
| Edwards et al. | using sequence database compression strategies to reduce EST database size by approximately 35-fold | (36) |
| Menon et al. | three-frame translation of mRNA sequences from the ECgene and ENSEMBL databases | (33),34 |
| Ramakrishnan et al. | using expression information from microarray to assist protein identification | (52) |
| Ning et al. | six-frame translation of novel junction mRNA sequence identified by RNA-Seq | (38) |
| Wang et al. | customized database from RNA-Seq data | (49),59 |
| Chen et al. | generating database for missense SNVs and RNA edits from genomic sequencing and RNA-Seq data | (48) |
| Sheynkman et al. | deriving novel splice-junction peptides from RNA-Seq data | (39) |
| Evan et al. | de novo assembly of transcriptomes from RNA-Seq data | (51) |
| Menschaert et al. | A custom protein database built from both Swiss-Prot and RIBO-seq derived translation products | (61) |
| Woo et al. | A proteogenomic database from large scale RNA-Seq data | (26) |
| Sheynkman et al. | detection of variant peptides from RNA-Seq data | (62) |
| Network Information | ||
|---|---|---|
| Li et al. | protein–protein interaction network-assisted protein assembly through clique enumeration and enrichment analysis | (54) |
| Ramakrishnan et al. | improving protein identification by considering information on functional associations from a gene function network | (56) |
| Goh et al. | using functional clusters to expand protein lists | (63) |
| Nusinow et al. | using a network-based inference tool, SNIPE, to select proteins that are likely to be active but undetectable in shotgun proteomics | (55) |
2. Improving Peptide Identification
Bottom-up proteomics technologies rely on peptide identification to infer protein presence. Integrating genomic and transcriptomic information allows the identification of peptides derived from novel protein coding genes, splice variants, and sequence variations, leading to a more comprehensive proteomic characterization of biological and clinical samples.
2.1. Novel Protein-Coding Genes
The database searching strategy relies on complete genomes and thorough protein coding gene annotations. Although whole genome sequencing data for human and other model organisms have been available for a decade, genome annotation remains incomplete even for the human genome. Several studies have demonstrated the potential of shotgun proteomics in the discovery of novel protein-coding genes in human and mouse using a variety of approaches.8,10−12
The most intuitive approach to enable the identification of novel protein-coding genes by shotgun proteomics is to use a database containing a six-frame translation of the whole genome. Right after the release of the initial human genome draft sequence, Choudhary and colleagues searched an LC–MS/MS data set containing peptides from at least 22 human proteins against a curated protein database, an expressed sequence tag (EST)-derived database, and a genome-derived database.13 Although the data set was small and the majority of proteins were found much more rapidly using the curated protein database, the study pioneered the use of genome translated databases for shotgun proteomics. A six-frame translation database does not depend on gene models and therefore contains all possible protein forms except for peptides spanning the exon junction regions. The strategy has been widely used in microbial studies because microbial genomes are small and lack alternative splicing.14,15 Since 98% of the human genome is not protein coding,16 this method dramatically increases the searching space and computational time. Tools have been developed to automate this strategy and make it practical for mammalian genomes.17 However, a major concern is the large amount of background noise introduced by this strategy. Therefore, extra efforts are needed when applying this method to large, complex mammalian genomes.
Methods have been developed to constrain the database size and complexity before the search. For experiments that use immobilized pH gradient strips to fractionate peptides, each fraction only contains peptides of a narrow isoelectric point (pI) range. This information has been used for the development of GENQUEST,18 a method that restricts the peptide search space based on the pI range. Specifically, after the six-frame translation of the genome, each putative protein is in silico digested with trypsin and the pI is calculated for each peptide. Peptides are then grouped together based on their pIs. Spectra generated from a specific peptide fraction are only searched against the subset of peptides with pIs in the same range. It has been shown that this method resulted in accurate and sensitive results comparable to searching a curated protein database. Another method utilizes a series of prescreening searches against databases translated from individual chromosomes to identify and eliminate nonmatching entries, and then a second search is performed against all the matched entries combined with a curated protein database. This method dramatically reduces the database size for individual searches and has been successfully used to identify novel peptides in two human cell lines.12
Methods have also been developed to control the peptide false discovery rate (FDR) after the search. Ferman et al. searched a data set from the Human Proteome Organization Plasma Proteome Project against a six-frame translation of the entire human genome to identify novel blood proteins.19 They used a Poisson model, which brings into consideration the number of spectra searched, score threshold applied to accepting a match, the size of the target sequence database, and the length of the matched protein sequence, to estimate the confidence of peptide identifications. A detailed analysis showed that among the 2309 high quality intragenic peptides, 73% were completely contained within annotated exons, 6% partially overlapped with annotated exons, and 21% were aligned to nonexonic regions.
Ever since the emergence of the RNA sequencing (RNA-Seq) technology, RNA-Seq data have been widely used to facilitate proteomics studies of nonmodel organisms that do not have a fully sequenced and well-annotated genome, such as many microorganisms and plants.20−23 In human and model organism studies, RNA-Seq has revealed a large number of transcribed unannotated regions,24,25 and some of them may represent novel protein-coding regions. Because gene expression changes over time and conditions, and each data set is associated with sequencing errors and mapping errors, combining different RNA-Seq data sets from an organism can lead to a more comprehensive and accurate reference protein database for the organism. A recent study in Caenorhabditis elegans generated an aggregated database from public C. elegans RNA-Seq data sets, allowing the identification of hundreds of novel genes in a MS/MS data set from 11 developmental stage of C. elegans.26
2.2. Novel Splice Variants
The incompleteness of genome annotation can also arise from unknown isoforms. Alternative splicing isoforms amplify the coding diversity and thus enable the functional repertoire of genes. A typical exon in the human genome is short with more than three-quarters of the exons having a length less than 200 bp,27 which means a relatively large number of peptides span the exon boundary. Because of incomplete genome annotation, many splice junctions might be missing in the public databases. A more comprehensive splicing annotation will certainly improve peptide identification in proteomics, as exemplified by a study demonstrating a 7% increase in peptide identification when using ENSEMBL database with explicit isoform entries rather than the nonredundant Swiss-Prot database.28
As mentioned above, one major limitation of the six-frame genome translation method is the failure to detect junction-spanning peptides. This limitation can be partially overcome by the generation of an exon–exon junction database. Mo et al. designed a theoretical exon–exon junction protein database to account for all possible combination of exons for each gene in the ENSEMBL database while keeping the frame of translation.29 They only took 25 amino acid residues from each exon and used X!Tandem and SEQUEST to identify exon junctions in a human liver secretome MS/MS data set. By combing search results from the two tools, they identified 488 nonredundant peptides corresponding to 395 ENSEMBL genes. Another study by Power et al. used a similar method to construct a database harboring peptide sequences derived from all hypothetical exon–exon junctions in the human genome.30 The strategy, named SkipE, employs two main steps for database construction. First, it includes a “full-length transcript”, which is the longest predicted exon sequence, for each gene. Overlapping exons are merged into a longer one. Second, entirely noncontiguous junction peptides are created from exon–exon junction-spanning sequences by cleaving the trypsin sites on both faces. Compared to the database generated by Mo et al. (873024 peptides), this method helped reduce the database size by more than half (307030 peptides).
One intrinsic limitation of using only genomic data (exon model) to generate exon–exon junction databases is that many predicted alternative splicing events do not occur at the transcriptional level, and therefore a large amount of noise is introduced. To address this limitation, some studies have used EST data to reduce the size of a putative junction database. ESTs are short sequences from complementary DNA (cDNA) sequences and can indicate gene expression. Tanner et al. have developed algorithms that combine genomic data and EST data to construct an exon graph, which is a compact representation of all putative exons, splice variants, and polymorphisms. By searching a large collection of 18.5 million tandem MS spectra from human proteomic samples against the database, they confirmed the translation of 224 hypothetical human proteins and over 40 alternative splicing events.31 Other studies use three-frame translation of mRNA sequences from ECgene, a comprehensive alternative splicing sequence database with splice variants predicted by EST clustering,32 to generate databases for integrating with the ENSEMBL database.33,34 Since alternative splice variants contribute to a number of diseases including cancer, these studies have been performed to identify both novel and known splice variants in cancer samples.
Using EST data could largely reduce the number of putative junctions and introduce novel proteins. However, this approach is limited by the (1) large and redundant data size; (2) inability to cover all genes; and (3) presence of unprocessed and truncated transcripts as well as genomic contaminants.31,35,36 Because of these limitations, some researchers even argue against using EST data for proteomics studies.37 Further efforts are required to overcome these limitations. Edwards et al. have introduced several sequence database compression strategies to maintain the high quality ESTs, thus reducing database size by approximately 35-fold. These strategies include: (1) limiting EST sequences to those mapping to the vicinity of known genes; (2) requiring a minimum peptide length of 30-mer; and (3) including only peptides supported by at least two ESTs. This approach brings the database size closer to the commonly used protein sequence databases and allows the discovery of novel peptides in a variety of public data sets.36 The GENQUEST method mentioned earlier can also be used to reduce the complexity of EST databases.18 Although very helpful, these approaches cannot overcome other above-mentioned limitations.
Compared to EST libraries, RNA-Seq provides a more advanced way to comprehensively identify alternative splicing events. Ning et al. performed a preliminary analysis using RNA-Seq data to derive a six-frame translated novel junction sequence database for MS/MS data search, with a focus on the identification of novel alternative splicing forms.38 Although the study only provided proteomic evidence for a few novel alternative splicing forms, it helped demonstrate the feasibility of using RNA-Seq data to facilitate the identification of junction peptides. In a more recent study, Sheynkman et al. built an unannotated splice-junction peptide database with more than 30000 peptides based on RNA-Seq data, allowing the identification of 57 novel splice junction peptides.39 Neither of these studies identified as many novel junction peptides as one would expect, which might be explained by the low expression level of the novel transcripts and the limited sequence coverage of proteomics data.
2.3. Sequence Variations
Tremendous progress has been made in the identification of disease or drug-response associated DNA sequence variations over the past decade. Validation of these variations at the protein level may lead to novel opportunities for disease diagnosis, prognosis, and treatment. Shotgun proteomics provides a high-throughput solution for the protein-level validation of genomic variations if such information is included in the sequence database used for the search.
An early study by Gatlin et al. used SEQUEST-SNP to identify sequence variations in human hemoglobin proteins.40 Their algorithm dynamically generates all possible SNPs and translates them into peptides for proteomics search. This strategy is only possible for data sets with one or several genes because the number of dynamically introduced variations can grow exponentially with increased number of genes. Several other studies incorporated SNPs derived from EST data to protein databases.31,36
More efforts have been made to enable the identification of protein sequence variations through incorporating genomic variation information from databases such as dbSNP and COSMIC. These works address two key challenges: how to include possible variations into a database and how to control the FDR in the search results with expanded databases.
Bunger et al. presented a refined two-step approach.41 First, LC–MS/MS data are searched against the reference protein database and a separate SNP database created from dbSNP. Next, search results are compared to get reliable SNP-containing peptides. They pointed out that searching for SNP-peptides carry a high risk of false positives due to small mass changes and post-translational modification or peptide modifications that result in similar mass shifts as amino acid substitutions. To control false positives, they proposed two strategies. First, a decoy database can be created by random substitution of reference peptides with similar size. Second, a more stringent match score cutoff can be applied for identifying SNP peptides. The score cutoff can be empirically identified to balance false-positives and false-negatives. Their study identified 36 alternative SNP alleles which were not included in the reference IPI database.
Nijveen et al. designed a Human Short Peptide Variation Database (HSPVdb) dedicated to minor histocompatibility antigens (MiHAs) and demonstrated the value of the database by identifying the majority of published polymorphic SNP or alternative reading frames (ARFs)-derived epitopes in a proteomics study.35 They generated the database by introducing SNPs into corresponding mRNA sequence fragments from RefSeq and then translated them using three reading frames. The database consists of 20-mer peptides. Further improvements were made to remove nonpolymorphic SNPs in dbSNP, which improved the elucidation of MiHAs.
A primary drawback of searching normal database and variant database separately is the loss of competition between normal and variant peptides. A single combined database is preferred because a spectrum that matches well to a peptide in one database may have a better match to a different peptide in another database. This cannot be resolved unless all candidate sequences are considered in a single database.42 Therefore, Su et al. added a “validation” phase after searching spectra against a variation database from SNPs.43
To build a combined database, Schandorff et al. developed MSIPI, in which each IPI protein sequence entry is appended with additional peptide sequences such as theoretical N-terminal peptides and variant peptides from coding SNPs.37 MSIPI allows the identification of N-terminal peptides and of cSNPs in proteomic samples, with an only 10% increase in database size. Along the same line, Xi et al. built a database named SysPIMP that adds human disease-related mutated proteins from OMIM, PMD, and Swiss-Prot to a reference database.44 More recently, we have developed CanProVar, which comprehensively integrates information on protein sequence variations from various public resources, with a focus on cancer-related variations.45 We have also developed a bioinformatics workflow to address several critical challenges in using such databases for identifying variant peptides from shotgun proteomics data, including FDR estimation, efficient storage of variation information, compatibility with different search engines, and result interpretation.46 Applying CanProVar and this workflow to proteomics data sets of human cancer cell lines and tumor samples identified hundreds of variant peptides. More importantly, genomic sequencing confirmed around 90% of the variant peptides randomly selected from the identified ones.
With the aid of the Next Generation Sequencing (NGS) technologies, large amounts of new SNPs and mutations are continually being identified, and the above-mentioned methods are both blessed and cursed. Significantly expanded databases inevitably lead to higher requirements on data storage, longer search time, and higher risk in false identifications. One particularly promising approach is to derive personalized databases for individual samples based on matching DNA or RNA sequencing data.
In an integrative personal omics profiling (iPOP) study, expanding a protein database with variations identified from DNA and RNA sequencing data allowed the identification of variant peptides resulted from single nucleotide variants (SNVs) and RNA edits.48 Using RNA-Seq and shotgun proteomics data from two colorectal cancer cell lines, we showed that customized protein sequence databases derived from RNA-Seq data can enable the detection of known and novel peptide variants.49 In an integrated genomics and proteomics analysis of rat liver, variants derived from genome and transcriptome variation were appended to the ENSEMBL rat database, allowing the detection of variant peptides in the proteomic data.50 Evan et al. used RNA-Seq reads generated from adenovirus-infected human HeLa cells for the de novo assembly of the entire (host and virus) transcriptome and then built a protein database by six-frame translation of the predicted transcripts for proteomics search.51 The proteomics informed by transcriptomics (PIT) technique identified more than 99% of the proteins identified using a traditional protein database with annotated human and adenovirus proteins. These studies demonstrate the great potential of integrative proteogenomic studies for an accurate and comprehensive characterizing of individual proteome.
3. Improving Protein Identification
Inferring proteins from identified peptides is a critical step in shotgun proteomics. Methods have been developed to enhance protein inference by integrating mRNA expression or protein–protein interaction data.
3.1. mRNA Expression
Most protein assembly tools assume that all proteins are equally likely to be present in a sample, even though this assumption is oversimplistic. Ramakrishnan et al. incorporated mRNA abundance estimated from microarray gene expression profiling as prior knowledge of protein presence to improve protein identification in shotgun proteomics experiments.52 Their approach, MSpresso, calculates a protein identification probability by combing direct measure of protein presence from proteomics data and the inferential evidence from microarray data. In their study, the method improves protein identification by ∼40% at a fixed error rate. This work clearly demonstrated the value of incorporating mRNA expression data as prior knowledge in protein identification.
An underlying assumption of the MSpresso approach is a good correlation between mRNA and protein abundance. However, recent studies have shown that mRNA and protein abundance are only moderately correlated. On the basis of a more realistic assumption that mRNA expression is a prerequisite for protein expression, we proposed an alternative method by refining proteomics search space based on RNA-Seq data from the same sample. Specifically, a transcript abundance cutoff is set to remove unexpressed transcripts or lowly expressed transcripts that are unlikely to be detected at the protein level. Using RNA-Seq and shotgun proteomics data from two colorectal cancer cell lines, we showed that this approach not only increases the number of identified protein groups but also the number of identifiable spectra,49 and the latter can help enhance spectral counting-based protein quantification.
3.2. Protein–Protein Interaction
Most biological functions arise from interactions among proteins; however, traditional protein assembly pipelines treat proteins as independent entities. To ensure the reliability of protein identification, these pipelines usually eliminate a large number of possible but nonconfident proteins, including many low-abundant proteins that may be vital for the understanding of biological systems. On the basis of the observation that proteins involved in the same biological process or pathway tend to lie close to one another in the protein–protein interaction network,53 several methods have been developed to improve protein identification by incorporating protein–protein interaction network data. These methods can be broadly classified into three categories: module-based approach, direct neighborhood approach, and diffusion-based approach.
A representative implementation of the module-based approach is the clique-enrichment approach (CEA) developed by our group.54 After protein assembly, all identified proteins are grouped into confident proteins and nonconfident proteins and mapped to a protein–protein interaction network. Network modules defined as fully connected subnetworks (or cliques) are enumerated from the network and evaluated for the enrichment of confident proteins. Nonconfident proteins that coexist in a network module enriched with confident proteins are rescued. In several data sets tested, CEA increased protein identification by 8–23% with an estimated accuracy of 85%.54 Although clique enumeration is used in CEA for the identification of network modules, other network clustering algorithms can be similarly used in the module-based approach.
The direct neighborhood approach considers all direct neighbors of a protein as the neighborhood of the protein. One representative implementation is Software for Network Inference of Proteomics Experiments (SNIPE).55 In this method, spectral counts for all proteins are mapped to their nodes in a network. An updated score for each protein is re-estimated by adding up the scores of the protein and all its immediate neighbors. Permutation is then applied to assess the statistical significance of the updated scores for all proteins. Applying SNIPE to a tooth development data set correctly highlights several proteins that are not normally detected by shotgun proteomics analysis of complex protein samples from whole tissues.55
The diffusion-based approach takes into consideration the global network topological structure. This approach is closely related to Google’s PageRank algorithm. One representative implementation is MSNet.56 The MSNet score for a protein is the convex combination of two terms: the probability that the protein is present in the sample given evidence from a MS experiment, and the weighted average of MSNet scores of the protein’s immediate network neighbors. This is very similar to SNIPE, but in MSNet, the scores are updated iteratively so that evidence from indirect neighbors can be included. Applying MSNet to yeast and human samples increased protein identification by 8–29% and 37%, respectively.56
Previously, we compared the performance of these three approaches through cross-validation using a yeast cell culture data set.54 Our results suggest that the module-based approach is more effective and more robust. As a large number of proteomics data sets are available now, it is worth re-evaluating these methods using multiple data sets. All network-based approaches depend on the network coverage and quality. To increase coverage, one may consider functional association networks instead of protein–protein interaction networks, so that different types of functional relationships can be included in the network. These approaches may also be improved by using condition-specific networks, such as the tissue-specific protein–protein interaction networks. Moreover, in the module-based approach, functional modules can be more broadly defined by Gene Ontology, pathways in different databases, and known protein complexes, etc. A recent study by Goh et al. showed that these functional modules can also be used to improve protein identification in proteomics studies.57
These network and pathway-based approaches not only improve protein identification but also put identified proteins into their functional context.54 In comparative studies, this approach enables comparisons at the network level instead of individual protein level, allowing a systems level understanding of the difference between the samples.
4. Conclusion and Perspectives
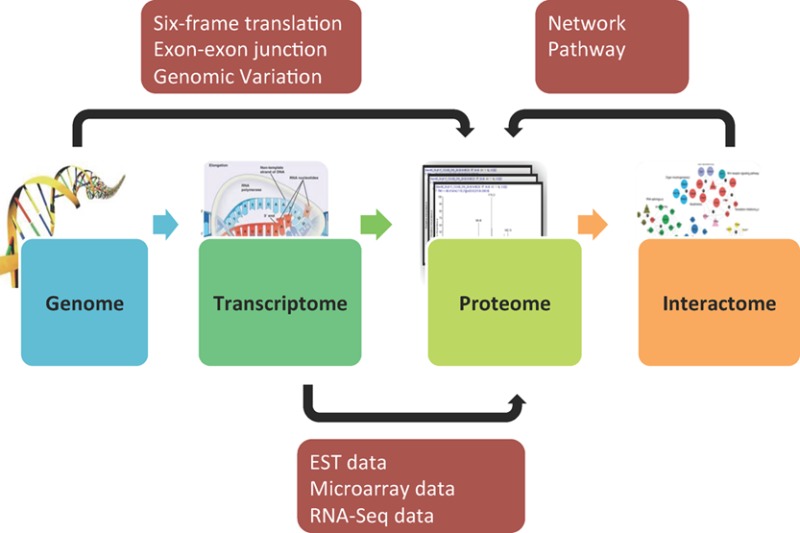
A major goal in proteomics is to comprehensively identify all proteins in biological and clinical samples. Following the information flow from DNA to RNA to protein and functional networks, genomic, transcriptomic, and interactome data can be applied to improve peptide and protein identification in shotgun proteomics (Figure 2).
Figure 2.
Orthogonal data assisted proteomics studies.
Despite substantial improvements in MS/MS data analysis, there remains a large number of unassigned spectra in a typical proteomics study, indicating a large unknown proteome territory.58 This unknown territory can be partly explained by unknown protein coding genes and different types of variations of known protein coding genes. Earlier efforts focus primarily on making databases more comprehensive using publicly available genomic and transcriptomic data. More recently, personalized protein databases derived from sample-specific genomic and transcriptomic data have emerged as an attractive approach.
Figure 3 summarizes major computational approaches to increasing database completeness using publicly available genomic and transcriptomics data. Combining six-frame translation and exon–exon junction predictions can theoretically enumerate all coding potentials of the genome. Further integrating sequence variation data from databases such as dbSNP and COSMIC allows the identification of variant peptides and proteins. Transcriptomics data from EST or RNA-Seq can be used to refine exon–exon junction predictions and filter for sequence variations with transcriptional evidence. Although these approaches can largely increase the completeness of protein databases, significantly expanded search space may introduce enormous background noise, reducing specificity, and sensitivity in peptide identification. In a recent study on ENCODE cell lines,8 shotgun proteomics data from two human cell lines K562 and GM12878 were searched against the GENCODE v7 protein database, the GENCODE v7 transcript-derived protein database, and the six-frame translation of the whole human genome. The GENCODE v7 protein search identified the largest number of peptides, despite of the smallest database size. In contrast, the whole genome search identified the smallest number of peptides. It is worth noting that each search identified a significant number of peptides that were missed by the other two searches, indicating different database constructing strategies are complementary and could be used in a joint way.8
Figure 3.

Methods for increasing database completeness using publicly available genomic and transcriptomic data.
With the recent advancements in DNA and RNA-sequencing technologies, deriving personalized protein databases from sample-specific genomic and transcriptomic data becomes a very attractive strategy. RNA-Seq is of particular interest because of its affordable cost and high information content, including information on novel transcribed regions, novel alternative splicing events, sequence variations resulted from genomic alteration and RNA editing events, and transcript presence and abundance. A sample-specific database taking into consideration all above information can better approximate the real protein pool in the sample and thus improves peptide and protein identification, and tools facilitating such integration, such as customProDB,59 have emerged.
Although the review focuses on using orthogonal data to improve shotgun proteomics studies, these integrative approaches are mutually beneficial. For example, proteomics can help refine genome annotations10,60 and confirm novel alternative splicing events predicted based on RNA-Seq data. Comprehensive identification of all proteins in biological samples can facilitate the reconstruction of sample-specific interactomes. The ability to identify sample-specific protein forms is critical for the emerging field of personalized proteomics, which could complement personalized genomics and lead to novel protein biomarkers and therapeutic targets. More importantly, comprehensive integration of information at DNA, RNA, protein, and network levels, including post-translational modification information that is not discussed in this review, will eventually lead to better understanding of cellular systems, comprehensive catalogue of disease-associated molecular alterations, and novel approaches to correct these alterations. A key to success is the continuous development of computational algorithms and tools that can help translate the large amount of multidimensional data into new knowledge that will eventually improve human health.
Acknowledgments
This work was supported by NIH (http://www.nih.gov/) Grants U24 CA159988, R01 CA126218, R01 GM088822, and contract 13XS029 from Leidos Biomedical Research, Inc.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Nagaraj N.; Wisniewski J. R.; Geiger T.; Cox J.; Kircher M.; Kelso J.; Paabo S.; Mann M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 2011, 7, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins D. N.; Pappin D. J.; Creasy D. M.; Cottrell J. S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20183551–35567. [DOI] [PubMed] [Google Scholar]
- Craig R.; Beavis R. C. TANDEM: matching proteins with tandem mass spectra. Bioinformatics 2004, 2091466–1467. [DOI] [PubMed] [Google Scholar]
- Tabb D. L.; Fernando C. G.; Chambers M. C. MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. J. Proteome Res. 2007, 62654–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z. Q.; Dasari S.; Chambers M. C.; Litton M. D.; Sobecki S. M.; Zimmerman L. J.; Halvey P. J.; Schilling B.; Drake P. M.; Gibson B. W.; Tabb D. L. IDPicker 2.0: Improved protein assembly with high discrimination peptide identification filtering. J. Proteome Res. 2009, 883872–3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotta D. J.; McFarland M. A.; Markey S. P. MassSieve: panning MS/MS peptide data for proteins. Proteomics 2010, 10163035–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii A. I.; Keller A.; Kolker E.; Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75174646–4658. [DOI] [PubMed] [Google Scholar]
- Khatun J.; Yu Y.; Wrobel J. A.; Risk B. A.; Gunawardena H. P.; Secrest A.; Spitzer W. J.; Xie L.; Wang L.; Chen X.; Giddings M. C. Whole human genome proteogenomic mapping for ENCODE cell line data: identifying protein-coding regions. BMC Genomics 2013, 14, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornblihtt A. R.; Schor I. E.; Allo M.; Dujardin G.; Petrillo E.; Munoz M. J. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat. Rev. Mol. Cell Biol. 2013, 143153–165. [DOI] [PubMed] [Google Scholar]
- Brosch M.; Saunders G. I.; Frankish A.; Collins M. O.; Yu L.; Wright J.; Verstraten R.; Adams D. J.; Harrow J.; Choudhary J. S.; Hubbard T. Shotgun proteomics aids discovery of novel protein-coding genes, alternative splicing, and “resurrected” pseudogenes in the mouse genome. Genome Res. 2011, 215756–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing X. B.; Li Q. R.; Sun H.; Fu X.; Zhan F.; Huang X.; Li J.; Chen C. L.; Shyr Y.; Zeng R.; Li Y. X.; Xie L. The discovery of novel protein-coding features in mouse genome based on mass spectrometry data. Genomics 2011, 985343–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitton D. A.; Smith D. L.; Connolly Y.; Scutt P. J.; Miller C. J. An integrated mass-spectrometry pipeline identifies novel protein coding-regions in the human genome. PLoS One 2010, 51e8949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary J. S.; Blackstock W. P.; Creasy D. M.; Cottrell J. S. Interrogating the human genome using uninterpreted mass spectrometry data. Proteomics 2001, 15651–667. [DOI] [PubMed] [Google Scholar]
- Tyers M.; Mann M. From genomics to proteomics. Nature 2003, 4226928193–197. [DOI] [PubMed] [Google Scholar]
- Baudet M.; Ortet P.; Gaillard J. C.; Fernandez B.; Guerin P.; Enjalbal C.; Subra G.; de Groot A.; Barakat M.; Dedieu A.; Armengaud J. Proteomics-based refinement of Deinococcus deserti genome annotation reveals an unwonted use of non-canonical translation initiation codons. Mol. Cell Proteomics 2010, 92415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander E. S.; Linton L. M.; Birren B.; Nusbaum C.; Zody M. C.; Baldwin J.; Devon K.; Dewar K.; Doyle M.; FitzHugh W.; Funke R.; Gage D.; Harris K.; Heaford A.; Howland J.; Kann L.; Lehoczky J.; LeVine R.; McEwan P.; McKernan K.; Meldrim J.; Mesirov J. P.; Miranda C.; Morris W.; Naylor J.; Raymond C.; Rosetti M.; Santos R.; Sheridan A.; Sougnez C.; Stange-Thomann N.; Stojanovic N.; Subramanian A.; Wyman D.; Rogers J.; Sulston J.; Ainscough R.; Beck S.; Bentley D.; Burton J.; Clee C.; Carter N.; Coulson A.; Deadman R.; Deloukas P.; Dunham A.; Dunham I.; Durbin R.; French L.; Grafham D.; Gregory S.; Hubbard T.; Humphray S.; Hunt A.; Jones M.; Lloyd C.; McMurray A.; Matthews L.; Mercer S.; Milne S.; Mullikin J. C.; Mungall A.; Plumb R.; Ross M.; Shownkeen R.; Sims S.; Waterston R. H.; Wilson R. K.; Hillier L. W.; McPherson J. D.; Marra M. A.; Mardis E. R.; Fulton L. A.; Chinwalla A. T.; Pepin K. H.; Gish W. R.; Chissoe S. L.; Wendl M. C.; Delehaunty K. D.; Miner T. L.; Delehaunty A.; Kramer J. B.; Cook L. L.; Fulton R. S.; Johnson D. L.; Minx P. J.; Clifton S. W.; Hawkins T.; Branscomb E.; Predki P.; Richardson P.; Wenning S.; Slezak T.; Doggett N.; Cheng J. F.; Olsen A.; Lucas S.; Elkin C.; Uberbacher E.; Frazier M.; Gibbs R. A.; Muzny D. M.; Scherer S. E.; Bouck J. B.; Sodergren E. J.; Worley K. C.; Rives C. M.; Gorrell J. H.; Metzker M. L.; Naylor S. L.; Kucherlapati R. S.; Nelson D. L.; Weinstock G. M.; Sakaki Y.; Fujiyama A.; Hattori M.; Yada T.; Toyoda A.; Itoh T.; Kawagoe C.; Watanabe H.; Totoki Y.; Taylor T.; Weissenbach J.; Heilig R.; Saurin W.; Artiguenave F.; Brottier P.; Bruls T.; Pelletier E.; Robert C.; Wincker P.; Smith D. R.; Doucette-Stamm L.; Rubenfield M.; Weinstock K.; Lee H. M.; Dubois J.; Rosenthal A.; Platzer M.; Nyakatura G.; Taudien S.; Rump A.; Yang H.; Yu J.; Wang J.; Huang G.; Gu J.; Hood L.; Rowen L.; Madan A.; Qin S.; Davis R. W.; Federspiel N. A.; Abola A. P.; Proctor M. J.; Myers R. M.; Schmutz J.; Dickson M.; Grimwood J.; Cox D. R.; Olson M. V.; Kaul R.; Shimizu N.; Kawasaki K.; Minoshima S.; Evans G. A.; Athanasiou M.; Schultz R.; Roe B. A.; Chen F.; Pan H.; Ramser J.; Lehrach H.; Reinhardt R.; McCombie W. R.; de la Bastide M.; Dedhia N.; Blocker H.; Hornischer K.; Nordsiek G.; Agarwala R.; Aravind L.; Bailey J. A.; Bateman A.; Batzoglou S.; Birney E.; Bork P.; Brown D. G.; Burge C. B.; Cerutti L.; Chen H. C.; Church D.; Clamp M.; Copley R. R.; Doerks T.; Eddy S. R.; Eichler E. E.; Furey T. S.; Galagan J.; Gilbert J. G.; Harmon C.; Hayashizaki Y.; Haussler D.; Hermjakob H.; Hokamp K.; Jang W.; Johnson L. S.; Jones T. A.; Kasif S.; Kaspryzk A.; Kennedy S.; Kent W. J.; Kitts P.; Koonin E. V.; Korf I.; Kulp D.; Lancet D.; Lowe T. M.; McLysaght A.; Mikkelsen T.; Moran J. V.; Mulder N.; Pollara V. J.; Ponting C. P.; Schuler G.; Schultz J.; Slater G.; Smit A. F.; Stupka E.; Szustakowski J.; Thierry-Mieg D.; Thierry-Mieg J.; Wagner L.; Wallis J.; Wheeler R.; Williams A.; Wolf Y. I.; Wolfe K. H.; Yang S. P.; Yeh R. F.; Collins F.; Guyer M. S.; Peterson J.; Felsenfeld A.; Wetterstrand K. A.; Patrinos A.; Morgan M. J.; de Jong P.; Catanese J. J.; Osoegawa K.; Shizuya H.; Choi S.; Chen Y. J. Initial sequencing and analysis of the human genome. Nature 2001, 4096822860–921. [DOI] [PubMed] [Google Scholar]
- Risk B. A.; Spitzer W. J.; Giddings M. C. Peppy: proteogenomic search software. J. Proteome Res. 2013, 1263019–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevinsky J. R.; Cargile B. J.; Bunger M. K.; Meng F.; Yates N. A.; Hendrickson R. C.; Stephenson J. L. Jr. Whole genome searching with shotgun proteomic data: applications for genome annotation. J. Proteome Res. 2008, 7180–88. [DOI] [PubMed] [Google Scholar]
- Fermin D.; Allen B. B.; Blackwell T. W.; Menon R.; Adamski M.; Xu Y.; Ulintz P.; Omenn G. S.; States D. J. Novel gene and gene model detection using a whole genome open reading frame analysis in proteomics. Genome Biol. 2006, 74R35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Casado G.; Covey P. A.; Bedinger P. A.; Mueller L. A.; Thannhauser T. W.; Zhang S.; Fei Z.; Giovannoni J. J.; Rose J. K. Enabling proteomic studies with RNA-Seq: The proteome of tomato pollen as a test case. Proteomics 2012, 126761–774. [DOI] [PubMed] [Google Scholar]
- Armengaud J. Microbiology and proteomics, getting the best of both worlds!. Environ. Microbiol 2013, 15112–23. [DOI] [PubMed] [Google Scholar]
- Song J.; Sun R.; Li D.; Tan F.; Li X.; Jiang P.; Huang X.; Lin L.; Deng Z.; Zhang Y. An improvement of shotgun proteomics analysis by adding next-generation sequencing transcriptome data in orange. PLoS One 2012, 76e39494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohien C. U.; Colquhoun D. R.; Mathias D. K.; Gibbons J. G.; Armistead J. S.; Rodriguez M. C.; Rodriguez M. H.; Edwards N. J.; Hartler J.; Thallinger G. G.; Graham D. R.; Martinez-Barnetche J.; Rokas A.; Dinglasan R. R. A bioinformatics approach for integrated transcriptomic and proteomic comparative analyses of model and non-sequenced anopheline vectors of human malaria parasites. Mol. Cell Proteomics 2013, 121120–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S.; Davis C. A.; Merkel A.; Dobin A.; Lassmann T.; Mortazavi A.; Tanzer A.; Lagarde J.; Lin W.; Schlesinger F.; Xue C.; Marinov G. K.; Khatun J.; Williams B. A.; Zaleski C.; Rozowsky J.; Roder M.; Kokocinski F.; Abdelhamid R. F.; Alioto T.; Antoshechkin I.; Baer M. T.; Bar N. S.; Batut P.; Bell K.; Bell I.; Chakrabortty S.; Chen X.; Chrast J.; Curado J.; Derrien T.; Drenkow J.; Dumais E.; Dumais J.; Duttagupta R.; Falconnet E.; Fastuca M.; Fejes-Toth K.; Ferreira P.; Foissac S.; Fullwood M. J.; Gao H.; Gonzalez D.; Gordon A.; Gunawardena H.; Howald C.; Jha S.; Johnson R.; Kapranov P.; King B.; Kingswood C.; Luo O. J.; Park E.; Persaud K.; Preall J. B.; Ribeca P.; Risk B.; Robyr D.; Sammeth M.; Schaffer L.; See L. H.; Shahab A.; Skancke J.; Suzuki A. M.; Takahashi H.; Tilgner H.; Trout D.; Walters N.; Wang H.; Wrobel J.; Yu Y.; Ruan X.; Hayashizaki Y.; Harrow J.; Gerstein M.; Hubbard T.; Reymond A.; Antonarakis S. E.; Hannon G.; Giddings M. C.; Ruan Y.; Wold B.; Carninci P.; Guigo R.; Gingeras T. R. Landscape of transcription in human cells. Nature 2012, 4897414101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K. A.; Ringler P.; Gu L.; Shen Q. J. RNA-sequencing reveals previously unannotated protein- and microRNA-coding genes expressed in aleurone cells of rice seeds. Genomics 2013, 103, 122–134. [DOI] [PubMed] [Google Scholar]
- Woo S.; Cha S. W.; Merrihew G.; He Y.; Castellana N.; Guest C.; Maccoss M.; Bafna V. Proteogenomic Database Construction Driven from Large Scale RNA-seq Data. J. Proteome Res. 2013, 13, 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakharkar M. K.; Chow V. T.; Kangueane P. Distributions of exons and introns in the human genome. In Silico Biol. 2004, 44387–393. [PubMed] [Google Scholar]
- Fei S. S.; Wilmarth P. A.; Hitzemann R. J.; McWeeney S. K.; Belknap J. K.; David L. L. Protein database and quantitative analysis considerations when integrating genetics and proteomics to compare mouse strains. J. Proteome Res. 2011, 1072905–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo F.; Hong X.; Gao F.; Du L.; Wang J.; Omenn G. S.; Lin B. A compatible exon-exon junction database for the identification of exon skipping events using tandem mass spectrum data. BMC Bioinformatics 2008, 9, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power K. A.; McRedmond J. P.; de Stefani A.; Gallagher W. M.; Gaora P. O. High-throughput proteomics detection of novel splice isoforms in human platelets. PLoS One 2009, 43e5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner S.; Shen Z.; Ng J.; Florea L.; Guigo R.; Briggs S. P.; Bafna V. Improving gene annotation using peptide mass spectrometry. Genome Res. 2007, 172231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim P.; Kim N.; Lee Y.; Kim B.; Shin Y.; Lee S. ECgene: genome annotation for alternative splicing. Nucleic Acids Res. 2005, 33Database issueD75–D79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon R.; Zhang Q.; Zhang Y.; Fermin D.; Bardeesy N.; DePinho R. A.; Lu C.; Hanash S. M.; Omenn G. S.; States D. J. Identification of novel alternative splice isoforms of circulating proteins in a mouse model of human pancreatic cancer. Cancer Res. 2009, 691300–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon R.; Omenn G. S. Proteomic characterization of novel alternative splice variant proteins in human epidermal growth factor receptor 2/neu-induced breast cancers. Cancer Res. 2010, 7093440–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijveen H.; Kester M. G. D.; Hassan C.; Viars A.; de Ru A. H.; de Jager M.; Falkenburg J. H. F.; Leunissen J. A. M.; van Veelen P. A. HSPVdb-the Human Short Peptide Variation Database for improved mass spectrometry-based detection of polymorphic HLA-ligands. Immunogenetics 2011, 633143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards N. J. Novel peptide identification from tandem mass spectra using ESTs and sequence database compression. Mol. Syst. Biol. 2007, 3, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schandorff S.; Olsen J. V.; Bunkenborg J.; Blagoev B.; Zhang Y.; Andersen J. S.; Mann M. A mass spectrometry-friendly database for cSNP identification. Nat. Methods 2007, 46465–6. [DOI] [PubMed] [Google Scholar]
- Ning K.; Nesvizhskii A. I. The utility of mass spectrometry-based proteomic data for validation of novel alternative splice forms reconstructed from RNA-Seq data: a preliminary assessment. BMC Bioinformatics 2010, 11Suppl 11S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheynkman G. M.; Shortreed M. R.; Frey B. L.; Smith L. M. Discovery and mass spectrometric analysis of novel splice-junction peptides using RNA-Seq. Mol. Cell Proteomics 2013, 12, 2341–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatlin C. L.; Eng J. K.; Cross S. T.; Detter J. C.; Yates J. R. 3rd Automated identification of amino acid sequence variations in proteins by HPLC/microspray tandem mass spectrometry. Anal. Chem. 2000, 724757–763. [DOI] [PubMed] [Google Scholar]
- Bunger M. K.; Cargile B. J.; Sevinsky J. R.; Deyanova E.; Yates N. A.; Hendrickson R. C.; Stephenson J. L. Jr. Detection and validation of non-synonymous coding SNPs from orthogonal analysis of shotgun proteomics data. J. Proteome Res. 2007, 662331–2340. [DOI] [PubMed] [Google Scholar]
- Elias J. E.; Gygi S. P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 43207–214. [DOI] [PubMed] [Google Scholar]
- Su Z. D.; Sun L.; Yu D. X.; Li R. X.; Li H. X.; Yu Z. J.; Sheng Q. H.; Lin X.; Zeng R.; Wu J. R. Quantitative detection of single amino acid polymorphisms by targeted proteomics. J. Mol. Cell Biol. 2011, 35309–315. [DOI] [PubMed] [Google Scholar]
- Xi H.; Park J.; Ding G.; Lee Y. H.; Li Y. SysPIMP: the web-based systematical platform for identifying human disease-related mutated sequences from mass spectrometry. Nucleic Acids Res. 2009, 37Database issueD913–D920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Duncan D. T.; Zhang B. CanProVar: a human cancer proteome variation database. Hum. Mutat. 2010, 313219–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Su Z.; Ma Z. Q.; Slebos R. J.; Halvey P.; Tabb D. L.; Liebler D. C.; Pao W.; Zhang B. A bioinformatics workflow for variant peptide detection in shotgun proteomics. Mol. Cell Proteomics 2011, 105M110 006536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth M. J.; Forbes A. J.; Boyne M. T. 2nd; Kim Y. B.; Robinson D. E.; Kelleher N. L. Precise and parallel characterization of coding polymorphisms, alternative splicing, and modifications in human proteins by mass spectrometry. Mol. Cell Proteomics 2005, 471002–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R.; Mias G. I.; Li-Pook-Than J.; Jiang L.; Lam H. Y.; Miriami E.; Karczewski K. J.; Hariharan M.; Dewey F. E.; Cheng Y.; Clark M. J.; Im H.; Habegger L.; Balasubramanian S.; O’Huallachain M.; Dudley J. T.; Hillenmeyer S.; Haraksingh R.; Sharon D.; Euskirchen G.; Lacroute P.; Bettinger K.; Boyle A. P.; Kasowski M.; Grubert F.; Seki S.; Garcia M.; Whirl-Carrillo M.; Gallardo M.; Blasco M. A.; Greenberg P. L.; Snyder P.; Klein T. E.; Altman R. B.; Butte A. J.; Ashley E. A.; Gerstein M.; Nadeau K. C.; Tang H.; Snyder M. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell 2012, 14861293–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Slebos R. J.; Wang D.; Halvey P. J.; Tabb D. L.; Liebler D. C.; Zhang B. Protein identification using customized protein sequence databases derived from RNA-Seq data. J. Proteome Res. 2012, 1121009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low T. Y.; van Heesch S.; van den Toorn H.; Giansanti P.; Cristobal A.; Toonen P.; Schafer S.; Hubner N.; van Breukelen B.; Mohammed S.; Cuppen E.; Heck A. J.; Guryev V. Quantitative and qualitative proteome characteristics extracted from in-depth integrated genomics and proteomics analysis. Cell Rep. 2013, 551469–1478. [DOI] [PubMed] [Google Scholar]
- Evans V. C.; Barker G.; Heesom K. J.; Fan J.; Bessant C.; Matthews D. A. De novo derivation of proteomes from transcriptomes for transcript and protein identification. Nat. Methods 2012, 9121207–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan S. R.; Vogel C.; Prince J. T.; Li Z.; Penalva L. O.; Myers M.; Marcotte E. M.; Miranker D. P.; Wang R. Integrating shotgun proteomics and mRNA expression data to improve protein identification. Bioinformatics 2009, 25111397–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharan R.; Ulitsky I.; Shamir R. Network-based prediction of protein function. Mol. Syst. Biol. 2007, 3, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Zimmerman L. J.; Park B. H.; Tabb D. L.; Liebler D. C.; Zhang B. Network-assisted protein identification and data interpretation in shotgun proteomics. Mol. Syst. Biol. 2009, 5, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusinow D. P.; Kiezun A.; O’Connell D. J.; Chick J. M.; Yue Y.; Maas R. L.; Gygi S. P.; Sunyaev S. R. Network-based inference from complex proteomic mixtures using SNIPE. Bioinformatics 2012, 28233115–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan S. R.; Vogel C.; Kwon T.; Penalva L. O.; Marcotte E. M.; Miranker D. P. Mining gene functional networks to improve mass-spectrometry-based protein identification. Bioinformatics 2009, 25222955–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh W. W.; Sergot M. J.; Sng J. C.; Wong L. Comparative network-based recovery analysis and proteomic profiling of neurological changes in valproic acid-treated mice. J. Proteome Res. 2013, 1252116–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning K.; Fermin D.; Nesvizhskii A. I. Computational analysis of unassigned high-quality MS/MS spectra in proteomic data sets. Proteomics 2010, 10142712–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Zhang B. customProDB: an R package to generate customized protein databases from RNA-Seq data for proteomics search. Bioinformatics 2013, 29243235–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascoigne D. K.; Cheetham S. W.; Cattenoz P. B.; Clark M. B.; Amaral P. P.; Taft R. J.; Wilhelm D.; Dinger M. E.; Mattick J. S. Pinstripe: a suite of programs for integrating transcriptomic and proteomic datasets identifies novel proteins and improves differentiation of protein-coding and non-coding genes. Bioinformatics 2012, 28233042–3050. [DOI] [PubMed] [Google Scholar]
- Menschaert G.; Van Criekinge W.; Notelaers T.; Koch A.; Crappe J.; Gevaert K.; Van Damme P. Deep proteome coverage based on ribosome profiling aids MS-based protein and peptide discovery and provides evidence of alternative translation products and near-cognate translation initiation events. Mol. Cell Proteomics 2013, 12, 1780–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheynkman G. M.; Shortreed M. R.; Frey B. L.; Scalf M.; Smith L. M. Large-scale mass spectrometric detection of variant peptides resulting from nonsynonymous nucleotide differences. J. Proteome Res. 2014, 131228–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh W. W.; Lee Y. H.; Zubaidah R. M.; Jin J.; Dong D.; Lin Q.; Chung M. C.; Wong L. Network-based pipeline for analyzing MS data: an application toward liver cancer. J. Proteome Res. 2011, 1052261–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]