

Abstract
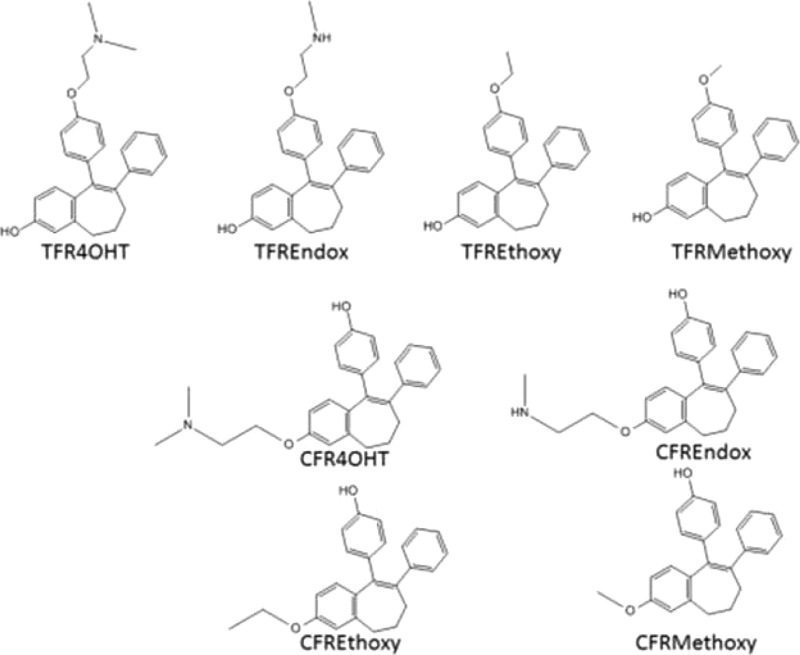
Tamoxifen has biologically active metabolites: 4-hydroxytamoxifen (4OHT) and endoxifen. The E-isomers are not stable in solution as Z-isomerization occurs. We have synthesized fixed ring (FR) analogues of 4OHT and endoxifen as well as FR E and Z isomers with methoxy and ethoxy side chains. Pharmacologic properties were documented in the MCF-7 cell line, and prolactin synthesis was assessed in GH3 rat pituitary tumor cells. The FR Z-isomers of 4OHT and endoxifen were equivalent to 4OHT and endoxifen. Other test compounds used possessed partial estrogenic activity. The E-isomers of FR 4OHT and endoxifen had no estrogenic activity at therapeutic serum concentrations. None of the newly synthesized compounds were able to down-regulate ER levels. Molecular modeling demonstrated that some compounds would each create a best fit with a novel agonist conformation of the ER. The results demonstrate modulation by the ER complex of cell replication or gene transcription in cancer.
Introduction
Tamoxifen remains an important, lifesaving medicine for the adjuvant treatment of early stage breast cancer.1−3 It is listed as an essential medicine in oncology by the World Health Organization and is available to prevent breast cancer in high risk women in both the United States and United Kingdom. The continued use of tamoxifen has profound effect on public health worldwide. For these reasons, it is appropriate to study the molecular pharmacology of tamoxifen and its metabolites and analogues. Indeed, the fact that tamoxifen will most likely be administered for 10 or more years for the treatment of breast cancer,3 and there is a role for long-term tamoxifen treatment in the prevention of breast cancer in healthy women,4 reinforces the value of understanding the molecular pharmacology of the medicine.
Tamoxifen ((Z)-1-(p-β-dimethylaminoethoxyphenyl-1,2-diphenylbut-1-ene) is the antiestrogenic Z-isomer of an estrogenic substituted triphenylethylene5 that is converted at the 4-position6 to two hydroxylated metabolites 4-hydroxytamoxifen (4OHT)7 and 4-hydroxy-N-desmethyltamoxifen (endoxifen),8,9 both of which have high binding affinity for the estrogen receptor (ER) found in estrogen target tissues or hormone-dependent tumors.7,10,11 The metabolites have similar pharmacology and activate or depress a similar gene profile in vitro.11−13 An interesting aspect of tamoxifen and its isomers is that the E-isomer (ICI 47 699) of tamoxifen (ICI 46 474) is an estrogen in vivo5 and in vitro.14 By contrast, the E-isomer of 4OHT is unstable and isomerizes to a mixture of E- and Z-isomers, displaying antiestrogenic activity both in vivo15 and in vitro.14 Subsequent studies examined fixed ring (FR) derivatives of the E- and Z-isomers of 4OHT16 using a previously reported synthetic pathway.17 The E-isomer was a weak antiestrogen.
We synthesized a series of FR analogues of the alkylaminoethoxy side chain of 4OHT to link molecular modeling with cell replication in breast cancer (MCF-7:WS8) and prolactin synthesis in the rat pituitary gland cancer cell line GH3. We took these approaches to study structure–function relationships: reducing the antiestrogenic side chain of 4OHT and comparing results with bisphenol (BPTPE) and trihydroxytriphenylethylene (3OHTPE),18 comparing E and Z FR isomers and finally the length and bulk of the antiestrogenic side chain of E-isomer of FR4OHT (EFR4OHT). Select ER-responsive genes (pS2, GREB1, and PgR) were measured following 48 h of incubation of all test compounds with MCF-7:WS8 cells as well as ER levels determined by Western blotting. Also we evaluated the impact of therapeutic concentrations of E-isomers of FR4OHT and FR endoxifen (FREndox) alone or in combination with therapeutic levels of Z-isomers on the growth of MCF-7:WS8 breast cancer cell line to estimate therapeutic relevance during breast cancer treatment with tamoxifen for tumor cell growth control by putative estrogenic metabolites.
Results
Chemistry
Five novel FR4OHT analogues (ZFRMethoxy, ZFREthoxy, EFREthoxy, ZFREndox, and EFREndox) were synthesized in a multistep sequence involving a Gringard reaction of a protected p-bromophenol with a substituted benzosuberone. Subsequent modifications provided two key intermediates that have a methoxy or heptafluorotolyl (HFT) protecting group on either of the phenolic groups. This versatile scaffold was important for the synthesis of the E- and Z-isomers of FRMethoxy, FREthoxy, FR4OHT, and FREndox compounds. Both isomers of FR4OHT (24 and 29 in Scheme 1) and CFRMethoxy (20) were synthesized according to McCague et al.,17 while compounds 3OHTPE and BPTPE were synthesized according to Maximov et al.18
Scheme 1. Synthesis of the Isomerically Stable Fixed Ring (FR) Isomers of Methoxy, Ethoxy Substituted Triphenylethylene Derivatives and Fixed Ring 4OHT and Endox.

Z Fixed Ring Methoxy. E and Z Fixed Ring Ethoxy Analogues (ZFRMethoxy and ZFR/EFREthoxy)
2-Methoxyheptenone (benzosuberone) was demethylated to 1 (Scheme 1) by refluxing with aluminum chloride in toluene.19,20 Phenol 1 was protected with octafluorotoluene using phase transfer reaction conditions to 2 or converted to the ethoxy analogue 3 using ethyl iodide and potassium carbonate in acetone. Both compounds were treated with the Grignard reagent of a protected p-bromophenol that resulted in the formation of the ethoxycycloheptene 5 and the methoxy analogue 6.17 For compounds 7 and 8, 4-bromophenyl 2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenyl ether 4 was obtained by the method of Jarman and McCague.21 This compound was converted to the Grignard reagent and reacted with suberone 3 which yielded 7 or reacted with 2-methoxybenzosuberone which led to 8. Bromine was introduced at the 8-position using pyridine hydrobromide perbromide (9–12) that was subsequently replaced with a phenyl moiety upon treatment with phenylzinc chloride and a palladium catalyst yielding compounds 13–16. These key intermediates were selectively deprotected to provide either E- or Z-isomer of FRMethoxy (18 and20) and FREthoxy (17 and 19) tamoxifen analogues.
E Fixed Ring Endoxifen (EFREndox)
The synthesis of EFREndox 27 was first attempted by selective demethlyation of EFR4OHT 24 using 1-chloroethyl chloroformate both with and without magnesium oxide,22 as well as vinyl chloroformate23 with no formation of product detected by LC/MS. In addition, demethylation using ruthenium chloride in methanol followed by treatment with hydrogen peroxide was also tried without success.24 Also, the attempted direct methyl amination of chloroethoxybenzocycloheptene 22 by heating with 33% methylamine in ethanol failed. Z-isomer of 4OHT (model compound) was converted to its N-oxide by stirring with 30% hydrogen peroxide in acetone but did not demethylate using selenium oxide.25 Alternatively, we investigated several methods for attaching the protected ethylamine side chain directly onto phenol 21. Methods included reaction of 21 with ethyl (2-bromoethyl)(methyl)carbamate22 by heating with cesium carbonate in DMF, heating with sodium hydride in DMF, and using phase transfer reaction conditions. All produced 25 in various yields with the last method giving the best overall yield. In the next step, the heptylfluorotolyl protecting group was removed using sodium methoxide in DMF to 26, followed by removal of the carbamate with pyridine HCl to 27. A faster route to EFREndox 27 could be achieved by starting with 14, where both protecting groups would be removed concurrently.
Z Fixed Ring Endoxifen (ZFREndox)
The synthesis of TFREndox 31 was attempted with selective demethlyation of the Z tamoxifen 29 using vinyl chloroformate23 with no product formation. The ZFREndox compound 30 was synthesized by heating compound 20 with ethyl (2-bromoethyl)(methyl)carbamate23 and cesium carbonate in DMF, but the reaction was not as efficient as using ethyl (2-hydroxyethyl)(methyl)carbamate,23 TPP, and DIAD in THF. Both the methoxide and carbamate protecting groups of 30 were removed simultaneously by heating with pyrdine·HCl to give ZFREndox 31.
Pharmacology
To assess estrogenic and antiestrogenic properties of the test compounds, we employed a DNA quantification assay with the ER positive human breast cancer cell line MCF-7:WS8 as described in Materials and Methods and have compared the results with the test compounds with previously described angular estrogens BPTPE and 3OHTPE.18 Estradiol (E2) induced growth of cells (Figure 2A) in a concentration-dependent manner with maximal stimulation starting at a concentration of 10–11 M. All of the test compounds are partial agonists and do not reach the same level of growth induction as with E2. It is therefore not appropriate to calculate EC50 against E2. However, they do cluster by their levels of growth induction. Compounds BPTPE, ZFRMethoxy, ZFREthoxy, and EFR4OHT induce the same levels of growth of MCF-7:WS8 cells at a concentration of 10–6 M with no statistical difference (P < 0.05). Thus, we estimated the potency of these compounds by comparing their EC50 concentrations (Figure 1). The results demonstrate that BPTPE is a much more potent partial agonist in MCF-7:WS8 cells (EC50 of 1.5 × 10–11 M) than other test compounds in this group (Figure 1). The ZFRMethoxy and ZFREthoxy compounds with the shortest side chains have EC50 of 3 × 10–10 M, while EFR4OHT compound has the highest EC50 in this group of compounds of 1.5 × 10–8 M (Figure 1). The next group of compounds (EFRMethoxy, EFREthoxy, and EFREndox) induce cell growth a little higher but statistically more significantly than the previous group (P < 0.05), so their EC50 concentrations can be estimated between these compounds (Figure 1). EFRMethoxy compound has an EC50 of 4 × 10–9 M, while EFREthoxy has EC50 of 2.7 × 10–9 M and EFREndox has EC50 of 2 × 10–8 M. The ZFR4OHT and ZFREndoxifen, like the structurally similar Z-4OHT and endoxifen, have no estrogenic properties over the whole concentration range of 10–12–10–6 M (Figure 1) (P > 0.05 for all concentration points when compared to each of their respective vehicle controls). Estrogenic properties on the growth of MCF-7:WS8 cells of 3OHTPE were previously described18 and are not shown here. The EC50 of 1.5 × 10–10 M is similar to that of BPTPE.
Figure 2.
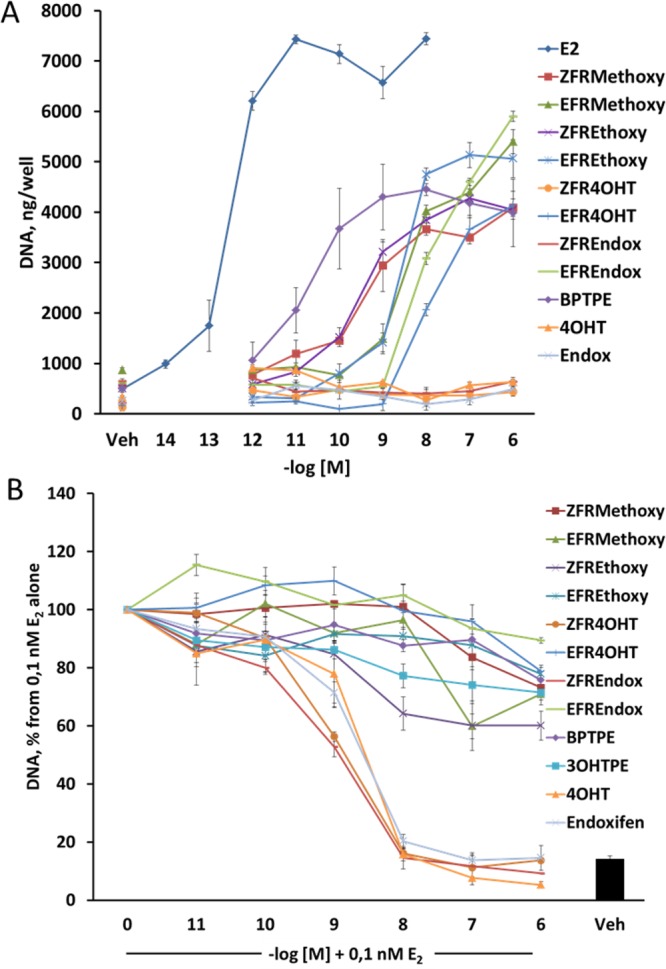
Assessment of estrogenic/antiestrogenic properties of the test compounds in MCF-7:WS8 ER-positive human breast cancer cell lines: (A) treatments of the MCF-7:WS8 cells with compounds alone; (B) treatments of MCF-7:WS8 cells with compounds in combination with 10–10 M E2. All DNA content was normalized to a corresponding 10–10 M E2 control of each of the experiments.
Figure 1.

Structures of compounds used in the current study that were purchased (E2, 4OHT, and Endox) or not synthesized (BPTPE and 3OHTPE).18
To test the antiestrogenic properties of test compounds, we employed the same DNA based growth assay with combination treatments with 10–10 M E2. The Z-isomers of the FR4OHT and FREndox produce an equivalent antiestrogenic effect (average IC50 of 3 × 10–9 M in MCF7:WS8 cells) inhibiting 10–10 M E2 completely (P > 0.05 at 10–6 M points when compared to vehicle control) like 4OHT and endoxifen (Figure 1B). ZFRMethoxy, ZFREthoxy, EFRMethoxy, EFREthoxy, EFR4OHT, ECFREndox, BPTPE, and 3OHTPE compounds all have very weak antiestrogenic properties (Figure 2B), inhibiting E2-stimulated cell growth by about 20% at top concentration (P < 0.05 compared to control); however, the ZFREthoxy compound seems to have a little more antiestrogenic properties than the rest of the group by about 20% (P < 0.05 at 10–6 M), and EFREndox inhibits only by about 10% compared to vehicle control (P < 0.05). All this is consistent with the intrinsic activity of test compounds alone (Figure 2A).
MCF-7:WS8 cells were treated with therapeutic concentrations of E- and Z-isomers of FR4OHT and endoxifen found in postmenopausal breast cancer patients treated with tamoxifen.26 Results show that pharmacological concentrations of tested E-isomers alone or in combination with Z-isomers were not able to induce significant cell growth (P > 0.05 compared to control), compared to cell proliferation induced by postmenopausal levels of estrogens (E1/E2) found in postmenopausal women taking tamoxifen (Figure 3) (P < 0.05 compared to control). The concentrations of estrogens corresponding to average levels of estrogens in postmenopausal women were 7.8 × 10–11 M for E1 and 4.7 × 10–11 M for E2 and were obtained from pervious publications.27,28 The levels for the test compounds corresponding to mean therapeutic levels of tamoxifen metabolites in breast cancer patients taking tamoxifen were the following: ZFR4OHT, 5.81 × 10–9 M; ZFREndox, 29.1 × 10–9 M; EFR4OHT, 0.56 × 10–9 M; EFREndox, 1.17 × 10–9 M.26
Figure 3.

Assessment of estrogenic properties of different stable isomers of tamoxifen’s metabolites 4OHT and endoxifen in MCF-7:WS8 at average therapeutic concentrations.26 The levels for the tested compounds corresponding to mean therapeutic levels of tamoxifen metabolites were the following: ZFR4OHT, 5.81 × 10–9 M; ZFREndox, 29.1 × 10–9 M; EFR4OHT, 5.6 × 10–9 M; EFREndox, 1.17 × 10–9 M.
Real-Time PCR
To assess the pharmacological properties the test compounds on estrogen responsive genes, we used real-time polymerase chain reaction (RT-PCR) in the ER positive rat pituitary tumor cell line GH3 to assess the modulation of the prolactin gene (Prl) and also in estrogen-responsive genes pS2, progesterone receptor (PgR), and GREB1 in MCF7:WS8 cells. All cells were first estrogen starved and then processed as described in Materials and Methods. Results of the Prl gene expression analysis show that the Prl gene in rat GH3 cells has elevated expression of mRNA in response to E2 in a concentration-dependent manner (Figure 4A) with maximal stimulation at 10–9 M (P < 0.05 compared to control). All of the test compounds had shallow partial agonist dose–response curves (Figure 4A). As a result of the inability of test compounds to induce maximal Prl gene actions higher than 40% of E2, it is inappropriate to estimate EC50. In combination with 1 nM E2 all test compounds exhibited antiestrogenic properties; however, only ZFR4OHT, ZFREndox, and 4OHT were able to completely inhibit 1 nM E2-induced Prl gene up-regulation to control levels at their top concentration of 10–6 M (P > 0.05) (Figure 4B). All other test compounds inhibited the effects of 1 nM E2 and the levels of the intrinsic activity of compounds alone (Figure 4B).
Figure 4.
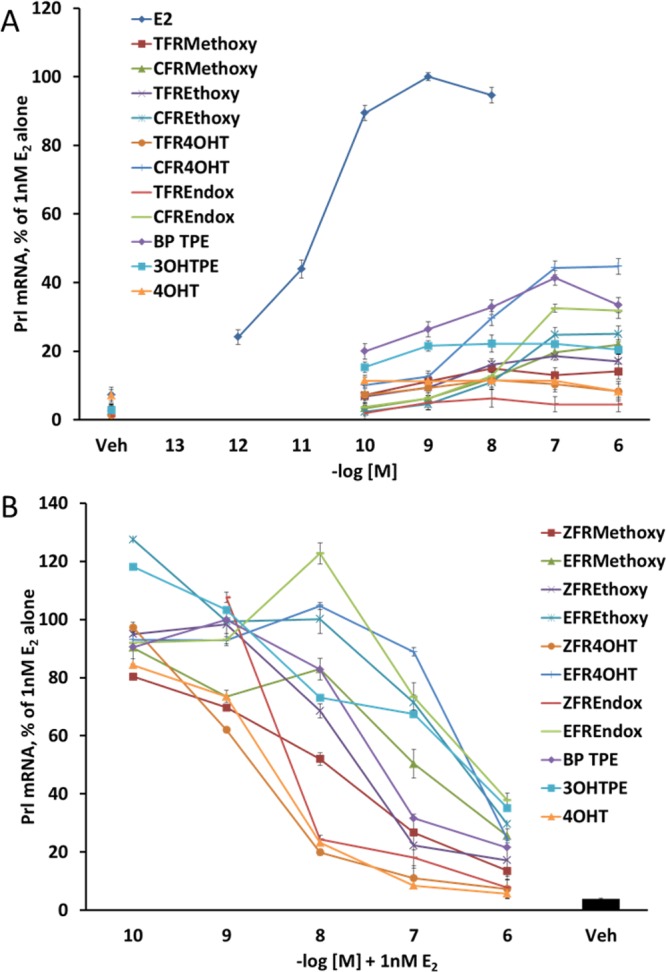
Assessment of estrogenic/antiestrogenic properties of the test compounds on inducing prolactin (Prl) gene’s mRNA expression in GH3 rat pituitary tumor cells. (A) Treatments of the GH3 cells with compounds alone. The fold change of the mRNA was first calculated using the ΔΔCt method. The corresponding 10–10 M E2 control values were considered as 100%, and all other treatments were calculated accordingly. (B) Treatments of the GH3 cells with compounds in combination with 10–9 M E2. The fold change of the mRNA was first calculated using the ΔΔCt method. The corresponding 10–10 M E2 control values were considered as 100%, and all other treatments were calculated accordingly.
RT-PCR of estrogen regulated genes pS2, GREB1, and PgR in MCF-7:WS8 cells treated with test compounds show a differential effect based on the structure of the ligands. Estradiol (10–10 M) induced expression of all test genes compared to vehicle control (Figure 5) after 48 h of treatment (P < 0.05 for all genes). Treatments with 3OHTPE and BPTPE produced a partial estrogenic effect on all genes (P < 0.05 when comparing to E2 treatment or vehicle control) and no significant difference between each other (P > 0.05) in any of the genes. Treatments with isomers of FRMethoxy and FREthoxy compounds produced partial estrogenic effects in all estrogen-responsive genes (P < 0.05 when compared to vehicle control); however, E-isomers were able to produce a higher induction of expression in all studied genes compared with corresponding Z-isomers (P < 0.05). ZFR4OHT, ZFREndox, 4OHT, and Endox produced no significant effect on mRNA synthesis in pS2 and GREB1 genes (P > 0.05 when compared to vehicle control) and were similar to each other (P > 0.05) but did induce 3- to 4-fold increase in PgR mRNA levels (Figure 5C) compared to vehicle control (P < 0.05). EFR4OHT and EFREndox compounds were able to induce expression of all genes investigated (Figure 5), significantly higher than their Z-isomers (P < 0.05). Higher than therapeutic concentrations of test compounds, in particular isomers of FR4OHT and FREndox, were chosen to demonstrate their ability to regulate estrogen responsive genes at concentrations consistent with their inhibitory effects on the estrogen-induced cell proliferation (Figure 2B).
Figure 5.

Assessment of estrogenic/antiestrogenic properties of the test compounds on inducing estrogen-responsive gene’s mRNA expression in MCF-7:WS8 breast cancer cell line: (A) pS2 gene; (B) GREB1 gene; (C) PgR gene. Treatment with E2 was made at 10–10 M. All of the other test compounds were treated at 10–6 M. The fold change of the mRNA was first calculated using the ΔΔCt method. Corresponding 10–10 M E2 control values were considered as 100%, and all other treatments were calculated accordingly.
Immunoblotting
Immunoblotting was performed to assess the impact of the test compounds on the regulation of the ERα protein levels in MCF-7:WS8 cells. We starved the cells in the same way as estrogen starvation for cell proliferation assays. After 24 h of treatment with compounds, cells were harvested and processed as described in Materials and Methods. Results showed that 1 nM E2 reduces the level of ERα by about 60% as measured by densitometry. In contrast, 4OHT and endoxifen and their ZFR analogues all caused an up-regulation of the ERα protein. The estrogen-like E-isomers of FR4OHT and FREndox did not induce the down-regulation of the protein. Fulvestrant (ICI), which degrades ERα, was used as a positive control and was able to down-regulate the ERα by more than 90%. Interestingly, compounds with shorter side chains like FRMethoxy and FREthoxy E and Z isomers and BPTPE and 3OHTPE were not able to induce any degradation of the ERα like E2, despite their estrogenic properties in these cells, and actually up-regulated the protein levels (Figure 6).
Figure 6.
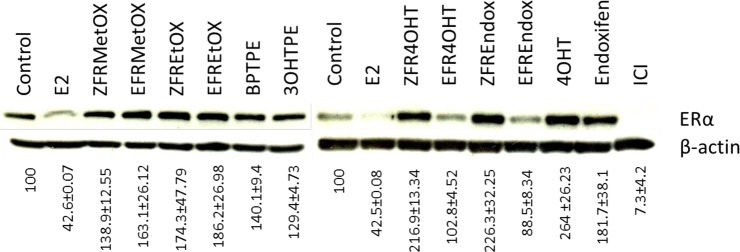
Immunoblotting results for test compounds after a 24 h treatment of MCF-7:WS8 breast cancer cells. Percent of control was calculated by comparison with the actin band. Immunoblotting experiments were repeated three times.
Molecular Modeling
To study the binding mode of FR derivatives of endoxifen and 4OHT in the ER binding pocket, flexible docking simulations were carried out against both conformations of ER ligand-binding domain (LBD), agonist (PDB codes 1GWR (ER LBD cocrystallized with E2),293ERD (ER LBD cocrystallized with DES),303Q97 (ER LBD cocrystallized with ethoxytriphenylethylene isomers),31 and antagonist (PDB codes 3ERT (ER LBD cocrystallized with 4OHT),301UOM (ER LBD cocrystallized with 2-phenyl-1-[4-(2-piperidin-1-ylethoxy)phenyl]-1,2,3,4-tetrahydroisoquinolin-6-ol, PTI),322OUZ (ER LBD cocrystallized with lasofoxifene)33). The X-ray structures to be used for docking were selected based on the shape similarity between the investigated compounds and cocrystallized ligands of ER LBD complexes from PDB. In the following, the most relevant results obtained in docking simulations run against antagonist conformation 3ERT (Figure 7A), and two agonist conformations 1GWR (Figure 7 B) and 3Q97 (Figure 7C) are discussed. We have selected this antagonist structure because the native ligand shows the highest structural similarity with the investigated compounds. The cocrystallized ligands were docked to their native experimental structures to evaluate the docking method efficiency. The best ranked docking poses of the native ligands recapitulate the binding mode of the ligand to the active site of the experimental structures, and the same interactions with the amino acids lining the binding pocket were found (Supporting Information Figures S1, S2, and S3).
Figure 7.

Representations of the experimental structures binding pockets used for modeling: (A) antagonist conformation of ER LBD cocrystallized with 4OHT (PDB code 3ERT); (B) agonist conformation of ER LBD cocrystallized with E2 (PDB code 1GWR); (C) the agonist conformation of ER LBD cocrystallized with a E-isomer of ethoxytriphenylethylene (PDB code 3Q97). The best docking poses of the Z-isomers to the LBD of ER (antagonist conformation; PDB code 3ERT) were (D) ZFREndox (yellow) and ZFR4OHT (magenta). E isomers of fixed ring 4OHT and endoxifen do not fit very well into the antagonist conformation (3ERT): (E) EFREndox (green) and EFR4OHT (purple). The best docking poses of the Z-isomers to the LBD of ER (agonist conformation, PDB code 3Q97) were (F) EFREndox (green) and EFR4OHT (purple).
The predicted binding mode of the ZFR4OHT and ZFREndox to the antagonist conformation of ER 3ERT is similar to that of 4OHT (Figure 7A). In these models the ligands are accommodated well in the binding pocket, the complex H-bond network involving amino acids Asp351, Glu353, and Arg394 is recapitulated, and similar hydrophobic interactions are encountered (Figure 7D). Conversely, the EFR4OHT and EFREndox are docked to the 3ERT binding site in a completely different alignment but forming the H-bonds with Asp351, Glu353, and Arg394 (Figure 7E). Although the E-isomers form the H-bond network, they do not fit the binding pocket of ER antagonist conformation as well as the Z-isomers, as can be seen from the docking scores (Table 1), especially the values for Emodel. E-Isomers do not fill the binding pocket and are not involved in hydrophobic interactions with the important amino acids of the binding site like the Z-isomers and 4OHT. These remarks are supported by the van der Waals (vdW) parameter which accounts for hydrophobic interactions and shows favorable values for Z-isomers (Table 1). This binding alignment has been recapitulated in docking experiments performed for other experimental structures of ER LBD in antagonist conformation, 1UOZ and 2OUM (data not shown). These results show that it is highly probable for the E-isomers to be accommodated in a different conformation of ER LBD. Docking runs performed at the agonist conformations of ER (the receptor cocrystallized to E2, PDB entry 1GWR (Figure 7B), and to DES, PDB entry 3ERD) have led to conflicting results; thus, no valid docking pose could be found. For this reason other experimental structures of ER in the agonist conformation were selected from PDB, based on the 3D similarity between the cocrystallized ligands and E-isomers. The structure showing the highest shape similarity between the native ligand and E-isomers was selected, namely, PDB entry 3Q97 (Figure 7C). Interestingly, this experimental structure contains two isomers corresponding to E- and Z-isomers of a triphenylethylene derivative, cocrystallized with ER LBD. The binding pocket of 3Q97 (Figure 7C) is wider and larger than the ones of 1GWR or 3ERD, and it can accommodate the E-isomers. The top ranked docking poses of EFR4OHT and EFREndox are shown in Figure 7F, and it can be seen that they fit in the binding pocket. The Z-isomers were ranked with lower docking scores and were docked in an orientation similar to that from the antagonist conformation of ER. It can be concluded from these findings that the predicted binding mode of Z-isomers is similar to that of 4OHT and other antagonists of ER, showing higher values of the docking scores when compared with E-isomers docked to antagonist conformation of ER LBD. The former compounds do not fit into the encapsulated binding pocket of ER, corresponding to agonist conformation of the receptor, even if some degree of flexibility has been allowed to the receptor. It is highly probable for E-isomers to induce conformational changes to the active site of ER upon binding which would be reflected in the repositioning of helix 12 to a conformation related to that of the experimental structure 3Q97.
Table 1. Docking Results for X-ray Structure 3ERTa.
| compd | GScore | H bond | vdW | Coul | Emodel | CvdW |
|---|---|---|---|---|---|---|
| ZFREndox | –14.22 | –1.5 | –48.6 | –15.4 | –92.6 | –64 |
| ZFR4OHT | –13.22 | –1.5 | –50 | –13.4 | –92.6 | –63.4 |
| EFREndox | –10.65 | –1.6 | 0.6 | –7.9 | 38.6 | –7.3 |
| EFR4OHT | –10.59 | –1.9 | 1.3 | –9.4 | 37.8 | –8.1 |
CvdW = Coul + vdW is the non-bonded interaction energy between the ligand and the receptor. Emodel is a specific combination of GScore. GlideScore (GScore in kcal/mol) is given by GScore = a × vdW + b × Coul + Lipo + Hbond + Metal + Rewards + RotB + Site, where vdW = van der Waals interaction energy, Coul = Coulomb interaction energy, Lipo = lipophilic contact plus phobic attractive term; Hbond = hydrogen-bonding term; Metal = metal-binding term (usually a reward); Rewards = various reward or penalty terms; RotB = penalty for freezing rotatable bonds; Site = polar interactions in the active site. The coefficients of vdW and Coul are a = 0.050, b = 0.150 for Glide 5.0 (the contribution from the Coulomb term is capped at −4 kcal/mol).
The Z- and E-isomers of FRMethoxy and FREthoxy compounds were also docked to the experimental structures of ER LBD in the agonist (PDB entries 1GWR and 3Q97) and antagonist (PDB entry 3ERT) conformations. Analysis of docking results shows Z-isomers being better accommodated in the agonist conformation of ER than the E-isomers (Figure 8B and Figure 8C). The Emodel and docking scores have higher values for Z-isomers (Table 2). Few details indicate that it is possible for these isomers to bind to a conformation of ER similar to that of 3Q97. Thus, in the agonist structure 1GWR the alkoxy substituent is involved in clashes with the side chains of Leu525 and Leu540 of helix12 while the fused rings system of the ZFREthoxy derivative is involved in clashes with Ile424 and Leu428 (Figure 8B). Thus, the best ranked docking poses of ZFRMethoxy and ZFREthoxy derivatives in the binding site of 3Q97 are free of these unfavorable contacts while a larger number of favorable interactions are formed with other hydrophobic amino acids of the binding site (Figure 8C). The binding site of the antagonist conformation, 3ERT, is larger and exposed to the solvent, and although the top ranked docking poses of Z-isomers form the H-bond network, the favorable hydrophobic contacts with Leu525 and Leu540 are missing (Figure 8A). As a result, it can be concluded that it is highly probable for Z-isomers to bind to a conformation of ER similar to the experimental structure 3Q97. Regarding the E-isomers, the binding mode most frequently predicted by the docking poses is similar for the antagonist conformation 3ERT (Figure 8D) and agonist conformation 1GWR (Figure 8E) with the methoxy and ethoxy substituents pointing toward the region of the binding pocket lined by amino acids Glu353 and Arg394. However, in this alignment clashes are encountered with these. Conversely, the top ranked docking poses at 3Q97 binding pocket show the alkoxy substituents oriented toward His524 in the opposite region of site and no H-bonds are formed (Figure 8F). Also, no clashes have been noticed with other amino acids of the binding site.
Figure 8.

Representations of the experimental structures binding pockets used for modeling: (A) best docking poses of the Z-isomers to the LBD of ER (antagonist conformation, PDB code 3ERT) of ZFRMethoxy (green) and ZFREthoxy (orange); (B) agonist conformation of ER LBD cocrystallized with E2 (PDB code 1GWR) of ZFRMethoxy (green) and ZFREthoxy (orange); (C) agonist conformation of ER LBD cocrystallized with a E-isomer of ethoxytriphenylethylene (PDB code 3Q97) of ZFRMethoxy (green) and ZFREthoxy (orange); (D) best docking poses of the Z-isomers to the LBD of ER (antagonist conformation, PDB code 3ERT) of EFRMethoxy (light pink) and EFREthoxy (magenta); (E) agonist conformation of ER LBD cocrystallized with E2 (PDB code 1GWR) of EFRMethoxy (light pink) and EFREthoxy (magenta); (F) agonist conformation of ER LBD cocrystallized with a E-isomer of ethoxytriphenylethylene (PDB code 3Q97) of EFRMethoxy (light pink) and EFREthoxy (magenta).
Table 2. Docking Scores for X-ray Structure 3Q97 (Binding Site Cocrystallized with Z-Isomer of Ethoxytriphenylethylene).
| compd | GScore | H bond | vdW | Coul | Emodel | CvdW | Intern |
|---|---|---|---|---|---|---|---|
| BPTPE | –11.96 | –1.5 | –44.7 | –5.9 | –85.4 | –50.6 | 2.1 |
| 3OHTPE | –12.76 | –2.2 | –37 | –14.7 | –86.4 | –51.7 | 2.9 |
| ZFRMethoxy | –12.76 | –1.5 | –48.2 | –4.8 | –89.4 | –53 | 3.6 |
| ZFREthoxy | –12.35 | –1.3 | –39.5 | –9.9 | –90.7 | –49.4 | 3 |
| EFRMethoxy | –10.8 | –1 | –37.1 | –2.4 | –67.6 | –39.5 | 0 |
| EFREthoxy | –10.41 | –0.7 | –30.1 | 0.7 | –44.2 | –29.4 | 6.9 |
Discussion and Conclusions
The goal of this investigation is to link estrogenic/antiestrogenic ligand structures of tamoxifen metabolites with the well documented estradiol responses of cell replication or an estrogen target gene activation in cancer and apply biological end points to molecular modeling of the ER complex. This study has its origins with original published reports16,17,34 of the synthesis and evaluation of E and Z isomers of FR4OHT. We now extend earlier work with this study of E and Z ER endoxifen, investigate new Z and E FRMethoxy and FREthoxy derivatives of triphenylethylene (TPE), and compare our results with the angular estrogens BPTPE and 3OHTPE.18 The biological end points used were cell replication in MCF-7:WS8 cells and the estrogen-regulated prolactin gene (Prl) in rat pituitary gland tumor GH3 cell line.
There are several important new findings with the structure–function relationship of new FR compounds. The length and positioning of the side chain of the new Z and E FR compounds govern estrogen-induced cell replication of MCF-7:WS8 cells (Figure 2A). The natural estrogen E2 is extremely active as a full agonist over the range 10–14–10–8 M; however, each Z FR derivative is a partial agonist, so comparative EC50 calculations are not appropriate. Nevertheless, BPTPE is a potent partial agonist (50% max of E2 curve) over the range 10–12–10–9 M. The Z FRMethoxy and FREthoxy partial agonist curve is displaced a log to the right, and the EFRMethoxy and EFREthoxy is displaced further. The E FR isomers of 4OHT and endoxifen are both low potency estrogens, and this is consistent with their lower ligand-binding activity of the ER.14 Only the nonsteroidal antiestrogens 4OHT and endoxifen and their ZFR derivatives were antiestrogenic on cell proliferation. By contrast, all compounds were antiestrogenic (Figure 4B) at 1 μM in the GH3 rat pituitary prolactin assay, i.e., down to the level of the partial agonist activity of each compound (Figure 4A). The inability of substituted angular estrogens to be unable to initiate prolactin gene synthesis fully but stimulate mouse vaginal cornification (which classifies them as estrogens) has been noted previously.35−37 The partial gene regulation (pS2, GREB1, and PgR) is also noted with BPTPE and 3OHTPE as well as the E and Z FRMethoxy and FRMEthoxy TPEs. It is interesting to note that at 1 μM EFREndox is particularly active in triggering pS2, GREB1, and PgR (Figure 5), so the ability of the E isomers of FR4OHT and Endox were tested at therapeutic concentrations26 to determine whether estrogen-induced cell replication could occur during therapy. None was noted (Figure 3).
Additionally, results from RT-PCR of the estrogen-responsive genes in MCF-7:WS8 cells show that the E-isomers are inducing higher expression of pS2, GREB1, and PgR genes mRNAs, and also Prl gene mRNA in rat GH3 cells. This contrasts with the Z-isomers. Considering all the results, it is possible to conclude that the E-isomers of the biologically active tamoxifen metabolites 4OHT and endoxifen have estrogenic properties in human breast cancer cells, but this is not of biological significance during therapy with tamoxifen.
The most important general observation was the sensitivity of all the different TPE structures to trigger cell replication (Figure 2A). This supersensitivity is clearly required for cancers to survive through relentless cell replication. Antiestrogenic activity blocking replication requires a correctly positioned alkylaminoethoxy side chain.38 By contrast, estrogen-regulated protein synthesis is much less successful with test compounds and the resulting complex is clearly less promiscuous, tending to create a biologically inert “antiestrogenic complex” (Figure 4B).
It is interesting to note that the accumulation of ER determined by Western blotting for all compounds is independent of estrogenic or antiestrogenic activity. The turnover of ER complexes is regulated by ubiquitinilation and proteosomal degradation,39 but it is clearly the shape of the ligand and the resulting conformation of the complex that determine accumulation or destruction. The shape of the ligand is critical; a planar class I (estradiol) ligand causes reduction of ER, whereas nonsteroidal antiestrogens such as 4OHT and endoxifen40 cause the ER complex to accumulate. The same is true of angular TPEs40 which are also all of the new FR compounds investigated here that bind to the ER. By contrast, fulvestrant (ICI 182,780) causes the rapid destruction of ER.41 A previous study by Wu et al.42 demonstrated that endoxifen also caused rapid destruction of ER, but this was not observed in this study. We used endoxifen obtained from the Mayo Clinic and the Z FR endoxifen, both of which had the same accumulation of the ER.
Molecular modeling demonstrates that most likely the positioning of the E-isomers in the ligand-binding cavity of the ER is different because of repositioned side chains, potentially reducing the affinity to the receptor. However, this structural change also alters the pharmacological properties of the E-isomers, as they are more estrogenic rather than antiestrogenic. The molecular modeling shows that the E-isomers fit better into the ER conformation when the receptor is bound to a structurally similar E-isomer of ethoxytriphenylethylene where X-ray crystallography (PDB entry 3Q97) shows that the H12 of the LBD is actually closed, which resembles the conformation induced by estrogens.30 This is also confirmed by the Western blotting results for the ER protein levels, which show that the Z-isomers of FR4OHT and endoxifen, being antiestrogens, are inducing up-regulation of the ER protein levels; however, the E-isomers are not inducing the same up-regulation, indicating their different properties (Figure 6). However, that is not the case with fixed-ring compounds with shorter side chains. In contrast, Z-isomers of FR 4OHT and endoxifen fit better into the antagonist conformation of the ER LBD.30 Compounds with shorter side chain fit better into the conformation of the ER LDB that accommodates their E-isomers, resulting in the H12 being closed. This results in estrogenic activity.
In summary, a well-defined series of compounds has been classified and characterized for cell growth and estrogen target protein synthesis. The important finding is that replication in the ER-positive breast cancer cell is extremely sensitive to stimulation by a broad range of synthetic estrogens. This supersensitivity to growth stimuli is the major survival mechanism of cancer. It is a simple principle based on growth to survive from any source through the ER signal transduction pathway. This promiscuous pathway is only stopped when the antiestrogenic side chain of antiestrogens interacts with Asp351 and Helix 12 is prevented from closing.43 By contrast, the transcription of RNA for estrogen target genes such as prolactin is highly selective with these new compounds synthesized in this study. The compounds tend to become antiestrogenic (Figure 6) possibly because the conformation of the ER complex cannot recruit all necessary transcription factors. The conformation of the complex is critical. However, it is also important to appreciate that X-ray crystallography of complex 3Q97, which appears to be estrogen-like, only gives a glimpse at that one moment of time of low energy crystallization. We anticipate that progressive changes occur over time as the estrogen ER complex adapts to the changing environment within the cell. Biological end points are correlated with the receptor docking of a new intermediate form of the ER ligand-binding domain (PDB entry 3Q97). These data will be used in the future to decipher and to advance the understanding of the molecular mechanisms of estrogen-induced apoptosis.44
Materials and Methods
Chemistry
The general schemes of synthesis are described in Scheme 1.
General Procedures
Unless stated otherwise, reactions were performed in heat-dried glassware under a positive pressure of nitrogen using solvents that were distilled from or stored over calcium hydride, LiAlH4, or molecular sieves. Commercial grade reagents and solvents were used without further purification except as stated. Thin layer chromatography (TLC) was performed on precoated silica gel 60 F254 plates and visualized by UV light (254). Flash column chromatography was performed on hand packed silica gel (230–400 mesh 60A) columns using the dry loading method. Automated column chromatography purifications were done using a Teledyne ISCO apparatus (CombiFlash Rf) with prepacked silica gel columns (4–40 g). 1H NMR was recorded on a Bruker Avance 300 MHz instrument. Chemical shifts were quoted in parts per million, and coupling constants were reported in hertz. 13C NMR was recorded at 75 MHz. HPLC–MS analyses and purifications were performed on a Waters HPLC system consisting of a model 2545 binary gradient pump, 2424 ELS detector, 2487 dual UV detector (254 and 365 nm), and a model 3100 single quadrupole mass spectrometer detector with electrospray and chemical ionization. Deltapak-C18 15 μm 300A reverse phase columns were used for analyses (3.9 mm × 30 cm) and preparative (30 mm × 30 cm) separations. The mobile phase was either a mixture of MeOH/H2O or CH3CN (0.05% FA)/H2O (0.05% FA) with a flow rate of 0.8 on the analytical side or 20 mL/min for preparative scale. Gradient system for analytical (15 m) and preparative (30 m) was a 5–95% linear gradient. For preparative runs, fractions were collected by hand using UV and MS detectors. High resolution MS results were obtained using an Acquity UPLC (ultraperformance liquid chromatography)–QTOF-MS (quadrupole time of flight mass spectrometry) Premiere system (Waters Corporation, USA). All final compounds were tested with a purity of more than 95% as analyzed by LC/MS.
Synthesis. Z-Fixed Ring Ethoxy (ZFREthoxy). 2-Hydroxy-6,7,8,9-tetrahydro-5H-benzocyclohepten-5-one (1)
2-Methoxyheptenone was demethylated according to the procedure of Lal et al. (adapted from Kahn et al.).20 The product was extracted with chloroform, resulting in a quantitative yield of 1. TLC (6% MeOH, 94% chloroform) Rf = 0.28. LC/MS tR = 13.10, (M + H+) 177. 1H NMR (CDCl3): δ = 1.79–190 (m, 4H); 2.74 (m, 2H); 2.90 (m, 2H); 6.68 (d, 1H, J = 2.4); 6.75 (dd, 1H, J = 2.4 and 8.4); 7.75 (d, 1H, J = 8.4).
2-(4-(2,3,5,6-Tetrafluoro-4-(trifluoromethyl)phenoxy)-6,7,8,9-tetrahydro-5H-benzocyclohepten-5-one (2)
Octafluorotoluene (807 mg, 484 μL, 3.42 mmol) and 2-hydroxy-6,7,8,9-tetrahydro-5H -benzocyclohepten-5-one (1) (587 mg, 3.33 mmol) were dissolved in dichloromethane (15 mL) and 1 N NaOH (15 mL). Then tetrabutylammonium hydrogen sulfate (572 mg) was added and the solution was stirred overnight at room temperature. The organic layer was separated, and the aqueous layer was extracted with dichloromethane (2 × 50 mL). The combined organic layers were washed with water and dried in vacuo. The product was purified from tetrabutylammonium hydrogen sulfate by flash column chromatography over silica (2.3 cm × 5 cm on 2.3 cm × 20 cm) and eluted with 400 mL of chloroform. Fractions (25 mL) 2–6 were combined to give 2 (923 mg, 71% yield). TLC (6% MeOH, 94% chloroform) Rf = 0.88. LC/MS tR = 19.37, (M + H+) 393. 1H NMR (CDCl3): δ = 1.81–195 (m, 4H); 2.74 (m, 2H); 2.94 (m, 2H); 6.83 (d, 1H, J = 2.4); 6.88 (dd, 1H, J = 2.4 and 8.4); 7.78 (d, 1H, J = 8.4).
4-Bromophenyl 2,3,5,6-Tetrafluoro-4-(trifluoromethyl)phenyl Ether (4)
Octafluorotoluene (30 g, 0.127 mol) and 4-bromophenol (21 g, 0.121 mol) were dissolved in dichloromethane (100 mL) and 1 N NaOH (100 mL). Then tetrabutylammonium hydrogen sulfate (10 g) was added and the solution was stirred at room temperature for 4 h. The dichloromethane layer was separated and the aqueous layer extracted with dichloromethane (2 × 50 mL). The combined organic layers were dried under reduced pressure. The residue was triturated with hexanes, and the insoluble tetrabutylammonium sulfate was filtered. The filtrate was evaporated under reduced pressure to give 4 (46.41 g, 98% yield). TLC (5% dichloromethane, 95% hexanes) Rf = 0.63. 1H NMR (CDCl3): δ = 6.87 (d, 2H, J = 8.7); 7.44 (d, 2H, J = 8.7).
3-(2,3,5,6-Tetrafluoro-4-(trifluoromethyl)phenoxy)-6,7-dihydro-9-(4-ethoxyphenyl)-5H-benzocycloheptene (5)
Bromophenetole (507 μL, 710 mg, 3.53 mmol) was dissolved in ether (20 mL) with stirring. Then magnesium turnings (125 mg) were added, followed by dropwise addition of 1,2-dibromoethane (142 μL) in diethyl ether (10 mL) over 1 h. Once the Grignard reagent had formed, 2-(4-(2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenoxy)-6,7,8,9-tetrahydro-5H -benzocyclohepten-5-one (2) (923 mg in 10 mL ether) was added, and the mixture was stirred overnight at room temperature. The next day, it was poured into 0.1 N hydrochloric acid solution (30 mL) and extracted with ether (3 × 50 mL). The ether was removed by evaporation under reduced pressure, and the residue was dissolved in ethanol (20 mL). Concentrated hydrochloric acid (5 mL) was added, and the solution was refluxed for 2 h. It was cooled, poured into water (50 mL), and extracted with ether (3 × 50 mL). The solvent was removed under reduced pressure, and the residue was purified by flash column chromatography (3.0 cm × 3.0 cm on 3.0 cm × 36.0 cm) over silica. The product was eluted with 250 mL of 100% hexanes, followed by 1 L of 10% dichloromethane, 95% hexanes. Fractions (25 mL) 37–70 contained the product and were combined and evaporated in vacuo to give 5 (608 mg, 52% yield). TLC (20% dichloromethane, 80% hexanes) Rf = 0.29. LC/MS tR = 22.45, (M + H+) 497. 1H NMR (CDCl3): δ = 1.41 (t, 3H, J = 6.9); 1.96 (m, 2H, J = 7.2); 2.16 (t, 2H, J = 7.2); 2.62 (t, 2H, J = 6.9); 4.03 (q, 2H, J = 6.9); 6.36 (t, 1H, J = 7.3); 6.75–7.24 (m, 7H).
3-(2,3,5,6-Tetrafluoro-4-(trifluoromethyl)phenoxy)-6,7-dihydro-8-bromo-9-(4-ethoxyphenyl)-5H-benzocycloheptene (9)
3-(2,3,5,6-Tetrafluoro-4-(trifluoromethyl)phenoxy)-6,7-dihydro-9-(4-ethoxyphenyl)-5H-benzocycloheptene (5) (608 mg, 1.224 mmol) and pyridine hydrobromide perbromide (428 mg) were stirred in dichoromethane (15 mL) at room temperature for 4 h. The orange solution was washed with 0.1 M HCl solution (25 mL) which contained sodium sulfite (20 mg), followed by water. Next the solvent was evaporated under reduced pressure to 9 (689 mg, 98%). TLC (10% toluene, 90% hexanes) Rf = 0.31. LC/MS tR = 22.39, (M + H+) 575. 1H NMR (CDCl3): δ = 1.42 (t, 3H, J = 6.9); 2.31 (m, 2H, J = 6.9); 2.58 (t, 2H, J = 6.9); 2.76 (t, 2H, J = 6.9); 4.05 (q, 2H, J = 6.9); 6.71–6.86 (m, 5H); 7.13 (d, 2H, J = 7.8).
3-(2,3,5,6-Tetrafluoro-4-(trifluoromethyl)phenoxy)-6,7-dihydro-8-phenyl-9-(4-ethoxyphenyl)-5H-benzocycloheptene (13)
Anhydrous zinc chloride (433 mg) was dissolved in THF (15 mL) with stirring. Phenyllithium in di-n-butyl ether (1.8 mL of 1.8 M solution) and THF (10 mL) were added dropwise over 15 min to the zinc chloride solution while it was cooled in an ice bath. After the solution was allowed to warm to room temperature, 3-(2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenoxy)-6,7-dihydro-8-bromo-9-(4-ethoxyphenyl)-5H-benzocycloheptene (9) (584 mg, 1.015 mmol) in THF (10 mL) was added dropwise followed by Pd(PPh3)4 (10 mg). The mixture was refluxed for 6 h and left to stir at room temperature overnight. The reaction mixture was poured into water (50 mL) and extracted with diethyl ether (3 × 50 mL). The combined ether extracts were dried in vacuo. Purification was performed with flash column chromatography over silica (2.3 cm × 4 cm on 2.3 cm × 20 cm). The column was equilibrated with 200 mL of 100% hexanes, and the product was eluted with 750 mL of 5% toluene, 95% hexanes. Fractions (25 mL) 23–41 were combined and evaporated in vacuo to give white solid 13 (372 mg, 64% yield). TLC (20% toluene, 80% hexanes) Rf = 0.33. LC/MS tR = 23.52, (M + H+) 573. 1H NMR (CDCl3): δ = 1.28 (t, 3H, J = 6.9); 2.12 (m, 2H); 2.30 (t, 2H, J = 6.9); 2.71 (t, 2H, J = 6.9); 3.85 (q, 2H, J = 6.9); 6.51–6.57 (m, 2H); 6.66–6.82 (m, 5H); 7.08 (m, 5H).
3-Ethoxy-6,7-dihydro-8-phenyl-9-(4-hydroxyphenyl)-5H-benzocycloheptene (17)
3-(2,3,5,6-Tetrafluoro-4-(trifluoromethyl)phenoxy)-6,7-dihydro-8-phenyl-9-(4-ethoxyphenyl)-5H-benzocycloheptene (13) (372 mg, 0.640 mmol) and sodium ethoxide (400 mg) in DMF (5 mL) were heated to 40 °C for 2 h with stirring. The brown solution was poured into saturated sodium bicarbonate solution (50 mL) and extracted with ether (3 × 50 mL). The combined ether layers were evaporated in vacuo. Purification was performed with silica column chromatography (2.3 cm × 3 cm on 2.3 cm × 21 cm) equilibrated with 200 mL of 100% hexanes. The product was eluted with 1.75 L of 50% dichloromethane, 50% hexanes. Fractions (25 mL) 34–68 were combined and evaporated in vacuo to give white solid 17 (203 mg, 89% yield). It was recrystallized in dichloromethane/hexanes (171 mg, mp 242–243 °C). TLC (50% dichloromethane, 50% hexanes) Rf = 0.13. LC/MS tR = 17.40, (M + H+) 357. 1H NMR (CDCl3): δ = 1.38 (t, 3H, J = 6.9); 2.16 (m, 2H, J = 7.2); 2.39 (t, 2H, J = 7.2); 2.76 (t, 2H, J = 7.2); 3.94 (q, 2H, J = 6.9); 6.59–6.63 (m, 3H); 6.74–6.82 (m, 4H); 7.15 (m, 5H). HRMS calculated for C25H24O2 (M + H)+ 357.1855; found 357.1859.
Z-Fixed Ring Methoxy (ZFRMethoxy). 3-(2,3,5,6-Tetrafluoro-4-(trifluoromethyl)phenoxy)-6,7-dihydro-9-(4-methoxyphenyl)-5H-benzocycloheptene(6)
2-(4-(2,3,5,6-Tetrafluoro-4-(trifluoromethyl)phenoxy)-6,7,8,9-tetrahydro-5H-benzocyclohepten-5-one (2) (6.198 g, 15.8 mmol) was dissolved in ether (20 mL) with stirring. Then 4-methoxyphenylmagnesium bromide (0.5 M solution in THF, 47.5 mL, 23.7 mmol) was added dropwise at room temperature, and it was stirred overnight. The next day, the orange solution was heated to reflux for 12 h. Then it was poured into 0.1 N hydrochloric acid solution (100 mL) and extracted with ether (3 × 100 mL). The ether was removed by evaporation under reduced pressure, and the residue was dissolved in ethanol (200 mL). Concentrated hydrochloric acid (5 mL) was added, and the solution was refluxed for 2 h. The solution turned from orange to green, forming a sticky tan precipitate. It was cooled and poured into water (200 mL). The product was extracted with ether (3 × 100 mL), and the combined ether layers were evaporated under reduced pressure (9.57 g). It was purified by column chromatography over silica on the CombiFlash Rf instrument. Compounds were eluted with ethyl acetate/hexanes gradient on a 40 g gold silica column. The sample was injected onto the column using a dry method with 50 g of silica. Fractions (25 mL) 3–10 were combined and evaporated in vacuo to give 6 (3.60 g, 47% yield). TLC (10% ethyl acetate, 90% hexanes) Rf = 0.69. LC/MS tR = 19.27, (M + H+) 483. 1H NMR (CDCl3): δ = 1.97 (m, 2H, J = 7.5); 2.17 (t, 2H, J = 7.2); 2.63 (t, 2H, J = 7.2); 3.81 (s, 3H); 6.37 (t, 1H, J = 7.5); 6.78–7.02 (m, 5H); 7.05–7.25 (m, 2H).
3-(2,3,5,6-Tetrafluoro-4-(trifluoromethyl)phenoxy)-6,7-dihydro-8-bromo-9-(4-methoxyphenyl)-5H-benzocycloheptene (10)
3-(2,3,5,6-Tetrafluoro-4-(trifluoromethyl)phenoxy)-6,7-dihydro-9-(4-methoxyphenyl)-5H-benzocycloheptene (6) (3.6 g, 8.4 mmol) and pyridine hydrobromide perbromide (2.94 g) were stirred in dichorormethane (50 mL) at room temperature for 20 h. The orange solution was washed with 0.1 M HCl solution (50 mL) which contained sodium sulfite (200 mg), followed by water. It was evaporated under reduced pressure and purified by column chromatography over silica on the CombiFlash Rf instrument. Compounds were eluted with ethyl acetate/hexanes gradient on a 40 g gold silica column. The sample was injected onto the column using a dry method with 25 g of silica. Fractions (25 mL) 5–16 were combined and evaporated in vacuo to give 10 (3.374g, 72% yield). TLC (10% ethyl acetate, 90% hexanes) Rf = 0.64. LC/MS tR = 19.47, (M + H+) 561. 1H NMR (CDCl3): δ = 2.30 (m, 2H, J = 7.2); 2.60 (t, 2H, J = 7.2); 2.74 (t, 2H, J = 7.2); 3.81 (s, 3H); 6.69–6.96 (m, 5H); 7.16 (d, 2H, J = 8.7).
3-(2,3,5,6-Tetrafluoro-4-(trifluoromethyl)phenoxy)-6,7-dihydro-8-phenyl-9-(4-ethoxyphenyl)-5H-benzocycloheptene (14)
Anhydrous zinc chloride (2.4 mg) was dissolved in THF (50 mL) with stirring. Then 1.8 M solution of phenyllithium in di-n-butyl ether (9.8 mL) in THF (10 mL) was added dropwise over 30 min to the zinc chloride solution while it was cooled in an ice bath below 0 °C. After allowing the mixture to warm to room temperature, 3-(2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenoxy)-6,7-dihydro-8-bromo-9-(4-methoxyphenyl)-5H-benzocycloheptene (10) (3.374 g, 6.01 mmol) in THF (10 mL) was added dropwise followed by Pd(PPh3)4 (57 mg). The reaction was refluxed for 3 h and then left to stir overnight at room temperature. The reaction mixture was poured into water (50 mL) and extracted with diethyl ether (3 × 50 mL). The combined ether extracts were evaporated under reduced pressure. The residue was purified by column chromatography over silica on the CombiFlash Rf instrument. Compounds were eluted with ethyl acetate/hexanes gradient on the 40 g gold silica column. Flow rate was 25 mL/min. The sample was injected onto the column using a dry method with 30 g of silica. Fractions (25 mL) 9–26 were combined and evaporated under reduced pressure to give 14 (2.90 g, 86% yield). TLC (5% EtOAc, 95% hexanes) Rf = 0.40. LC/MS tR = 19.95, (M + H+) 559. 1H NMR (CDCl3): δ = 2.21 (m, 2H, J = 7.2); 2.41 (t, 2H, J = 7.2); 2.81 (t, 2H, J = 7.2); 3.76 (s, 3H); 6.63–6.94 (m, 7H); 7.13–7.21 (m, 5H).
3-Methoxy-6,7-dihydro-8-phenyl-9-(4-hydroxyphenyl)-5H-benzocycloheptene (18)
3-(2,3,5,6-Tetrafluoro-4-(trifluoromethyl)phenoxy)-6,7-dihydro-8-phenyl-9-(4-ethoxyphenyl)-5H-benzocycloheptene (14) (2.90 g, 5.19 mmol) and sodium methoxide (3.6 g) in DMF (25 mL) were heated to 35 °C for 3 h. The orange solution was poured into saturated sodium bicarbonate solution (100 mL) and extracted with ether (3 × 100 mL). The combined ether layers were evaporated in vacuo, and the residue was purified by column chromatography over silica on the CombiFlash Rf instrument. Compounds were eluted with ethyl acetate/hexanes gradient of 0–100% ethyl acetate over 35 min on a 40 g gold silica column. Flow rate was 25 mL/min. The sample was injected onto the column using a dry method with 20 g of silica. Fractions (25 mL) 8–10 were combined and evaporated in vacuo to give 18 (300 mg, 17% yield). TLC (5% EtOAc, 95% hexanes) Rf = 0.30. LC/MS tR = 15.67, (M + H+) 343. 1H NMR (CDCl3): δ = 2.18 (m, 2H, J = 7.2); 2.41 (t, 2H, J = 7.2); 2.78 (t, 2H, J = 7.2); 3.75 (s, 2H); 6.62–6.89 (m, 7H); 7.15 (m, 5H). HRMS calculated for C24H22O2 (M + H)+ 343.1698; found 343.1700.
E-Fixed Ring Ethoxy (EFREthoxy). 2-Ethoxy-6,7,8,9-tetrahydro-5H-benzocyclohepten-5-one (7-Ethoxy-1-benzosuberone) (3)
2-Hydroxy-6,7,8,9-tetrahydro-5H-benzocyclohepten-5-one (1) (2.171 g, 12.32 mmol) and anhydrous potassium carbonate (5.102 g, 15.21 mmol) were dissolved in acetone (50 mL). Then iodoethane (5.43 mL) was added and it was stirred at room temperature overnight. The reaction mixture was evaporated in vacuo and purified by flash chromatography (2.3 cm × 4 cm on 2.3 cm × 23 cm) over silica. The column was equilibrated with hexanes (200 mL), and the product was eluted in chloroform. Fractions (25 mL) containing product were combined and evaporated in vacuo to 3 (1.72 g, 68% yield). LC/MS tR = 13.35, (M + H+) 205. 1H NMR (CDCl3): δ = 1.32 (t, 3H, J = 7.2); 1.81–195 (m, 4H); 2.74 (m, 2H); 2.92 (m, 2H); 6.87 (m, 2H); 3.93 (q, 2H, J = 6.9); 7.77 (d, 1H, J = 8.4); 7.75 (d, 1H, J = 8.4).
3-Ethoxy-6,7-dihydro-9-(4-(2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenoxy)phenyl)-5H-benzocycloheptene (7)
4-Bromophenyl 2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenyl ether (6.244 g, 16.05 mmol) was dissolved in ether (50 mL) with stirring. Then magnesium turnings (500 mg) were added followed by dropwise addition of 1,2-dibromoethane (0.564 mL) in ether (5 mL) over 30 min. After the Grignard reagent formed, the mixture was heated for 1 h. Next, 2-ethoxy-6,7,8,9-tetrahydro-5H -benzocyclohepten-5-one (3) (1.72 g, 8.42 mmol) in ether (30 mL) was added and the solution was stirred overnight at room temperature. The next day, it was heated for 10 h. LC/MS analysis confirmed the formation of the intermediate tertiary alcohol. The reaction mixture was poured into 0.1 N hydrochloric acid solution (50 mL) and extracted with ether (3 × 50 mL). The ether was removed by evaporation under reduced pressure, and the residue was dissolved in ethanol (50 mL). Concentrated hydrochloric acid (10 mL) was added, and the solution was refluxed for 2 h. After cooling, it was poured into water (100 mL), extracted with ether (3 × 50 mL), and evaporated under reduced pressure. The product was purified by silica column chromatography (4.0 cm × 1.0 cm on 4.0 cm × 23.0 cm). The column was equilibrated with 500 mL of 100% hexanes, and the product was eluted with 2 L of 5% dichloromethane, 95% hexanes. Fractions (250 mL) 10–31 contained the product and were combined and evaporated in vacuo to give 7 (1.802 g, 43% yield). LC/MS tR = 19.67, (M + H+) 497. 1H NMR (CDCl3): δ = 1.31 (t, 3H, J = 7.2); 1.84 (m, 2H); 2.05 (t, 2H; J = 7.2); 2.51 (t, 2H; J = 6.9); 3.92 (q, 2H, J = 6.9); 6.21 (t, 1H, J = 8.4); 6.71–6.81 (m, 5H); 7.13 (d, 2H, J = 8.4).
3-Ethoxy-6,7-dihydro-8-bromo-9-(4-(2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenoxy)phenyl)-5H-benzocycloheptene (11)
3-Ethoxy-6,7-dihydro-9-(4-(2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenoxy)phenyl)-5H-benzocycloheptene (7) (1.802 g, 3.63 mmol) was dissolved in dichoromethane (20 mL) with stirring. Then pyridine hydrobromide perbromide (1.27 g) was added and the solution was stirred at room temperature for 4 h. The orange solution was washed with 0.1 M HCl solution (25 mL) containing sodium sulfite (0.1 g), followed by water. It was dried by evaporation under reduced pressure to give 11 (1.798 g, 86% yield). LC/MS tR = 19.77, (M + H+) 575. 1H NMR (CDCl3): δ = 1.38 (t, 3H, J = 7.2); 2.28 (m, 2H); 2.58 (t, 2H; J = 6.9); 2.73 (t, 2H; J = 6.9); 3.99 (q, 2H, J = 7.2); 6.61–6.79 (m, 3H); 6.94 (d, 2H, J = 8.4); 7.24 (d, 2H, J = 8.4).
3-Ethoxy-6,7-dihydro-8-phenyl-9-(4-(2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenoxy)phenyl)-5H-benzocycloheptene (15)
Anydrous zinc chloride (1.28 g) was added dropwise over 15 min to the zinc chloride solution while it was cooled in an ice bath. After it was allowed to warm to room temperature, 3-ethoxy-6,7-dihydro-8-bromo-9-(4-(2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenoxy)phenyl)-5H-benzocycloheptene (11) (1.798 g, 3.15 mmol) in THF (10 mL) was added dropwise followed by Pd(PPh3)4 (50 mg). It was heated to reflux for 3 h. The orange solution was poured into water (40 mL) and extracted with diethyl ether (3 × 40 mL). The combined ether extracts were dried over sodium sulfate, filtered, and evaporated under reduced pressure. The product was purified on a silica gel column (3 cm × 3 cm on 3 cm × 30 cm) that was eluted with 250 mL of hexanes, 500 mL of 5% dichloromethane, 95% hexanes, and 1.5 L of 10% dichloromethane 90% hexanes. Fractions (25 mL) 41–78 were combined and evaporated in vacuo to give 15 (1.523 g). TLC (30% dichloromethane, 70% hexanes) Rf = 0.47. LC/MS tR = 20.35, (M + H+) 573. 1H NMR (CDCl3): δ = 1.42 (t, 3H, J = 7.2); 2.19 (m, 2H); 2.40 (t, 2H; J = 6.9); 2.77 (t, 2H; J = 6.9); 4.05 (q, 2H, J = 7.2); 6.58–6.96 (m, 7H); 7.12–7.19 (m, 5H).
3-Ethoxy-6,7-dihydro-8-phenyl-9-(4-hydroxyphenyl)-5H-benzocycloheptene (19)
3-Ethoxy-6,7-dihydro-8-phenyl-9-(4-(2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenoxy)phenyl)-5H-benzocycloheptene (15) (414 mg, 1.01 mmol) and sodium methoxide (1.6 g) were dissolved in DMF (25 mL) with stirring. The mixture was stirred at room temperature for 4 h. The solution was poured into saturated sodium bicarbonate solution (50 mL), extracted with ether (3 × 40 mL), and dried in vacuo. Purification was performed using silica gel column chromatography (3.0 cm × 3 cm on 3.0 cm × 25 cm), equilibrating with 200 mL of 100% hexanes. The product was eluted with 500 mL of 25% dichloromethane, 75% hexanes; 500 mL of 50% dichloromethane, 50% hexanes; and 1 L of 75% dichloromethane, 25% hexanes. Fractions (25 mL) 16–22 were combined and evaporated in vacuo to give white solid 19 (240 mg, 67% yield). TLC (75% dichloromethane, 25% hexanes) Rf = 0.17. LC/MS tR = 17.40, (M + H+) 357. 1H NMR (CDCl3): δ = 1.43 (t, 3H, J = 6.9); 2.18 (m, 2H); 2.38 (t, 2H, J = 7.2); 2.77 (t, 2H, J = 7.2); 4.05 (q, 2H, J = 6.9); 6.52 (d, 2H, J = 8.7); 6.72–6.81 (m, 5H); 7.01–7.17 (m, 5H). 13C NMR (CDCl3): δ = 14.8; 32.4; 33.6; 34.2; 63.3; 111.6; 114.2; 114.6; 125.8; 127.7; 129.4; 130.2; 132.6; 134.8; 135.6; 137.8; 138.2; 142.9; 143.7; 154.2; 157.5. HRMS calculated for C25H24O2 (M + H)+ 357.1855; found 357.1859.
E-Fixed Ring Endoxifen (EFREndox). 6,7-Dihydro-8-phenyl-9-(4-(2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenoxy)phenyl)-5H-benzocyclohepten-3-ol (21)
3-Methoxy-6,7-dihydro-8-phenyl-9-(4-(2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenoxy)phenyl)-5H-benzocycloheptene (16) (929 mg, 1.663 mmol) was suspended in 33% HBr in acetic acid solution (20 mL) in a flask fitted with a condenser and drying tube. It was refluxed for 6 h and analyzed by LC/MS which determined the reaction was incomplete. Additional 48% HBr in water (4 mL) and AcOH (4 mL) were added, and the mixture was refluxed for an additional 4 h. The orange reaction mixture was poured into water (100 mL), and 1 N sodium hydroxide was added until the solution was basic to pH paper. Then saturated sodium bicarbonate (50 mL) was added, and the product was extracted with ether (2 × 100 mL). The combined ether layers were washed with water, dried over sodium sulfate, and evaporated under reduced pressure to give 21 (905 mg, 100% yield). LC/MS (MeOH) tR = 21.00, (M – H+) 543. 1H NMR (CDCl3): δ = 2.18 (m, 2H); 2.41 (t, 2H, J = 6.9); 2.77 (t, 2H, J = 6.9); 6.61–6.97 (m, 7H); 7.15 (m, 5H).
Ethyl Methyl-(2-((8-phenyl-9-(4-(2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenoxy)phenyl)-6,7-dihydro-5H-benzocyclohepten-3-yl)oxy)ethyl)carbamate (25)
6,7-Dihydro-8-phenyl-9-(4-(2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenoxy)phenyl)-5H-benzocyclohepten-3-ol (21) (259 mg, 0.476 mmol), ethyl (2-bromoethyl)(methyl)carbamate (600 μL), 1.5 N sodium hydroxide (2 mL), dichloromethane (2 mL), and tetrabutylammonium hydrogen sulfate (600 mg) were stirred at room temperature for 16 h. The aqueous layer was extracted with chloroform (3 × 50 mL), and the combined organic layers were dried in vacuo. The residue was purified by preparative HPLC using a CH3CN/H2O gradient. The sample was injected in THF (2 mL). Fractions 36–40 min were collected and dried in vacuo to give 25 (35 mg, 11% yield). LC/MS (CH3CN) tR = 20.25, (M + H+) 674. 1H NMR (CDCl3): δ = 1.22 (m, 3H); 2.18 (m, 2H); 2.38 (m, 2H); 2.77 (m, 2H); 3.04 (s, 3H); 3.63 (t, 2H, J = 4.8); 4.11 (m, 4H); 6.68–6.89 (m, 7H); 7.13 (m, 5H).
Ethyl (2-((9-(4-Hydroxyphenyl)-8-phenyl-6,7-dihydro-5H-benzocycloehepten-3-yl)oxy)ethyl)(methyl)carbamate (26)
Ethyl methyl(2-((8-phenyl-9-(4-(2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenoxy)phenyl)-6,7-dihydro-5H -benzocyclohepten-3-yl)oxy)ethyl)carbamate (25) (120 mg, 178 mmol) and sodium methoxide (200 mg) in DMF (10 mL) were heated to 80 °C for 6 h with stirring. After cooling, the reaction mixture was evaporated under reduced pressure. It was purified by flash column chromatography over silica (4 g gold silica column) on the CombiFlash Rf instrument. The gradient was 0–50% ethyl acetate in hexanes over 30 min. Flow rate was 10 mL/min. The sample was injected onto the column using the solid loading option (10 g of silica). Product was collected in fractions (25 mL) 3–6 and dried in vacuo to give 26 (61 mg, 75% yield). LC/MS (CH3CN) tR = 16.32, (M + H+) 458. 1H NMR (CDCl3): δ = 1.24 (m, 3H); 2.20 (m, 2H); 2.36 (m, 2H); 2.77 (m, 2H); 3.06 (s, 3H); 3.67 (m, 2H); 3.89 (m, 2H); 4.15 (m, 2H); 6.56–7.26 (m, 12H).
4-(3-(2-(Methylamino)ethoxy)-8-phenyl-6,7-dihydro-5H-benzocyclohepten-9-yl)phenol (27)
Ethyl (2-((9-(4-hydroxyphenyl)-8-phenyl-6,7-dihydro-5H-benzocyclohepten-3-yl)oxy)ethyl)(methyl)carbamate (26) (61 mg, 0.133 mmol) and pyridine HCl (200 mg) were heated to 150 °C in an oil bath with stirring for 3 h. The black residue was purified by preparative HPLC with a CH3CN/H2O gradient. Sample was injected in 2 mL of MeOH. Fraction at 13–20 min was collected and dried in vacuo to give 25 (21 mg, 41% yield). LC/MS (CH3CN) tR = 12.82, (M + H+) 386. 1H NMR (MeOD): δ = 2.11 (m, 2H); 2.33 (t, 2H, J = 6.9); 2.78 (m, 2H); 2.81 (s, 3H); 3.40 (m, 2H); 4.27 (m, 2H); 6.43–6.95 (m, 7H); 7.23 (m, 5H). HRMS calculated for C26H27NO2 (M + H)+ 386.2120; found 386.2122.
Z-Fixed Ring Endoxifen (ZFREndox). Ethyl (2-(4-(3-Methoxy-8-phenyl-6,7-dihydro-5H-benzocyclohepten-9-yl)phenoxy)ethyl)(methyl)carbamate (30)
3-Methoxy-6,7-dihydro-8-phenyl-9-hydroxyphenol-5H-benzocycloheptene (20) (208 mg, 0.607 mmol), 2-hydroxyethylmethylcarbamate (114 mg, 0.774 mmol) and triphenylphosphine (164 mg, 1.544 mmol) were stirred in tetrahydrofuran (20 mL). The reaction vial was cooled to below 0 °C, and diisopropyl azodicarboxylate (500 μL) was added dropwise over 5 min. The reaction mixture was stirred at room temperature for 3 days. The solution changed from yellow to orange during this time and was dried in vacuo. The compound was purified by preparative HPLC with a gradient of 5–75% in 30 min, 75% hold until 45 min, 75%–100% at 60 min in MeOH/H2O system. The sample was injected in 3 mL of MeOH. Fraction at 58–64 min was collected and evaporated in vacuo to give 30 (111 mg, 39% yield). LC/MS (CH3CN) tR = 19.12, (M + H+) 472. 1H NMR (CDCl3): δ = 1.26 (m, 3H); 2.14 (m, 4H); 2.38 (t, 2H, J = 6.9); 2.78 (t, 2H, 6.9); 2.99 (s, 3H); 3.58 (t, 2H); 3.82 (s, 3H); 4.11 (q, 2H, J = 7.2); 6.57–6.83 (m, 7H); 7.14 (m, 5H).
9-(4-(2-(Methylamino)ethoxy)phenyl)-8-phenyl-6,7-dihydro-5H-benzocyclohepten-3-ol (31)
Ethyl (2-(4-(3-methoxy-8-phenyl-6,7-dihydro-5H-benzocyclohepten-9-yl)phenoxy)ethyl)(methyl)carbamate (30) (156 mg, 0.331 mmol) and pyridine hydrochloride (600 mg) were heated in an oil bath to 180 °C with stirring for 3 h. The black solid was dissolved in methanol and purified by preparative HPLC using a CH3CN/H2O gradient. Sample was injected in 2 mL of MeOH. Fraction at 15–21 min was collected and dried in vacuo (61 mg). This was purified further by flash column chromatography over silica (2.3 cm × 1.0 cm on 2.3 cm × 23 cm). It was equilibrated with 200 mL of dichloromethane and eluted with 400 mL of 10% MeOH, 90% CH2Cl2, followed by 600 mL of 15% MeOH, 85% CH2Cl2. Fractions (25 mL) 15–36 were combined and evaporated in vacuo to give 31 (52 mg, 41% yield). TLC (15% MeOH, 85% dichloromethane) Rf = 0.31. LC/MS (CH3CN) tR = 15.42, (M + H+) 386. 1H NMR (MeOH): δ = 2.12 (m, 2H); 2.35 (t, 2H, J = 6.9); 2.61 (t, 2H, 6.9); 2.74 (s, 3H); 3.18 (t, 2H, J = 5.1); 4.09 (t, 2H, J = 5.1); 6.56–6.83 (m, 7H); 7.11 (m, 5H). HRMS calculated for C26H27NO2 (M + H)+ 386.2120; found 386.2114.
Cell Culture
The ER positive MCF-7:WS8 and GH3 cell lines were used in this study. The human ER positive breast cancer cells MCF-7:WS8 are hypersensitive to estrogens and were cloned from wild type MCF-7 cells and were maintained in phenol-red RPMI 1640 medium containing 10% fetal bovine serum (FBS), 2 mM glutamine, penicillin at 100 U/mL, streptomycin at 100 μg/mL, 1× nonessential amino acids (all from Life Technologies, Carlsbad, CA), and bovine insulin at 6 ng/mL (Sigma-Aldrich, St. Louis, MO). Rat pituitary GH3 cells were obtained from American Type Culture Collection (ATCC, Rockville, MD) and were maintained in DMEM medium supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, penicillin at 100 U/mL, streptomycin at 100 μg/mL, 1× nonessential amino acids, and bovine insulin at 6 ng/mL. All cells were cultured in T185 flasks (Nalge Nunc International, Rochester, NY) and passaged twice a week. All cell lines were grown in 5% CO2 at 37 °C.
Pharmacological Evaluation
All the biological properties of the synthesized compounds were tested by assessing the cell proliferation of the ER positive MCF-7:WS8 cells. Before the start of the experiment cells were estrogen starved by splitting them into RPMI 1640 medium without phenol red, and containing 10% charcoal stripped fetal serum (estrogen free), for 3 days. Cells were seeded into 24-well plates at a density of 10 000 cells per well. Next day after seeding (day 1) cells were treated with serial dilutions of the tested drugs in estrogen-free medium. The medium was changed every 2 days for a total of 7 days. All concentration points were performed in triplicate. On the last day the cells were harvested by medium aspiration and washed in cold PBS (Life Technologies, Carlsbad, CA) once and analyzed with fluorescent DNA quantification kit (Bio-Rad, Hercules, CA) according to the manufacturer’s instructions, and samples were read in a Mirthas LB540 fluorimiter/luminometer (Berthold Technologies, Oak Ridge, TN) in black wall 96-well plates (Nalge Nunc International, Rochester, NY).
Real-Time PCR
Real-time polymerase chain reaction (RT-PCR) was performed on all cells after a 3-day starvation in estrogen free medium. Cells were seeded the day prior to treatment in six-well plates at a density of 300 000 cells per well. Cells were treated with all treatments for 48 h, after which they were harvested in Trizol reagent (Invitrogen, Carlsbad, CA) and then frozen at −80 °C. RNA was isolated using RNeasy mini kit (Quiagen, Valencia, CA) according to the manufacturer’s instructions. cDNA was synthesized using high capacity cDNA reverse transcription kit (Applied Bioscience, Carlsbad, CA) according to the manufacturer’s instructions and using 1 μg of purified RNA. Synthesized cDNA was diluted in nuclease-free water and used for RT-PCR. For RT-PCR a Power SYBR green PCR master mix was used (Applied Bioscience, Carlsbad, CA) according to the manufacturer’s instructions. RT-PCR was run using a 7900HT fast real time PCR system thermocycler (Applied Bioscience, Carlsbad, CA). Primers sequences that were used for human pS2 cDNA amplification are 5′-CATCGACGTCCCTCCAGAAGA-3′ sense and 5′-CTCTGGGACTAATCACCGTGCTG-3′ anti-sense; human progesterone receptor (PgR), 5′-CGTGCCTATCCTGCCTCTCAA-3′ sense and 5′-CCGCCGTCGTAACTTTCGT-3′ anti-sense; human GREB1 gene, 5′-CAAAGAATAACCTGTTGGCCCTGC-3′ sense and 5′-GACATGCCTGCGCTCTCATACTTA-3′ anti-sense; the reference gene 36B4, 5′-GTGTCCGACAATGGCAGCAT-3′ sense and 5′-GACACCCTCCAGGAAGCGA-3′ anti-sense. All primers were obtained from Integrated DNA Technologies Inc. (IDT, Coralville, IA) and were tested by plotting dissociation curves which gave single peaks for all primer pairs. The fold changes of the mRNA after treatments to vehicle controls were calculated using ΔΔCt method and then normalized, including standard deviations, to each of the corresponding E2 control values for each of the experiments.
Immunoblotting
MCF-7:WS8 cells were seeded on 10 cm Petri dishes at a density of 3 million cells per plate after being estrogen starved in phenol red-free RPMI 1640 medium for 3 days. The cells were treated for 24 h with the tested compounds, and the cells were subsequently washed with cold PBS (Life Technologies, Carlsbad, CA) and were lysed using 1× lysis buffer (Cell Signaling Technology Inc., Danvers, MA), which contained 1× Complete Mini protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN) and 1× phosphatase inhibitors (Calbiochem, Gibbstown, NJ). The cells were lysed for 60 min on ice and subsequently centrifuged at 12 000 rpm for 20 min. Supernatants were transferred in fresh tubes and stored at −20 °C. The concentration of proteins in the lysates were measured using a Pierce BCA protein assay (Thermo Scientific, Rockfold, IL) according to the manufacturer’s instructions. An amount of 20 μg of each protein sample, diluted in a NuPAGE loading dye (Life technologies, Carlsbad, CA), was loaded and separated on NuPAGE 4–12% Bis-Tris gel (Life technologies, Carlsbad, CA). After electrophoresis the samples were transferred onto Hybond enhanced chemiluminescence (ECL) nitrocellulose membranes (Amersham Biosciences, Piscataway, NJ), which were subsequently blocked with blocking solution with TBS-T (Tris-Bis saline with Tween 20:50 nM Tris-HCl, pH 7.5, 150 nM NaCl, 0.1% Tween-20), containing 5% skim milk for 1 h at room temperature. The membranes were subsequently probed with primary antibodies anti-ERα, (Santa Cruz Biotechnology, Santa Cruz, CA) and with anti-β-actin (Sigma-Aldrich, St. Louis, MO) diluted in blocking buffer at ratios recommended by the supplier at 4 °C overnight. The membranes were washed three times for 10 min with TBS-T buffer and subsequently incubated with the appropriate horseradish peroxidase (HRP) linked secondary antibodies (anti-mouse or anti-rabbit from Cell Signaling Technology Inc., Danvers, MA) diluted in blocking buffer for 1 h at room temperature. The membranes were washed again as described above with TBS-T buffer, and the signal was visualized using ECL Western blotting detection reagents (PerkinElmer, Waltham, MA). All results were replicated in three independent experiments, and each result was analyzed by densitometry using Image J imaging software (NIH). Pixel intensities of all lanes were normalized to their corresponding β-actin lanes with background intensity subtracted and were normalized to vehicle control as 100%.
Molecular Modeling. Ligand Preparation
The three-dimensional structures of the ligands to be docked were generated and prepared for docking using the LigPrep utility (LigPrep, version 2.5; Schrödinger, LLC: New York, NY, 2011). In this stage a series of treatments are applied to the structures. For example, conversions are performed and then corrections are applied to the structures, ionization states (pH 7 ± 0.4) and tautomers are generated, and finally the geometries are optimized using OPLS_2005 force field.
Proteins Selection and Preparation
The experimental X-ray structures of ERα LBD to be used for docking were selected from Protein Databank45 based on the three-dimensional shape similarity between the compounds to be docked and cocrystallized ligands extracted from the receptor–ligand complexes. The three-dimensional shape similarity was computed using the ROCS utility of Openeye. As query data set, the ligands of interest were used while the screening library was compiled from the ligands extracted from all the available crystal structures of human ERα deposited in PDB. Shape Tanimoto parameter was used for scoring with a cutoff value of 0.8, and four ligands met this criterion. The 3D coordinates of the corresponding ERα complexes were extracted from PDB entries 3ERT,301UOM,322OUZ33 (antagonist conformations of the receptor) and 3Q9731 (agonist conformation). For comparison reasons, the other two experimental structures of the agonist conformation of ERα were extracted, PDB entries 1GWR (ERα cocrystallized with E2)29 and 3ERD (the receptor cocrystallized with diethylstilbestrol, DES).30
Subsequently, the structures were prepared for docking using the Protein Preparation Workflow (Schrödinger, LLC, New York, NY, 2011) accessible from within the Maestro program (Maestro, version 9.2; Schrödinger, LLC: New York, NY, 2011). Shortly, the hydrogens were properly added to the complexes, water molecules beyond 5 Å from a heteroatom were deleted, bond corrections were applied to the cocrystallized ligands, and the orientation of hydroxyl groups, Asn, Gln, and the protonation state of His were optimized to maximize hydrogen bonds formation. All Asp, Glu, Arg, and Lys residues were left in their charged state. In the final stage a restrained minimization on the ligand–protein complexes was carried out with the OPLS_2001 force field and the default value for rmsd of 0.30 Å for non-hydrogen atoms was used. Docking simulations were performed with Glide software (Glide, version 5.7; Schrödinger, LLC: New York, NY, 2011), a grid-based docking method that can be run rigid or fully flexible for the ligand.46,47 To some extent, a degree of flexibility was allowed to the X-ray structures of ERα in agonist conformation by scaling down the van der Waals radii of nonpolar atoms with a scale factor of 0.8 and allowing the free rotation of hydroxyl groups. The van der Waals radii of ligands nonpolar atoms were kept to the default value of the scaling factor of 0.8. The receptor grids were generated using the prepared proteins, with the docking grids centered on the center of the bound ligand for each receptor. The binding sites were enclosed in a grid box of 10 Å3 with default parameters and without constrains. The generated ligand poses were evaluated with Schrodinger’s proprietary version of ChemScore empirical scoring function, GlideScore.47 This algorithm recognizes favorable hydrophobic, hydrogen-bonding, and metal-ligation interactions like ChemScore but adds a steric-clash term and buried polar terms to penalize electrostatic discrepancies. However, the composite energy scoring function, Emodel, was used to select the best-docked pose for each ligand.47,48 This energy function is a combination of the ligand–receptor molecular mechanics interaction energy, the binding affinity predicted by GlideScore, and the ligand strain energy (for flexible docking). For each receptor and docking run five poses were retrieved and the best ones were selected based on the Emodel score.
Reagents and Supplies
Estradiol (E2), 4-hydroxytamoxifen (4OHT), endoxifen (Z-isomer), bovine insulin, and mouse anti-β-actin antibodies were all obtained from Sigma-Aldrich, St. Louis, MO. Fetal bovine serum (FBS), 2 mM glutamine, penicillin at 100 U/mL, streptomycin at 100 μg/mL, 1× nonessential amino acids, RPMI 1640 with phenol red and without media, DMEM media with and without phenol red, PBS buffer, Trizol reagent, NuPAGE loading dye, and NuPAGE 4–12% Bis-Tris gel were all obtained from Life Technologies, Carlsbad, CA. Fluorescent DNA quantification kit was obtained from Bio-Rad, Hercules, CA. RNeasy Mini isolation kits were obtained from Quiagen, Valencia, CA. High capacity cDNA reverse transcription kit and Power SYBR green PCR master mix were obtained from Applied Bioscience, Carlsbad, CA. All primers were obtained from Integrated DNA Technologies Inc., Coralville, IA. The 1× lysis buffer and anti-mouse and anti-rabbit horseradish peroxidase (HRP) linked secondary antibodies were purchased from Cell Signalling Technology Inc., Danvers, MA. The 1× Complete Mini protease inhibitor cocktail were from Roche Diagnostics, Indianapolis, IN. The 1× phosphatase inhibitors were from Calbiochem, Gibbstown, NJ. Pierce BCA protein assay was obtained from Thermo Scientific, Rockfold, IL. Hybond enhanced chemiluminescence (ECL) nitrocellulose membranes were from Amersham Biosciences, Piscataway, NJ. Primary rabbit anti-ERα antibodies were obtained from Santa Cruz Biotechnology, Santa Cruz, CA. ECL Western blotting detection reagents were from PerkinElmer, Waltham, MA.
Statistical Analysis
Statistical analysis of the data was performed for each of the repeated experiments separately using standard t test, paired and two-tailed in Microsoft Excel. P values less than 0.05 were considered significant.
Acknowledgments
This work was supported by the Department of Defense Breast Program (Award W81XWH-06-1-0590) Center of Excellence, the Susan G. Komen for the Cure Foundation (Award SAC100009), and the Lombardi Comprehensive Cancer Center Support Grant (Core Grant NIH P30 CA051008). The views and opinions of the author do not reflect those of the U.S. Army or the Department of Defense. R.F.C. is supported by UEFISCU-CNCSIS, Grant PN-II-RU-PD_502 No. 174/2010.
Glossary
Abbreviations Used
- FR
fixed ring
- ER
estrogen receptor
- pS2
trefoil factor 1 gene, aka TFF1
- GREB1
growth regulation by estrogen in breast cancer 1 gene
- PgR
progesterone receptor gene
- Prl
prolactin gene
- TPP
triphenylphosphine
- DIAD
diisopropyl azodicarboxylate
- RT-PCR
real time polymerase chain reaction
- E2
17β-estradiol
- vdW
van der Waals parameter
Supporting Information Available
Figures showing structure and docking poses. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Maximov P. Y.; McDaniel R. E.; Jordan V. C.. Tamoxifen: Pioneering Medicine in Breast Cancer; Springer: Basel, Switzerland, 2013. [Google Scholar]
- Davies C.; Godwin J.; Gray R.; Clarke M.; Cutter D.; Darby S.; McGale P.; Pan H. C.; Taylor C.; Wang Y. C.; Dowsett M.; Ingle J.; Peto R. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet 2011, 378, 771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies C.; Pan H.; Godwin J.; Gray R.; Arriagada R.; Raina V.; Abraham M.; Medeiros Alencar V. H.; Badran A.; Bonfill X.; Bradbury J.; Clarke M.; Collins R.; Davis S. R.; Delmestri A.; Forbes J. F.; Haddad P.; Hou M. F.; Inbar M.; Khaled H.; Kielanowska J.; Kwan W. H.; Mathew B. S.; Mittra I.; Muller B.; Nicolucci A.; Peralta O.; Pernas F.; Petruzelka L.; Pienkowski T.; Radhika R.; Rajan B.; Rubach M. T.; Tort S.; Urrutia G.; Valentini M.; Wang Y.; Peto R. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 2013, 381, 805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher B.; Costantino J. P.; Wickerham D. L.; Cecchini R. S.; Cronin W. M.; Robidoux A.; Bevers T. B.; Kavanah M. T.; Atkins J. N.; Margolese R. G.; Runowicz C. D.; James J. M.; Ford L. G.; Wolmark N. Tamoxifen for the prevention of breast cancer: current status of the National Surgical Adjuvant Breast and Bowel Project P-1 study. J. Natl. Cancer Inst. 2005, 97, 1652–1662. [DOI] [PubMed] [Google Scholar]
- Harper M. J.; Walpole A. L. Contrasting endocrine activities of cis and trans isomers in a series of substituted triphenylethylenes. Nature 1966, 212, 87. [DOI] [PubMed] [Google Scholar]
- Allen K. E.; Clark E. R.; Jordan V. C. Evidence for the metabolic activation of non-steroidal antioestrogens: a study of structure–activity relationships. Br. J. Pharmacol. 1980, 71, 83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan V. C.; Collins M. M.; Rowsby L.; Prestwich G. A monohydroxylated metabolite of tamoxifen with potent antioestrogenic activity. J. Endocrinol. 1977, 75, 305–316. [DOI] [PubMed] [Google Scholar]
- Lien E. A.; Solheim E.; Kvinnsland S.; Ueland P. M. Identification of 4-hydroxy-N-desmethyltamoxifen as a metabolite of tamoxifen in human bile. Cancer Res. 1988, 48, 2304–2308. [PubMed] [Google Scholar]
- Lien E. A.; Solheim E.; Lea O. A.; Lundgren S.; Kvinnsland S.; Ueland P. M. Distribution of 4-hydroxy-N-desmethyltamoxifen and other tamoxifen metabolites in human biological fluids during tamoxifen treatment. Cancer Res. 1989, 49, 2175–2183. [PubMed] [Google Scholar]
- Borgna J. L.; Rochefort H. Hydroxylated metabolites of tamoxifen are formed in vivo and bound to estrogen receptor in target tissues. J. Biol. Chem. 1981, 256, 859–868. [PubMed] [Google Scholar]
- Johnson M. D.; Zuo H.; Lee K. H.; Trebley J. P.; Rae J. M.; Weatherman R. V.; Desta Z.; Flockhart D. A.; Skaar T. C. Pharmacological characterization of 4-hydroxy-N-desmethyl tamoxifen, a novel active metabolite of tamoxifen. Breast Cancer Res. Treat. 2004, 85, 151–159. [DOI] [PubMed] [Google Scholar]
- Lim Y. C.; Desta Z.; Flockhart D. A.; Skaar T. C. Endoxifen (4-hydroxy-N-desmethyl-tamoxifen) has anti-estrogenic effects in breast cancer cells with potency similar to 4-hydroxy-tamoxifen. Cancer Chemother. Pharmacol. 2005, 55, 471–478. [DOI] [PubMed] [Google Scholar]
- Lim Y. C.; Li L.; Desta Z.; Zhao Q.; Rae J. M.; Flockhart D. A.; Skaar T. C. Endoxifen, a secondary metabolite of tamoxifen, and 4-OH-tamoxifen induce similar changes in global gene expression patterns in MCF-7 breast cancer cells. J. Pharmacol. Exp. Ther. 2006, 318, 503–512. [DOI] [PubMed] [Google Scholar]
- Lieberman M. E.; Gorski J.; Jordan V. C. An estrogen receptor model to describe the regulation of prolactin synthesis by antiestrogens in vitro. J. Biol. Chem. 1983, 258, 4741–4745. [PubMed] [Google Scholar]
- Jordan V. C.; Haldemann B.; Allen K. E. Geometric isomers of substituted triphenylethylenes and antiestrogen action. Endocrinology 1981, 108, 1353–1361. [DOI] [PubMed] [Google Scholar]
- Jordan V. C.; Koch R.; Langan S.; McCague R. Ligand interaction at the estrogen receptor to program antiestrogen action: a study with nonsteroidal compounds in vitro. Endocrinology 1988, 122, 1449–1454. [DOI] [PubMed] [Google Scholar]
- McCague R.; Leclercq G.; Jordan V. C. Nonisomerizable analogues of (Z)- and (E)-4-hydroxytamoxifen. Synthesis and endocrinological properties of substituted diphenylbenzocycloheptenes. J. Med. Chem. 1988, 31, 1285–1290. [DOI] [PubMed] [Google Scholar]
- Maximov P. Y.; Myers C. B.; Curpan R. F.; Lewis-Wambi J. S.; Jordan V. C. Structure–function relationships of estrogenic triphenylethylenes related to endoxifen and 4-hydroxytamoxifen. J. Med. Chem. 2010, 53, 3273–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal B.; Khanna J. M.; Anand N. Phenethylamine in a rigid framework. 2,3-Substituted cis- and trans-6-amino-6,7,8,9-tetrahydro-5H-benzocyclohepten-5-ols. J. Med. Chem. 1972, 15, 23–27. [DOI] [PubMed] [Google Scholar]
- Kahn A. M.; Proctor G. R.; Rees L. Novel aromatic systems. Part IV. Synthesis and dehydrogenation of 4′-hydroxy-1,2-benzocycloheptatrienes. J. Chem. Soc. 1966, 990–994. [Google Scholar]
- Jarman M.; McCague R. Heptafluoro-p-tolyl and tetrafluoro-4-pyridyl as novel and selective protecting groups for phenolic and alcoholic functions: synthesis and cleavage of perfluoroaryl ethers of steroids. J. Chem. Res., Synop. 1985, 114–115. [Google Scholar]
- Fauq A. H.; Maharvi G. M.; Sinha D. A convenient synthesis of (Z)-4-hydroxy-N-desmethyltamoxifen (endoxifen). Bioorg. Med. Chem. Lett. 2010, 20, 3036–3038. [DOI] [PubMed] [Google Scholar]
- Stearns V.; Johnson M. D.; Rae J. M.; Morocho A.; Novielli A.; Bhargava P.; Hayes D. F.; Desta Z.; Flockhart D. A. Active tamoxifen metabolite plasma concentrations after coadministration of tamoxifen and the selective serotonin reuptake inhibitor paroxetine. J. Natl. Cancer Inst. 2003, 95, 1758–1764. [DOI] [PubMed] [Google Scholar]
- Murahashi S. I.; Naota T.; Miyaguchi N.; Nakato T. Ruthenium-catalyzed oxidation of tertiary-amines with hydrogen-peroxide in the presence of methanol. Tetrahedron Lett. 1992, 33, 6991–6994. [Google Scholar]
- Lee H. W.; Ahn J. B.; Lee J. H.; Kang S. K.; Ahn S. K.; Ha D. C. Selective N-demethylation of tertiary aminofumagillols with selenium dioxide via a non-classical Polonovski type reaction. Heterocycles 2006, 68, 915–932. [Google Scholar]
- Murdter T. E.; Schroth W.; Bacchus-Gerybadze L.; Winter S.; Heinkele G.; Simon W.; Fasching P. A.; Fehm T.; Eichelbaum M.; Schwab M.; Brauch H. Activity levels of tamoxifen metabolites at the estrogen receptor and the impact of genetic polymorphisms of phase I and II enzymes on their concentration levels in plasma. Clin. Pharmacol. Ther. 2011, 89, 708–717. [DOI] [PubMed] [Google Scholar]
- Jordan V. C.; Fritz N. F.; Tormey D. C. Endocrine effects of adjuvant chemotherapy and long-term tamoxifen administration on node-positive patients with breast cancer. Cancer Res. 1987, 47, 624–630. [PubMed] [Google Scholar]
- Ravdin P. M.; Fritz N. F.; Tormey D. C.; Jordan V. C. Endocrine status of premenopausal node-positive breast cancer patients following adjuvant chemotherapy and long-term tamoxifen. Cancer Res. 1988, 48, 1026–1029. [PubMed] [Google Scholar]
- Warnmark A.; Treuter E.; Gustafsson J. A.; Hubbard R. E.; Brzozowski A. M.; Pike A. C. Interaction of transcriptional intermediary factor 2 nuclear receptor box peptides with the coactivator binding site of estrogen receptor alpha. J. Biol. Chem. 2002, 277, 21862–21868. [DOI] [PubMed] [Google Scholar]
- Shiau A. K.; Barstad D.; Loria P. M.; Cheng L.; Kushner P. J.; Agard D. A.; Greene G. L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95, 927–937. [DOI] [PubMed] [Google Scholar]
- Rajan S. S.; Kim Y.; Vanek K.; Joachimiak A.; Jordan C.; Greene G. L., Crystal structure of human estrogen receptor alpha LBD in complex with GRIP peptide and two ismoers of ethoxy triphenylethylene. Manuscript to be published.
- Renaud J.; Bischoff S. F.; Buhl T.; Floersheim P.; Fournier B.; Halleux C.; Kallen J.; Keller H.; Schlaeppi J. M.; Stark W. Estrogen receptor modulators: identification and structure–activity relationships of potent ERalpha-selective tetrahydroisoquinoline ligands. J. Med. Chem. 2003, 46, 2945–2957. [DOI] [PubMed] [Google Scholar]
- Vajdos F. F.; Hoth L. R.; Geoghegan K. F.; Simons S. P.; LeMotte P. K.; Danley D. E.; Ammirati M. J.; Pandit J. The 2.0 Å crystal structure of the ERalpha ligand-binding domain complexed with lasofoxifene. Protein Sci. 2007, 16, 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy C. S.; Langan-Fahey S. M.; McCague R.; Jordan V. C. Structure–function relationships of hydroxylated metabolites of tamoxifen that control the proliferation of estrogen-responsive T47D breast cancer cells in vitro. Mol. Pharmacol. 1990, 38, 737–743. [PubMed] [Google Scholar]
- Jordan V. C.; Lieberman M. E. Estrogen-stimulated prolactin synthesis in vitro. Classification of agonist, partial agonist, and antagonist actions based on structure. Mol. Pharmacol. 1984, 26, 279–285. [PubMed] [Google Scholar]
- Jordan V. C.; Lieberman M. E.; Cormier E.; Koch R.; Bagley J. R.; Ruenitz P. C. Structural requirements for the pharmacological activity of nonsteroidal antiestrogens in vitro. Mol. Pharmacol. 1984, 26, 272–278. [PubMed] [Google Scholar]
- Jordan V. C.; Koch R.; Mittal S.; Schneider M. R. Oestrogenic and antioestrogenic actions in a series of triphenylbut-1-enes: modulation of prolactin synthesis in vitro. Br. J. Pharmacol. 1986, 87, 217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy C. S.; Parker C. J.; McCague R.; Jordan V. C. Structure–activity relationships of nonisomerizable derivatives of tamoxifen: importance of hydroxyl group and side chain positioning for biological activity. Mol. Pharmacol. 1991, 39, 421–428. [PubMed] [Google Scholar]
- Lonard D. M.; Nawaz Z.; Smith C. L.; O’Malley B. W. The 26S proteasome is required for estrogen receptor-alpha and coactivator turnover and for efficient estrogen receptor-alpha transactivation. Mol. Cell 2000, 5, 939–948. [DOI] [PubMed] [Google Scholar]
- Obiorah I. E.; Sengupta S.; Curpan R.; Jordan V. C. Defining the conformation of the estrogen receptor complex that controls estrogen induced apoptosis in breast cancer. Mol. Pharmacol. 2014, 85, 789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson R. I.; Gee J. M.; Manning D. L.; Wakeling A. E.; Montano M. M.; Katzenellenbogen B. S. Responses to pure antiestrogens (ICI 164384, ICI 182780) in estrogen-sensitive and -resistant experimental and clinical breast cancer. Ann. N.Y. Acad. Sci. 1995, 761, 148–163. [DOI] [PubMed] [Google Scholar]
- Wu X.; Hawse J. R.; Subramaniam M.; Goetz M. P.; Ingle J. N.; Spelsberg T. C. The tamoxifen metabolite, endoxifen, is a potent antiestrogen that targets estrogen receptor alpha for degradation in breast cancer cells. Cancer Res. 2009, 69, 1722–1727. [DOI] [PubMed] [Google Scholar]
- Liu H.; Lee E. S.; Deb Los Reyes A.; Zapf J. W.; Jordan V. C. Silencing and reactivation of the selective estrogen receptor modulator–estrogen receptor alpha complex. Cancer Res. 2001, 61, 3632–3639. [PubMed] [Google Scholar]
- Jordan V. C. The 38th David A. Karnofsky Lecture: The paradoxical actions of estrogen in breast cancer—survival or death?. J. Clin. Oncol. 2008, 26, 3073–3082. [DOI] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Murphy R. B.; Repasky M. P.; Frye L. L.; Greenwood J. R.; Halgren T. A.; Sanschagrin P. C.; Mainz D. T. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.