

Abstract
Endotoxin tolerance was first described in a study that exposed animals to a sublethal dose of bacterial endotoxin. The animals subsequently survived a lethal injection of endotoxin. This refractory state is associated with the innate immune system and, in particular, with monocytes and macrophages, which act as the main participants. Several mechanisms are involved in the control of endotoxin tolerance; however, a full understanding of this phenomenon remains elusive. A number of recent reports indicate that clinical examples of endotoxin tolerance include not only sepsis but also diseases such as cystic fibrosis and acute coronary syndrome. In these pathologies, the risk of new infections correlates with a refractory state. This review integrates the molecular basis and clinical implications of endotoxin tolerance in various pathologies.
Introduction
The mammalian innate immune system (IIS) is able to detect and respond to danger signals from various sources such as bacteria, tumor processes, and tissue damage [1]. The IIS constitutes the first line of defense for preventing pathogen colonization and tumor growth. The principal players in this system are the innate immune cells: monocytes/macrophages (MΦs), neutrophils, and natural killers. Although the IIS was initially believed to be nonspecific, the discovery of Toll-like receptors (TLRs) in drosophila, and subsequently in mammalian innate immune cells, changed the paradigm of the scarce specificity of the IIS [2]. The expression of different receptor families in IIS cells accounts for the finely tuned specificity of the IIS. Kumar and colleagues [3] conducted an in-depth review of a number of these families, including membrane-bound receptors (for example, TLRs and C-type lectin receptors) and intracellular receptors such as RIG-like and NOD-like receptors [4]. The most studied among these receptors are TLRs, particularly TLR4, which is involved in the recognition of Gram-negative bacteria and their associated endotoxins such as lipopolysaccharide (LPS).
Upon detecting danger signals, IIS cells trigger a robust inflammatory response. However, this reaction must be closely regulated because uncontrolled inflammation leads to clinical complications (for example, septic shock, cancer, and autoimmune diseases). In the event of bacterial colonization, a number of mechanisms regulate the inflammation and protect against shock. One of the most important mechanisms for protecting the host is endotoxin tolerance (ET). Under ET, cells and organisms exhibit a transient state in which they are unable to respond to endotoxin challenges [5]. The phenomenon has been described as a type of tolerance in which exposure to low concentrations of endotoxins reprograms IIS cells. In other words, the innate response to further endotoxin challenges is compromised. However, this does not constitute an ‘immunoparalysis’ but rather an alternative activation that triggers other mechanisms that have yet to be described. The presence of ET has been studied and reported in several pathologies such as sepsis [6] cystic fibrosis (CF) [7], acute coronary syndrome (ACS) [8], and trauma and pancreatitis [9,10] (Figure 1). In the past few years, a growing number of studies have focused on the main characteristics of ET, revealing much about its mechanisms. This review discusses the main characteristics of ET, ET models, and the molecules involved in this process and its clinical significance.
Figure 1.
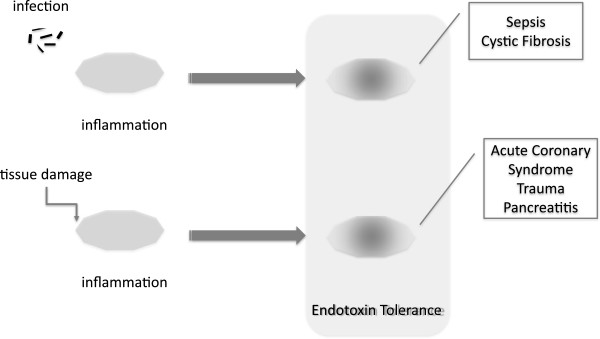
Endotoxin tolerance has been reported in several pathologies such as sepsis, cystic fibrosis, acute coronary syndrome, trauma, and pancreatitis. Cells from the innate immune system develop a refractory state after infection or tissue damage.
From the clinic to the bench
The first reported observation of ET was made by Paul Beeson in 1946. He reported that the repeated inoculation of rabbits with typhoid shots caused a significant reduction in the vaccine-induced fever [11]. This phenomenon also occurred in humans who were recovering from malaria; when rechallenged with endotoxins, the patients also presented reduced fever [12]. This response suggested that inoculation with live Salmonella typhosa led to reduced symptoms in response to the endotoxin or killed bacteria [13]. A similar observation was reported in volunteers inoculated with Plasmodium cynomolgi through mosquito bites [14]. This finding indicates that there is also a cross-tolerization among various stimuli. Other early examples of ET were found in patients with pyelonephritis [15] and in patients recovering from typhoid and paratyphoid fever [16]. These reports indicated the widespread existence of this phenomenon in the clinical setting.
Later experiments in mice showed that once the animals had received a sublethal dose of LPS, they were protected from a subsequent lethal dose of the endotoxin. These studies also demonstrated the crucial role of MΦs in ET in vivo[6]. Murine macrophages and human monocytes are unable to mount a standard inflammatory response after an endotoxin challenge if they have been previously exposed to the endotoxin [5].
In vitro and in vivo models
Several models have been established to study the ET phenomenon in depth (Figure 2). The majority attempt to reproduce the effect of two consecutive pathogen infections, either in vitro or in vivo.
Figure 2.
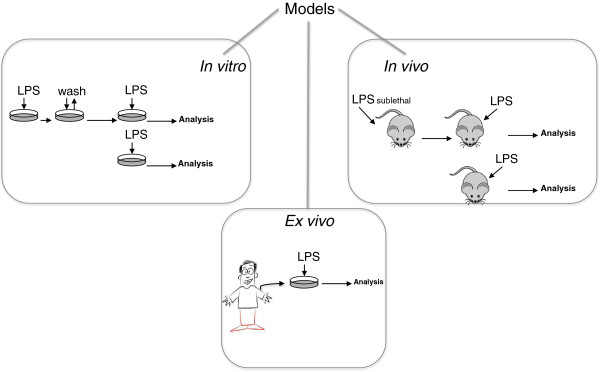
Models established to study the endotoxin tolerance phenomenon. LPS, lipopolysaccharide.
The classic in vitro human ET models are based on a culture of human blood cells stimulated twice with LPS [17]. First, a low dose of endotoxin is administered (0.001 to 0.01 μg/mL); the cells are washed, and a second high dose of LPS (0.1 to 1 μg/mL) is administered [18,19]. This experimental approach has been applied to myeloid cell lines such as the promonocytic THP-1 [2], circulating MΦs [6], and total peripheral blood mononuclear cells [4]. However, a number of these models do not adequately approximate the clinical situation. For example, patients who experience bacteremia subsequently develop an endotoxin tolerance episode during which their IIS does not respond to new pathogens as it did prior to the bacteremia [20]. This situation can be modeled by two consecutive LPS treatments, with the first and the second challenge separated in time. Data from a study that used this type of model indicated that only 1 hour of LPS exposure was required to induce an ET. However, this refractory state is not permanent, and after 5 days, the cells reverted to a proinflammatory phenotype in response to endotoxin stimulation [17]. These findings imply that IIS cells possess a type of ‘memory’ that may be governed by epigenetic changes [21]. Other characteristics of ET, such as high phagocytosis activity of ET monocytes and their inability to step up the adaptive response, have been discovered by using the abovementioned model [17,22]. Both characteristics were verified in patients experiencing a patent endotoxin tolerance such as CF [17].
Mouse in vivo models have been developed to further confirm the physiological relevance of the tolerance process and have helped confirm in vitro-obtained data [23,24] and address the contribution of various cell types to the overall ET outcome [25]. However, an elegant work based on genomics has pointed out how mouse models reproduce weakly inflammatory responses in humans [26]. It does not mean that results obtained from murine models are not useful; rather, it means that these very models should fit the symptoms and etiology of human diseases, in order to provide pathophysiological relevant data [27].
Perhaps the most relevant data, especially in regard to specific gene expression patterns, resulted from human in vivo ET studies, in which volunteers were injected with defined doses of LPS (approximately 1 ng/kg of body weight) and their response monitored [28-30]. These studies observed reductions in proinflammatory cytokines (TNF-α and IL-6) and anti-inflammatory cytokines (IL-10 and IL-1RA), suggesting that, under in vivo ET conditions, the overall response is much more complex than indicated by in vitro studies. In vivo data reveal the relevance of antimicrobial mechanisms displayed by cells that are not commonly studied in vitro in an ET context, such as neutrophils [25].
The ex vivo approach is an intermediate experimental solution that improves researcher control over the secondary endotoxin stimulus. In these models, cells of interest are re-exposed to LPS in vitro after an initial challenge with the endotoxin in vivo[31,32]. Data obtained from this approach are important for highlighting the differences between the more common in vitro data and the complex full in vivo findings [11]. This type of assay is also useful for determining whether patients’ blood cells exhibit ET [6,7,17]. As Kox and colleagues [11] have pointed out, there is, however, a difference between ex vivo and in vivo that needs to be taken into account. Although a patient’s circulatory cells may exhibit an inflammatory phenotype ex vivo, the patient may be in a tolerant status [11]. These authors demonstrated that when volunteers were injected with endotoxin, their cells exhibited an inflammatory profile when isolated for 1 week and then challenged ex vivo with LPS. In contrast, the cells showed significant attenuation of inflammation in vivo when the volunteers were re-injected with the endotoxin 1 week after the first treatment.
In any case, the approach based on using clinically relevant samples from patients ‘locked’ into an ET state seemingly provides highly valuable data, especially regarding the description of molecular mechanisms (for example, in CF [7], ACS [8], and tumoral processes [33,34]).
In Table 1, we have reviewed the main ET characteristics found in various tissues according to the models used.
Table 1.
Main endotoxin tolerance features found in different tissues according to the models used
| Tissue/cell type | Model | Stimuli a | Most relevant phenotype | Reference |
|---|---|---|---|---|
|
bPeritoneal macrophage |
In vitro |
LPS + LPS |
TNF-α, IL-1β, GMCSF, CCL2, CCL4, CXCL10, CXCL2 |
[35] |
|
bPeritoneal macrophage |
In vitro |
LPS + LPS |
↓ TNF-α, IL-1β, IL-12p40, COX2, CXCL10, ↓ TNF-α, IL-1β, IL-12p40, COX2, CXCL10 = TNF-α, IL-1β, ↓ IL-12p40, COX2, CXCL10, ↓ TNF-α, IL-1β, IL-12p40, COX2, ↑CXCL10 = TNF-α, ↓ CXCL10, ↓ TNF-α, ↑ CXCL10 |
[36] |
| MALP2 + MALP2 | ||||
| LPS + MALP2 | ||||
| MALP2 + LPS | ||||
| Poli(I:C) + LPS | ||||
| R848 + LPS | ||||
|
bPeritoneal macrophage |
In vitro |
LPS + LPS |
↓ TNF-α, IFN-β |
[37] |
| Pam3Cys + Pam3Cys |
↓ TNF-α , = TNF-α |
|||
| LPS + Pam3Cys |
↑ TNF-α, = IFN-β |
|||
| Pam3Cys + LPS | ||||
|
bBMMs |
In vitro |
LPS + LPS |
↓ inflammatory genes (IL-6, IL-1β, MMP3) |
[28]c |
| ↑ antimicrobial genes (Fpr1, Oasl1) | ||||
|
bBMMs |
In vitro |
LipidA + LipidA |
↓ TNF-α, CCL3, ↑ IFN-β, IL-10 |
[38] |
| Pam3Cys + Pam3Cys |
↓ TNF-α, CCL3 |
|||
| LipidA + Pam3Cys |
= TNF-α, CCL3, IFN-β |
|||
| = TNF-α, CCL3, IFN-β | ||||
| Pam3Cys + LipidA |
↓ TNF-α, CCL3, ↑ IFN-β, IL-10 |
|||
| Poli(I:C) + LipidA | ||||
|
dMonocytes |
In vitro |
LPS + LPS |
↓ inflammatory cytokines/chemokines |
[17]c |
| ↑ IFN-β, TGF-β | ||||
| ↑ Scavenger receptors | ||||
| ↑ Phagocytic capacity | ||||
| ↓ Antigen presentation potential | ||||
|
dPBMCs |
In vitro |
LPS + LPS |
↓ TNF-α, IL−12, ↑ IL-10, COX2 |
[19] |
|
ePBMCs |
In vitro |
LPS + LPS |
↓ TNF-α, IL-12, ↑ IL-10 |
[39] |
|
dSeptic monocytes |
Ex vivo |
Monocytes from patients with sepsis + LPS |
↓ TNF-α, IL6, IL-1β |
[6] |
|
dWhole blood |
Ex vivo |
LPS injection + ex vivo restimulation |
↓ TNF-α, IL6, IL-10 |
[11] |
|
bPeritoneal/splenic macrophage |
Ex vivo |
LPS injection + ex vivo restimulation |
↓ TNF-α, IL6, CXCL8 = IL-10, IL-1RA |
[40] |
|
fMammary tissue |
In vivo |
LPS + E. coli injection |
↓ TNF-α, IL6, CXCL8, CCL20, ↑ IL-10 |
[41] |
|
dSerum |
In vivo |
5 consecutive LPS injections |
↓ elastase, TNF-α, IL-6, IL-10, IL-1RA = TGF-β |
[42] |
| dPlasma | In vivo | 2 consecutive LPS inoculations | ↓ TNF-α, IL6, IL-10, IL-1RA, TGF-β | [11] |
aLipopolysaccharide (LPS) – TLR4 ligand; MALP2 – TLR2 ligand; Poli(I:C) – TLR3 ligand; R848 – TLR7/8 ligand; LipidA – TLR4 ligand; bmurine; cgenome-wide analysis; dhuman; eequine; fbovine. BMM, bone marrow-derived macrophage; IL, interleukin; MALP2, macrophage-activating lipopeptide-2; PBMC, peripheral blood mononuclear cell; TGF-β, transforming growth factor-beta; TLR, Toll-like receptor; TNF, tumor necrosis factor.
Mechanism implicated: main controllers and pathways involved
Relevant information on a number of mechanisms involved in ET development has been obtained from the previously mentioned models. Initially, the soluble mediators generated during the first endotoxin challenge were considered essential for ET. Therefore, we can state that the role of the immunomodulatory cytokine IL-10 was paradigmatic [43]. The neutralization of IL-10 by blocking antibodies during the first LPS stimulation induced non-tolerant TNF-α production in response to a second endotoxin challenge [44]. However, IL-10-deficient mice still developed ET [45], suggesting the existence of alternative mechanisms involved in this process.
In-depth studies of ET development in gene-deficient mice have analyzed the participation of intracellular molecules in this process and have established the roles of SHIP-1, A20, and IRAK-M, roles that have been observed occasionally in different models [22,40]. The pseudokinase IRAK-M could be considered a ‘master regulator’ of ET because it is one of the genes that is consistently induced into ET [46,47] and has been implicated in a number of human pathologies – such as sepsis [6], cancer [34], ACS [8], and asthma [48] – during which ET manifests. This protein belongs to the four-member interleukin-1 receptor-associated kinase (IRAK) family. Its systematic nomenclature (IRAK-3) is usually substituted with the more common one (IRAK-M) because of its restricted expression pattern in the myeloid cell compartment [49]. Owing to a mutation in its kinase domain, IRAK-M is unable to exert a kinase activity [50]. Kobayashi and colleagues [30] reported the first link between ET and IRAK-M; they described IRAK-M-deficient mice as unable to develop ET in vivo. As indicated by genome-wide data, IRAK-M is not expressed in myeloid cells under steady-state conditions, and its expression is rapidly induced by the first LPS challenge [6]. Both structural analysis and indirect evidence suggest that the mechanism by which IRAK-M regulates the LPS response is related to the inhibition of the signaling pathway downstream of TLR4 [51,52].
A number of studies have supported the importance of IRAK-M induction during ET development. In a human in vitro model of ET, rapid IRAK-M upregulation was described, as was its expression in freshly isolated monocytes from patients with sepsis [6]. Similar results were obtained with LPS from Porphyromonas gingivalis in THP-1 promonocytic cells [53] and in Kupffer cells [54]. More importantly, IRAK-M upregulation was associated with higher mortality after Gram-negative-induced sepsis [47]. We reviewed the role of IRAK-M in ET in detail in an earlier study [52].
Other molecules – such as the orphan immunoreceptor TREM-1 (triggering receptor expressed on myeloid cells 1), which sustains cell surface expression in human monocytes – have been identified as important participants in ET development. Through metalloproteinase inhibition, TREM-1 counteracts the well-characterized downregulation of several proinflammatory cytokines during the ET time frame [55]. Moreover, its expression is notably downregulated in monocytes that are isolated from patients with ET, in contrast to those extracted from healthy volunteers, as discussed below [7]. This orphan immunoreceptor magnifies inflammation after TLR activation in myeloid cells and is implicated in a number of inflammatory pathologies [56].
Given that microRNAs (miRNAs) are able to regulate gene expression at the pro-transcriptional level, these factors have been studied in the context of ET. Several authors have described the activation of a number of miRNAs during ET, including miR-146a, miR-221, miR-579, miR-125b, miR-155, let-7e, and miR-98 [57]. These molecules represent new targets for modulating the development of ET.
Other pathways and molecules are involved in controlling ET [22,58] and a number of them require in-depth study to verify their potential role in this phenomenon. Figure 3 summarizes the main ET mechanisms reported in MΦs.
Figure 3.
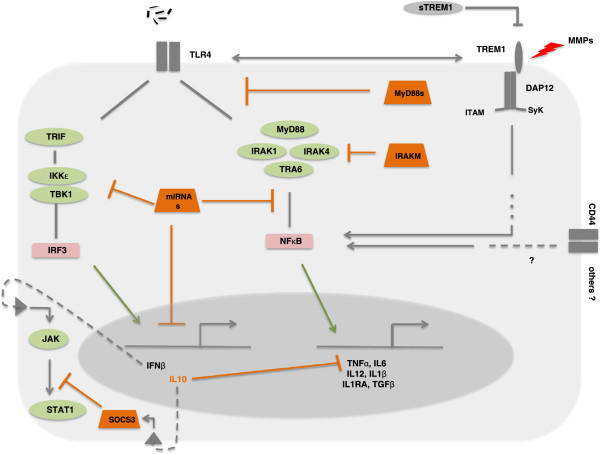
Molecular mechanisms implicated in endotoxin tolerance. DAP12, DNAX activation protein of 12 kDa; IFNβ, interferon-beta; IL, interleukin; IKK, IκB kinase; IRAK, interleukin-1 receptor-associated kinase; IRF3, interferon regulatory transcription factor 3; ITAM, immunoreceptor tyrosine-based activation motif; JAK, Janus kinase; miRNA, microRNA; MMP, matrix metalloproteinase; MyD88, myeloid differentiation primary response gene 88; NF-κB, nuclear factor-kappa-B; SOCS3, suppressor of cytokine signaling proteins 3; STAT1, signal transducer and activator of transcription 1; sTREM1, soluble triggering receptor expressed on myeloid cells 1; SyK, Spleen tyrosine kinase; TBK1, TANK-binding kinase 1; TGF-β, transforming growth factor-beta; TLR4, Toll-like receptor 4; TNF-α, tumor necrosis factor-alpha; TRAF6, TNF receptor associated factor (TRAF) protein family 6; TREM1, triggering receptor expressed on myeloid cells 1; TRIF, TIR-domain-containing adapter-inducing interferon-β.
From the bench to the clinic
At the bedside
By the end of the 20th century, ET had been described in patients, and research on ET moved from the clinic to the laboratory. As we mentioned earlier, a number of models were constructed and data on ET began to emerge. The use of these models, however, revealed other previously unreported characteristics, and researchers had to return to the clinic to verify the models. In addition, important innate immune response occurred not only in sepsis but also in several other pathologies in which the ET phenotype had been reported.
Sepsis: where endotoxin tolerance was first described
A clinically relevant example of ET was observed in patients with sepsis [59]. This complex pathology results from a deregulated inflammatory response by the IIS following a systemic bacterial infection. The evolution of the ET includes two phases. First, patients suffer from an overt inflammation that leads to an immunocompromised phase [60,61]. During the second stage, the patients’ innate immune cells show a patent ET [6,18]. Clinically, this state is correlated with a high risk of secondary infection and mortality [62]. Monocytes from patients with sepsis exhibit numerous characteristics of ET. For example, after an ex vivo challenge with LPS, the monocytes fail to produce proinflammatory cytokines, such as TNF-α, IL-12, IL-23, and IL-6 [42,62-64], in contrast to monocytes from healthy volunteers. Downregulation of major histocompatibility (MHC) class II, CD86, and class II transactivator (CIITA) has also been observed in circulating cells from patients with sepsis [65,66]. However, the expression of anti-inflammatory factors such as IL-10 is controversial in this context. Several authors have pointed out that IL-10 plays a crucial role in the control of ET [19,67], whereas others have reported a weak effect for IL-10 in sepsis-induced tolerance [68]. Studies have validated IL-10 as a predictor of poor prognosis in sepsis and have shown that IL-10 upregulation was correlated with higher mortality in patients with sepsis [69]. In addition, it has been reported that IL-10 induces miR-187, which is a negative regulator of TNF-α, IL-6, and IL-12 expression in TLR4-stimulated monocytes [70]. However, as we mentioned earlier, IL-10 knockout mice reproduced an ET phenotype when they were twice exposed to LPS [45]. Expression of other anti-inflammatory factors, such as transforming growth factor-beta, IL-1RA, SLPI, and glucocorticoids, was reported in septic cells and is believed to contribute to ET [5,62,71]. The failure of anti-inflammatory therapies in sepsis and the high mortality of immunocompromised patients indicate the importance of the immunosuppressive phase of sepsis and, consequently, the ET phenotype of IIS cells.
Cystic fibrosis: a ‘marriage’ of convenience
CF is another complex disease in which ET has been described. CF essentially affects all exocrine epithelia [72] and results from abnormalities in the gene that codifies the chloride channel known as the CF transmembrane conductance regulator, which belongs to the family of ATP-binding cassette transporter ATPases [72]. The clinical features of CF include primary organ damage (pancreas, sinus, liver, intestines, and exocrine pancreas) and secondary complications such as malnutrition and diabetes. However, the morbidity and mortality of patients with CF are usually the result of chronic lower airway bacterial infections and inflammation of the lungs [73]. Repeated episodes of polymicrobial infection in these patients cause a progressive deterioration of lung tissue, a decline in pulmonary function, and, ultimately, respiratory failure and death in 90% of patients with CF [74]. The high frequency of pathogen colonization observed in these patients points to a significant deficiency in their IIS [75,76]. In previous studies, we have reported a patent ET status in circulating monocytes isolated from patients with CF [7,17]. In addition to the inability of these cells to mount a standard inflammatory response after an ex vivo endotoxin challenge, other primary features of ET were observed (for example, high phagocytosis ability, downregulation of MHC-II and CIITA, and poor antigen presentation) [7,17]. Low expression of TREM-1 at the cell surface has also been detected in circulating CF-MΦs [7]. The low levels of TREM-1 expression in circulating CF-MΦs partially explain the non-responsive state in patients with CF. Moreover, high LPS concentrations have been detected in CF plasma [76]. The amount of endotoxin in plasma is correlated to the inability of the patients’ monocytes to generate inflammation ex vivo. This contributes decisively to the permanent ET status in patients with CF. These data indicate that patients with CF are ‘locked’ into an ET state, which results from an endotoxin diffusion in the bloodstream. This condition could be beneficial for the lifespan of patients with CF because it allows them to mount a moderate innate response against pathogen colonization. In this context, patients with CF avoid a permanent ‘cytokine storm’ that could compromise their lives.
Acute coronary syndrome: an example of heterotolerance
ACS includes a range of thrombotic coronary artery diseases such as unstable angina, ST-elevation myocardial infarction (STEMI), and non-ST-elevation myocardial infarction (NSTEMI). The IIS plays a key role in the progression of the atherosclerotic lesions and in the remodeling process after myocardial infarction (MI) [77-79]. In this context, the activation of the innate immune response mediated by MΦs leads to the release of factors that cause inflammation [80], tissue damage, and plaque instability [78]. Circulating MΦs in patients with ACS after 1 to 3 hours of MI (STEMI and NSTEMI) showed a proinflammatory phenotype, with upregulation of TNF-α [8]. These MΦs had high levels of IRAK-M, thus providing negative feedback regulation for the proinflammatory response. This is a classic paradigm of ET producing a hyporesponsive state following an LPS challenge [6,52]. Moreover, when MΦs were analyzed several hours after MI, a shift from its initial proinflammatory response to its anti-inflammatory phenotype had occurred. Despite the absence of any previous infection, the MΦs showed a diminished response ex vivo to a subsequent endotoxin challenge in addition to other main characteristics of an ET status (for example, low expression of MHC-II and poor antigen presentation). In addition, patients with a marked ET phenotype were rehospitalized with infection over the course of the 3 months following enrolment.
This is an example of heterotolerance, a concept that could be defined as the induction of a tolerogenic response to a determined stimulus by a different one. For instance, TLR2 agonists induce heterotolerance to TLR4 ligands [37]. In the context of ACS, the IIS fails to respond to an external pathogen but with no prior exposure to infection, thus indicating that a stimulus quite different from that of the pathogen has induced the tolerant state. Several internal factors could act as initial stimuli in this respect; molecules known as danger-associated molecular patterns (DAMPs) [81] such as hyaluronic acid, high-mobility group box 1 (HMGB1), and heat shock protein are candidates. However, in the case of MI, there is a correlation between plasma mitochondrial DNA concentrations and ET grading [82]. MI may be recognized by the patient’s IIS by using its pattern recognition receptors that detect bacteria. Note that mitochondria are evolutionary endosymbionts derived from bacteria and contain DNA similar to bacterial DNA [83]. This may explain why responses to these mitochondrial DAMPs can mimic sepsis and also substantiate MΦs, developing ET under sterile conditions.
The cancer paradigm and others pathologies: open questions
Situations similar to ET have been described in other pathologies such as cancer. Several authors have reported that IIS cells that infiltrate into solid tumor such as tumor-associated macrophages (TAMs) exhibit the same characteristics of ET-MΦs in vivo[29,84]. Moreover, the exposure of monocytes to tumor cells induces a transient state in which these cells are refractory to further contact to cancer in vitro[34,85]. This phenomenon, termed ‘tumor tolerance’, reminds us of ET and is characterized by a decreased production of proinflammatory cytokines mediated by IRAK-M upregulation and TLR4 and CD44 activation [34]. In addition, these cells show a marked downregulation of MHC-II molecules as well as the MHC-II master regulator, CIITA, and combine an impaired capability for antigen presentation with potent phagocytic activity [86]. All of these features were found in circulating monocytes isolated from patients with chronic lymphocytic leukemia (CLL) [86]. Note that CLL-MΦs are in contact with tumor cells and the patients enrolled did not experience a prior infection. These data lead to the following question: could the interaction between tumor and IIS cells induce endotoxin tolerance? This would not be the first time that a ‘sterile’ pathology induces ET (see the ACS case discussed earlier). This possibility might explain the high risk of infections found in patients with CCL [87]. Besides, the correlation between the presence and functionality of TAM and a worse prognosis of many different cancers has been reported [88-90]. Indeed, targeting TAM is proposed as a feasible anti-tumor strategy [91,92].
Other pathologies might be candidates to study the ET phenomenon. In particular, stroke and Alzheimer’s disease are illnesses in which patients show a significant risk of pathogen colonization [93,94]. As far as stroke is concerned, one-third of patients die as a result of no direct factors, including infections [95]; this fact points to an impaired host defense in the context of strokes [96]. This pathology is characterized by severe tissue damage that increases the presence of DAMPs (for example, the nuclear protein HMGB1) [97]. Interestingly, HMGB1 has been described as an inducer of ET [98]. Consequently, this protein could be regulating the IIS by boosting an ET state that would dampen the antibacterial responses. In this line, different therapeutic strategies are being proposed for stroke treatment that is based on HMGB1 blockade or inhibition [99,100].
In addition, the immunosuppressive effects of some anesthetics such as morphine are gaining much attention. Its administration has been described as having a regulatory impact on LPS-induced responses [101]. In addition, chronic morphine administration is a boosting event for sepsis [102,103]. Some of the mechanisms involved in this process, it should be highlighted, are beginning to be elucidated and concur with some of the processes described as being responsible for ET, such as modulation of miR-146a expression [57,104]. All of these findings suggest that those diverse pathological contexts that generate a regulation IIS response might be revisited under the light of a putative tolerant process.
Conclusions
An endotoxin-tolerant state is a clinical phenomenon not restricted to sepsis but has been observed for a number of pathologies such as ACS, CF, and even cancer. Although ET has been thought of as a protective mechanism against septic shock and ischemia, its incidence is associated with high risks of secondary infections. Several studies have also shown some common mechanistic paradigms in ET across different diseases.
Abbreviations
ACS: Acute coronary syndrome; CF: Cystic fibrosis; CIITA: Class II transactivator; CLL: Chronic lymphocytic leukemia; DAMP: Danger-associated molecular pattern; ET: Endotoxin tolerance; HMGB1: High-mobility group box 1; IIS: Innate immune system; IL: Interleukin; IRAK: Interleukin-1 receptor-associated kinase; LPS: Lipopolysaccharide; MΦ: Monocyte/macrophage; MHC: Major histocompatibility; MI: Myocardial infarction; miRNA: MicroRNA; NSTEMI: Non-ST-elevation myocardial infarction; STEMI: ST-elevation myocardial infarction; TAM: Tumor-associated macrophage; TLR: Toll-like receptor; TNF-α: Tumor necrosis factor-alpha; TREM-1: Triggering receptor expressed on myeloid cells 1.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Eduardo López-Collazo, Email: elopezc@salud.madrid.org.
Carlos del Fresno, Email: carlos.delfresno@cnic.es.
References
- Goldszmid RS, Trinchieri G. The price of immunity. Nat Immunol. 2012;17:932–938. doi: 10.1038/ni.2422. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;17:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. 2011;17:16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- Ramos HJ, Gale M Jr. RIG-I like receptors and their signaling crosstalk in the regulation of antiviral immunity. Curr Opin Virol. 2011;17:167–176. doi: 10.1016/j.coviro.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaillon JM, Adib-Conquy M. Bench-to-bedside review: Endotoxin tolerance as a model of leukocyte reprogramming in sepsis. Crit Care. 2006;17:233. doi: 10.1186/cc5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escoll P, del Fresno C, García L, Vallés G, Lendínez MJ, Arnalich F, López-Collazo E. Rapid up-regulation of IRAK-M expression following a second endotoxin challenge in human monocytes and in monocytes isolated from septic patients. Biochem Biophys Res Commun. 2003;17:465–472. doi: 10.1016/j.bbrc.2003.10.019. [DOI] [PubMed] [Google Scholar]
- del Fresno C, Gómez-Piña V, Lores V, Soares-Schanoski A, Fernández-Ruiz I, Rojo B, Alvarez-Sala R, Caballero-Garrido E, García F, Veliz T, Arnalich F, Fuentes-Prior P, García-Río F, López-Collazo E. Monocytes from cystic fibrosis patients are locked in an LPS tolerance state: down-regulation of TREM-1 as putative underlying mechanism. PLoS One. 2008;17:e2667. doi: 10.1371/journal.pone.0002667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Fresno C, Soler-Rangel L, Soares-Schanoski A, Gómez-Piña V, González-León MC, Gómez-García L, Mendoza-Barberá E, Rodríguez-Rojas A, García F, Fuentes-Prior P, Arnalich F, López-Collazo E. Inflammatory responses associated with acute coronary syndrome up-regulate IRAK-M and induce endotoxin tolerance in circulating monocytes. J Endotoxin Res. 2007;17:39–52. doi: 10.1177/0968051907078623. [DOI] [PubMed] [Google Scholar]
- Lin CY, Tsai IF, Ho YP, Huang CT, Lin YC, Lin CJ, Tseng SC, Lin WP, Chen WT, Sheen IS. Endotoxemia contributes to the immune paralysis in patients with cirrhosis. J Hepatol. 2007;17:816–826. doi: 10.1016/j.jhep.2006.12.018. [DOI] [PubMed] [Google Scholar]
- Cavaillon JM, Adrie C, Fitting C, Adib-Conquy M. Reprogramming of circulatory cells in sepsis and SIRS. J Endotoxin Res. 2005;17:311–320. doi: 10.1179/096805105X58733. [DOI] [PubMed] [Google Scholar]
- Kox M, de Kleijn S, Pompe JC, Ramakers BP, Netea MG, van der Hoeven JG, Hoedemaekers CW, Pickkers P. Differential ex vivo and in vivo endotoxin tolerance kinetics following human endotoxemia. Crit Care Med. 2011;17:1866–1870. doi: 10.1097/CCM.0b013e3182190d5d. [DOI] [PubMed] [Google Scholar]
- Perry JA, Olver CS, Burnett RC, Avery AC. Cutting edge: the acquisition of TLR tolerance during malaria infection impacts T cell activation. J Immunol. 2005;17:5921–5925. doi: 10.4049/jimmunol.174.10.5921. [DOI] [PubMed] [Google Scholar]
- Greisman SE, Hornick RB, Wagner HN Jr, Woodward WE, Woodward TE. The role of endotoxin during typhoid fever and tularemia in man. IV. The integrity of the endotoxin tolerance mechanisms during infection. J Clin Invest. 1969;17:613–629. doi: 10.1172/JCI106020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein M, Mulholland JH, Jeffery GM, Wolff SM. Malaria induced endotoxin tolerance. Proc Soc Exp Biol Med. 1965;17:283–287. doi: 10.3181/00379727-118-29820. [DOI] [PubMed] [Google Scholar]
- McCabe WR. Endotoxin tolerance. II. Its occurrence in patients with pyelonephritis. J Clin Invest. 1963;17:618–625. doi: 10.1172/JCI104752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neva FA, Morgan HR. Tolerance to the action of endotoxins of enteric bacilli in patients convalescent from typhoid and paratyphoid fevers. J Lab Clin Med. 1950;17:911–922. [PubMed] [Google Scholar]
- Fuentes-Prior P, Biswas SK, López-Collazo E, Prados C, Arnalich F, Gutierrez Del Arroyo A, García-Rio F, Gómez-Piña V, Soares-Schanoski A, Fernández-Ruíz I, Jurado T, Kajiji T, Shu C, Marín E, del Fresno C. Potent phagocytic activity with impaired antigen presentation identifying lipopolysaccharide-tolerant human monocytes: demonstration in isolated monocytes from cystic fibrosis patients. J Immunol. 2009;17:6494–6507. doi: 10.4049/jimmunol.0803350. [DOI] [PubMed] [Google Scholar]
- Landoni VI, Chiarella P, Martire-Greco D, Schierloh P, Van-Rooijen N, Rearte B, Palermo MS, Isturiz MA, Fernandez GC. Tolerance to lipopolysaccharide promotes an enhanced neutrophil extracellular traps formation leading to a more efficient bacterial clearance in mice. Clin Exper Immunol. 2012;17:153–163. doi: 10.1111/j.1365-2249.2012.04560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes ME, Baggio-Zappia GL, Brunialti MK, Fernandes Mda L, Rapozo MM, Salomao R. Differential expression of toll-like receptor signaling cascades in LPS-tolerant human peripheral blood mononuclear cells. Immunobiology. 2011;17:285–295. doi: 10.1016/j.imbio.2010.07.008. [DOI] [PubMed] [Google Scholar]
- Chen X, El Gazzar M, Yoza BK, McCall CE. The NF-kappaB factor RelB and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance. J Biol Chem. 2009;17:27857–27865. doi: 10.1074/jbc.M109.000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenberg MA, Galanos C. Induction of tolerance to lipopolysaccharide (LPS)-D-galactosamine lethality by pretreatment with LPS is mediated by macrophages. Infect Immun. 1988;17:1352–1357. doi: 10.1128/iai.56.5.1352-1357.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009;17:475–487. doi: 10.1016/j.it.2009.07.009. [DOI] [PubMed] [Google Scholar]
- Karp CL, Wysocka M, Ma X, Marovich M, Factor RE, Nutman T, Armant M, Wahl L, Cuomo P, Trinchieri G. Potent suppression of IL-12 production from monocytes and dendritic cells during endotoxin tolerance. Eur J Immunol. 1998;17:3128–3136. doi: 10.1002/(SICI)1521-4141(199810)28:10<3128::AID-IMMU3128>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Wolk K, Docke WD, von Baehr V, Volk HD, Sabat R. Impaired antigen presentation by human monocytes during endotoxin tolerance. Blood. 2000;17:218–223. [PubMed] [Google Scholar]
- Lopez-Collazo E, Gomez-Pina V, Arnalich F. Understanding immune dysfunctions in sepsis patients. Crit Care. 2010;17:435. doi: 10.1186/cc9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, López CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, Tompkins RG. Inflammation and Host Response to Injury, Large Scale Collaborative Research Program: Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013;17:3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza N. Model organisms: mouse models challenged. Nat Methods. 2013;17:288. doi: 10.1038/nmeth.2429. [DOI] [PubMed] [Google Scholar]
- Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;17:972–978. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- Pena OM, Pistolic J, Raj D, Fjell CD, Hancock RE. Endotoxin tolerance represents a distinctive state of alternative polarization (M2) in human mononuclear cells. J Immunol. 2011;17:7243–7254. doi: 10.4049/jimmunol.1001952. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Hernandez LD, Galan JE, Janeway CA Jr, Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;17:191–202. doi: 10.1016/S0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- Peng Q, O'Loughlin JL, Humphrey MB. DOK3 negatively regulates LPS responses and endotoxin tolerance. PLoS One. 2012;17:e39967. doi: 10.1371/journal.pone.0039967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, Brownstein BH, Cobb JP, Tschoeke SK, Miller-Graziano C, Moldawer LL, Mindrinos MN, Davis RW, Tompkins RG, Lowry SF. Inflammation and Host Response to Injury, Large Scale Collaborative Research Program: A network-based analysis of systemic inflammation in humans. Nature. 2005;17:1032–1037. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
- Dominguez-Soto A, Sierra-Filardi E, Puig-Kroger A, Perez-Maceda B, Gomez-Aguado F, Corcuera MT, Sanchez-Mateos P, Corbi AL. Dendritic cell-specific ICAM-3-grabbing nonintegrin expression on M2-polarized and tumor-associated macrophages is macrophage-CSF dependent and enhanced by tumor-derived IL-6 and IL-10. J Immunol. 2011;17:2192–2200. doi: 10.4049/jimmunol.1000475. [DOI] [PubMed] [Google Scholar]
- del Fresno C, Otero K, Gómez-García L, González-León MC, Soler-Ranger L, Fuentes-Prior P, Escoll P, Baos R, Caveda L, García F, Arnalich F, López-Collazo E. Tumor cells deactivate human monocytes by up-regulating IL-1 receptor associated kinase-M expression via CD44 and TLR4. J Immunol. 2005;17:3032–3040. doi: 10.4049/jimmunol.174.5.3032. [DOI] [PubMed] [Google Scholar]
- Medvedev AE, Kopydlowski KM, Vogel SN. Inhibition of lipopolysaccharide-induced signal transduction in endotoxin-tolerized mouse macrophages: dysregulation of cytokine, chemokine, and toll-like receptor 2 and 4 gene expression. J Immunol. 2000;17:5564–5574. doi: 10.4049/jimmunol.164.11.5564. [DOI] [PubMed] [Google Scholar]
- Sato S, Takeuchi O, Fujita T, Tomizawa H, Takeda K, Akira S. A variety of microbial components induce tolerance to lipopolysaccharide by differentially affecting MyD88-dependent and -independent pathways. Int Immunol. 2002;17:783–791. doi: 10.1093/intimm/dxf046. [DOI] [PubMed] [Google Scholar]
- Dobrovolskaia MA, Medvedev AE, Thomas KE, Cuesta N, Toshchakov V, Ren T, Cody MJ, Michalek SM, Rice NR, Vogel SN. Induction of in vitro reprogramming by Toll-like receptor (TLR)2 and TLR4 agonists in murine macrophages: effects of TLR ‘homotolerance’ versus ‘heterotolerance’ on NF-kappa B signaling pathway components. J Immunol. 2003;17:508–519. doi: 10.4049/jimmunol.170.1.508. [DOI] [PubMed] [Google Scholar]
- Biswas SK, Bist P, Dhillon MK, Kajiji T, Del Fresno C, Yamamoto M, Lopez-Collazo E, Akira S, Tergaonkar V. Role for MyD88-independent, TRIF pathway in lipid A/TLR4-induced endotoxin tolerance. J Immunol. 2007;17:4083–4092. doi: 10.4049/jimmunol.179.6.4083. [DOI] [PubMed] [Google Scholar]
- Frellstedt L, McKenzie HC, Barrett JG, Furr MO. Induction and characterization of endotoxin tolerance in equine peripheral blood mononuclear cells in vitro. Vet Immunol Immunopathol. 2012;17:97–102. doi: 10.1016/j.vetimm.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Medvedev AE. Induction of endotoxin tolerance in vivo inhibits activation of IRAK4 and increases negative regulators IRAK-M, SHIP-1, and A20. J Leukoc Biol. 2011;17:1141–1148. doi: 10.1189/jlb.0611273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petzl W, Gunther J, Pfister T, Sauter-Louis C, Goetze L, von Aulock S, Hafner-Marx A, Schuberth HJ, Seyfert HM, Zerbe H. Lipopolysaccharide pretreatment of the udder protects against experimental Escherichia coli mastitis. Innate Immun. 2012;17:467–477. doi: 10.1177/1753425911422407. [DOI] [PubMed] [Google Scholar]
- Draisma A, Pickkers P, Bouw MP, van der Hoeven JG. Development of endotoxin tolerance in humans in vivo. Crit Care Med. 2009;17:1261–1267. doi: 10.1097/CCM.0b013e31819c3c67. [DOI] [PubMed] [Google Scholar]
- Grutz G. New insights into the molecular mechanism of interleukin-10-mediated immunosuppression. J Leukoc Biol. 2005;17:3–15. doi: 10.1189/jlb.0904484. [DOI] [PubMed] [Google Scholar]
- Randow F, Syrbe U, Meisel C, Krausch D, Zuckermann H, Platzer C, Volk HD. Mechanism of endotoxin desensitization: involvement of interleukin 10 and transforming growth factor beta. J Exp Med. 1995;17:1887–1892. doi: 10.1084/jem.181.5.1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg DJ, Kuhn R, Rajewsky K, Muller W, Menon S, Davidson N, Grunig G, Rennick D. Interleukin-10 is a central regulator of the response to LPS in murine models of endotoxic shock and the Shwartzman reaction but not endotoxin tolerance. J Clin Invest. 1995;17:2339–2347. doi: 10.1172/JCI118290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Poll T, de Vos AF, van den Pangaart PS, van Zoelen MA, de Kruif M, Birjmohun RS, Stroes ES, Van't Veer C. Induction of IRAK-M is associated with lipopolysaccharide tolerance in a human endotoxemia model. J Immunol. 2007;17:7110–7120. doi: 10.4049/jimmunol.179.10.7110. [DOI] [PubMed] [Google Scholar]
- Wiersinga WJ, van't Veer C, van den Pangaart PS, Dondorp AM, Day NP, Peacock SJ, van der Poll T. Immunosuppression associated with interleukin-1R-associated-kinase-M upregulation predicts mortality in Gram-negative sepsis (melioidosis) Crit Care Med. 2009;17:569–576. doi: 10.1097/CCM.0b013e318194b1bf. [DOI] [PubMed] [Google Scholar]
- Stiehm M, Peters K, Wiesmuller KH, Bufe A, Peters M. A novel synthetic lipopeptide is allergy-protective by the induction of LPS-tolerance. Clin Exp Allergy. 2013;17:785–797. doi: 10.1111/cea.12116. [DOI] [PubMed] [Google Scholar]
- Wesche H, Gao X, Li X, Kirschning CJ, Stark GR, Cao Z. IRAK-M is a novel member of the Pelle/interleukin-1 receptor-associated kinase (IRAK) family. J Biol Chem. 1999;17:19403–19410. doi: 10.1074/jbc.274.27.19403. [DOI] [PubMed] [Google Scholar]
- Janssens S, Beyaert R. Functional diversity and regulation of different interleukin-1 receptor-associated kinase (IRAK) family members. Mol Cell. 2003;17:293–302. doi: 10.1016/S1097-2765(03)00053-4. [DOI] [PubMed] [Google Scholar]
- Mendoza-Barberá E, Corral-Rodríguez MA, Soares-Schanoski A, Velarde M, Macieira S, Messerschmidt A, López-Collazo E, Fuentes-Prior P. Contribution of globular death domains and unstructured linkers to MyD88.IRAK-4 heterodimer formation: an explanation for the antagonistic activity of MyD88s. Biochem Biophys Res Commun. 2009;17:183–187. doi: 10.1016/j.bbrc.2009.01.069. [DOI] [PubMed] [Google Scholar]
- López-Collazo E, Fuentes-Prior P, Arnalich F, del Fresno C. Pathophysiology of interleukin-1 receptor-associated kinase-M: implications in refractory state. Curr Opin Infect Dis. 2006;17:237–244. doi: 10.1097/01.qco.0000224817.35105.7d. [DOI] [PubMed] [Google Scholar]
- Domon H, Honda T, Oda T, Yoshie H, Yamazaki K. Early and preferential induction of IL-1 receptor-associated kinase-M in THP-1 cells by LPS derived from Porphyromonas gingivalis. J Leukoc Biol. 2008;17:672–679. doi: 10.1189/jlb.0607432. [DOI] [PubMed] [Google Scholar]
- Liu ZJ, Yan LN, Li XH, Xu FL, Chen XF, You HB, Gong JP. Up-regulation of IRAK-M is essential for endotoxin tolerance induced by a low dose of lipopolysaccharide in Kupffer cells. J Surg Res. 2008;17:34–39. doi: 10.1016/j.jss.2007.12.759. [DOI] [PubMed] [Google Scholar]
- Gómez-Piña V, Martínez E, Fernández-Ruíz I, Del Fresno C, Soares-Schanoski A, Jurado T, Siliceo M, Toledano V, Fernández-Palomares R, García-Rio F, Arnalich F, Biswas SK, López-Collazo E. Role of MMPs in orchestrating inflammatory response in human monocytes via a TREM-1-PI3K-NF-kappaB pathway. J Leukoc Biol. 2012;17:933–945. doi: 10.1189/jlb.0711340. [DOI] [PubMed] [Google Scholar]
- Arts RJ, Joosten LA, van der Meer JW, Netea MG. TREM-1: intracellular signaling pathways and interaction with pattern recognition receptors. J Leukoc Biol. 2013;17:209–215. doi: 10.1189/jlb.0312145. [DOI] [PubMed] [Google Scholar]
- Quinn EM, Wang J, Redmond HP. The emerging role of microRNA in regulation of endotoxin tolerance. J Leukoc Biol. 2012;17:721–727. doi: 10.1189/jlb.1111571. [DOI] [PubMed] [Google Scholar]
- Broad A, Jones DE, Kirby JA. Toll-like receptor (TLR) response tolerance: a key physiological ‘damage limitation’ effect and an important potential opportunity for therapy. Curr Med Chem. 2006;17:2487–2502. doi: 10.2174/092986706778201675. [DOI] [PubMed] [Google Scholar]
- Moller K. Of cells and men: ex vivo and in vivo tolerance to lipopolysaccharide. Crit Care Med. 2011;17:1997–1998. doi: 10.1097/CCM.0b013e3182249931. [DOI] [PubMed] [Google Scholar]
- Buras JA, Holzmann B, Sitkovsky M. Animal models of sepsis: setting the stage. Nat Rev Drug Discov. 2005;17:854–865. doi: 10.1038/nrd1854. [DOI] [PubMed] [Google Scholar]
- van der Poll T, Opal SM. Host-pathogen interactions in sepsis. Lancet. 2008;17:32–43. doi: 10.1016/S1473-3099(07)70265-7. [DOI] [PubMed] [Google Scholar]
- Monneret G, Venet F, Pachot A, Lepape A. Monitoring immune dysfunctions in the septic patient: a new skin for the old ceremony. Mol Med. 2008;17:64–78. doi: 10.2119/2007-00102.Monneret. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz C, Carlet J, Fitting C, Misset B, Blériot JP, Cavaillon JM. Dysregulation of in vitro cytokine production by monocytes during sepsis. J Clin Invest. 1991;17:1747–1754. doi: 10.1172/JCI115493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz C, Misset B, Fitting C, Bleriot JP, Carlet J, Cavaillon JM. Dissociation between plasma and monocyte-associated cytokines during sepsis. European J Immunol. 1991;17:2177–2184. doi: 10.1002/eji.1830210928. [DOI] [PubMed] [Google Scholar]
- Pachot A, Lepape A, Vey S, Bienvenu J, Mougin B, Monneret G. Systemic transcriptional analysis in survivor and non-survivor septic shock patients: a preliminary study. Immunol Lett. 2006;17:63–71. doi: 10.1016/j.imlet.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Manjuck J, Saha DC, Astiz M, Eales LJ, Rackow EC. Decreased response to recall antigens is associated with depressed costimulatory receptor expression in septic critically ill patients. J Lab Clin Med. 2000;17:153–160. doi: 10.1067/mlc.2000.104306. [DOI] [PubMed] [Google Scholar]
- Wysocka M, Robertson S, Riemann H, Caamano J, Hunter C, Mackiewicz A, Montaner LJ, Trinchieri G, Karp CL. IL-12 suppression during experimental endotoxin tolerance: dendritic cell loss and macrophage hyporesponsiveness. J Immunol. 2001;17:7504–7513. doi: 10.4049/jimmunol.166.12.7504. [DOI] [PubMed] [Google Scholar]
- Murphey ED, Sherwood ER. Bacterial clearance and mortality are not improved by a combination of IL-10 neutralization and IFN-gamma administration in a murine model of post-CLP immunosuppression. Shock. 2006;17:417–424. doi: 10.1097/01.shk.0000226343.70904.4f. [DOI] [PubMed] [Google Scholar]
- Abe R, Hirasawa H, Oda S, Sadahiro T, Nakamura M, Watanabe E, Nakada TA, Hatano M, Tokuhisa T. Up-regulation of interleukin-10 mRNA expression in peripheral leukocytes predicts poor outcome and diminished human leukocyte antigen-DR expression on monocytes in septic patients. J Surg Res. 2008;17:1–8. doi: 10.1016/j.jss.2007.07.009. [DOI] [PubMed] [Google Scholar]
- Rossato M, Curtale G, Tamassia N, Castellucci M, Mori L, Gasperini S, Mariotti B, De Luca M, Mirolo M, Cassatella MA, Locati M, Bazzoni F. IL-10-induced microRNA-187 negatively regulates TNF-alpha, IL-6, and IL-12p40 production in TLR4-stimulated monocytes. Proc Natl Acad Sci U S A. 2012;17:E3101–3110. doi: 10.1073/pnas.1209100109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C. Points of control in inflammation. Nature. 2002;17:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- Ratjen F, Doring G. Cystic fibrosis. Lancet. 2003;17:681–689. doi: 10.1016/S0140-6736(03)12567-6. [DOI] [PubMed] [Google Scholar]
- Jain K, Smyth AR. Current dilemmas in antimicrobial therapy in cystic fibrosis. Expert Rev Respir Med. 2012;17:407–422. doi: 10.1586/ers.12.39. [DOI] [PubMed] [Google Scholar]
- Pier GB. The challenges and promises of new therapies for cystic fibrosis. J Exp Med. 2012;17:1235–1239. doi: 10.1084/jem.20121248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl D, Gaggar A, Bruscia E, Hector A, Marcos V, Jung A, Greene C, McElvaney G, Mall M, Döring G. Innate immunity in cystic fibrosis lung disease. J Cyst Fibros. 2012;17:363–382. doi: 10.1016/j.jcf.2012.07.003. [DOI] [PubMed] [Google Scholar]
- del Campo R, Martínez E, del Fresno C, Alenda R, Gómez-Piña V, Fernández-Ruíz I, Siliceo M, Jurado T, Toledano V, Arnalich F, García-Río F, López-Collazo E. Translocated LPS might cause endotoxin tolerance in circulating monocytes of cystic fibrosis patients. PLoS One. 2011;17:e29577. doi: 10.1371/journal.pone.0029577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross R. Atherosclerosis - an inflammatory disease. N Engl J Med. 1999;17:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;17:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- Shishido T, Nozaki N, Yamaguchi S, Shibata Y, Nitobe J, Miyamoto T, Takahashi H, Arimoto T, Maeda K, Yamakawa M, Takeuchi O, Akira S, Takeishi Y, Kubota I. Toll-like receptor-2 modulates ventricular remodeling after myocardial infarction. Circulation. 2003;17:2905–2910. doi: 10.1161/01.CIR.0000101921.93016.1C. [DOI] [PubMed] [Google Scholar]
- Mach F, Schonbeck U, Bonnefoy JY, Pober JS, Libby P. Activation of monocyte/macrophage functions related to acute atheroma complication by ligation of CD40: induction of collagenase, stromelysin, and tissue factor. Circulation. 1997;17:396–399. doi: 10.1161/01.CIR.96.2.396. [DOI] [PubMed] [Google Scholar]
- Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. 2012;17:158–175. doi: 10.1111/j.1600-065X.2012.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnalich F, Codoceo R, Lopez-Collazo E, Montiel C. Circulating cell-free mitochondrial DNA: a better early prognostic marker in patients with out-of-hospital cardiac arrest. Resuscitation. 2012;17:e162–e163. doi: 10.1016/j.resuscitation.2012.03.032. [DOI] [PubMed] [Google Scholar]
- Schon EA, DiMauro S, Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat Rev Genet. 2012;17:878–890. doi: 10.1038/nrg3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;17:231–237. doi: 10.1016/j.coi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Allavena P, Garlanda C, Locati M. Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Hum Immunol. 2009;17:325–330. doi: 10.1016/j.humimm.2009.02.008. [DOI] [PubMed] [Google Scholar]
- Soares-Schanoski A, Jurado T, Córdoba R, Siliceo M, Fresno CD, Gómez-Piña V, Toledano V, Vallejo-Cremades MT, Alfonso-Iñiguez S, Carballo-Palos A, Fernández-Ruiz I, Cubillas-Zapata C, Biswas SK, Arnalich F, García-Río F, López-Collazo E. Impaired antigen presentation and potent phagocytic activity identifying tumor-tolerant human monocytes. Biochem Biophys Res Commun. 2012;17:331–337. doi: 10.1016/j.bbrc.2012.05.124. [DOI] [PubMed] [Google Scholar]
- Nosari A. Infectious complications in chronic lymphocytic leukemia. Mediterr J Hematol Infect Dis. 2012;17:e2012070. doi: 10.4084/MJHID.2012.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuhashi A, Goto H, Kuramoto T, Tabata S, Yukishige S, Abe S, Hanibuchi M, Kakiuchi S, Saijo A, Aono Y, Uehara H, Yano S, Ledford JG, Sone S, Nishioka Y. Surfactant protein A suppresses lung cancer progression by regulating the polarization of tumor-associated macrophages. Am J Pathol. 2013;17:1843–1853. doi: 10.1016/j.ajpath.2013.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera M, Herrera A, Domínguez G, Silva J, García V, García JM, Gómez I, Soldevilla B, Muñoz C, Provencio M, Campos-Martin Y, García De Herreros A, Casal I, Bonilla F, Peña C. Cancer-associated fibroblast and M2 macrophage markers together predict outcome in colorectal cancer patients. Cancer Sci. 2013;17:437–444. doi: 10.1111/cas.12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan C, Huang X, Lin S, Huang H, Cai Q, Wan T, Lu J, Liu J. Expression of M2-polarized macrophages is associated with poor prognosis for advanced epithelial ovarian cancer. Technol Cancer Res Treat. 2013;17:259–267. doi: 10.7785/tcrt.2012.500312. [DOI] [PubMed] [Google Scholar]
- Tang X, Mo C, Wang Y, Wei D, Xiao H. Anti-tumour strategies aiming to target tumour-associated macrophages. Immunology. 2013;17:93–104. doi: 10.1111/imm.12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica A, Rubino L, Mancino A, Larghi P, Porta C, Rimoldi M, Solinas G, Locati M, Allavena P, Mantovani A. Targeting tumour-associated macrophages. Expert Opin Ther Targets. 2007;17:1219–1229. doi: 10.1517/14728222.11.9.1219. [DOI] [PubMed] [Google Scholar]
- Miklossy J. Emerging roles of pathogens in Alzheimer disease. Expert Rev Mol Med. 2011;17:e30. doi: 10.1017/S1462399411002006. [DOI] [PubMed] [Google Scholar]
- Grau AJ, Buggle F, Heindl S, Steichen-Wiehn C, Banerjee T, Maiwald M, Rohlfs M, Suhr H, Fiehn W, Becher H, Hacke W. Recent infection as a risk factor for cerebrovascular ischemia. Stroke. 1995;17:373–379. doi: 10.1161/01.STR.26.3.373. [DOI] [PubMed] [Google Scholar]
- Koennecke HC, Belz W, Berfelde D, Endres M, Fitzek S, Hamilton F, Kreitsch P, Mackert BM, Nabavi DG, Nolte CH, Pöhls W, Schmehl I, Schmitz B, von Brevern M, Walter G, Heuschmann PU. Berlin Stroke Register Investigators. Factors influencing in-hospital mortality and morbidity in patients treated on a stroke unit. Neurology. 2011;17:965–972. doi: 10.1212/WNL.0b013e31822dc795. [DOI] [PubMed] [Google Scholar]
- Meisel C, Meisel A. Suppressing immunosuppression after stroke. N Engl J Med. 2011;17:2134–2136. doi: 10.1056/NEJMcibr1112454. [DOI] [PubMed] [Google Scholar]
- Schulze J, Zierath D, Tanzi P, Cain K, Shibata D, Dressel A, Becker K. Severe stroke induces long-lasting alterations of high-mobility group box 1. Stroke. 2013;17:246–248. doi: 10.1161/STROKEAHA.112.676072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aneja RK, Tsung A, Sjodin H, Gefter JV, Delude RL, Billiar TR, Fink MP. Preconditioning with high mobility group box 1 (HMGB1) induces lipopolysaccharide (LPS) tolerance. J Leukoc Biol. 2008;17:1326–1334. doi: 10.1189/jlb.0108030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Takahashi HK, Liu K, Wake H, Liu R, Maruo T, Date I, Yoshino T, Ohtsuka A, Mori S, Nishibori M. Anti-high mobility group box-1 monoclonal antibody protects the blood–brain barrier from ischemia-induced disruption in rats. Stroke. 2011;17:1420–1428. doi: 10.1161/STROKEAHA.110.598334. [DOI] [PubMed] [Google Scholar]
- Tong H, Tang Y, Chen Y, Yuan F, Liu Z, Peng N, Tang L, Su L. HMGB1 activity inhibition alleviating liver injury in heatstroke. J Trauma Acute Care Surg. 2013;17:801–807. doi: 10.1097/TA.0b013e31827e9a65. [DOI] [PubMed] [Google Scholar]
- Bryant HU, Bernton EW, Holaday JW. Morphine pellet-induced immunomodulation in mice: temporal relationships. J Pharmacol Exp Ther. 1988;17:913–920. [PubMed] [Google Scholar]
- Ocasio FM, Jiang Y, House SD, Chang SL. Chronic morphine accelerates the progression of lipopolysaccharide-induced sepsis to septic shock. J Neuroimmunol. 2004;17:90–100. doi: 10.1016/j.jneuroim.2003.12.016. [DOI] [PubMed] [Google Scholar]
- Breslow JM, Monroy MA, Daly JM, Meissler JJ, Gaughan J, Adler MW, Eisenstein TK. Morphine, but not trauma, sensitizes to systemic Acinetobacter baumannii infection. J Neuroimmune Pharmacol. 2011;17:551–565. doi: 10.1007/s11481-011-9303-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Meng J, Das S, Krishnan A, Haworth J, Charboneau R, Zeng Y, Ramakrishnan S, Roy S. Morphine induced exacerbation of sepsis is mediated by tempering endotoxin tolerance through modulation of miR-146a. Sci Rep. 1977;17:3. doi: 10.1038/srep01977. [DOI] [PMC free article] [PubMed] [Google Scholar]