

Abstract
Regulation of protein function through thiol-based redox switches plays an important role in the response and adaptation to local and global changes in the cellular levels of reactive oxygen species (ROS). Redox regulation is used by first responder proteins, such as ROS-specific transcriptional regulators, chaperones or metabolic enzymes to protect cells against mounting levels of oxidants, repair the damage and restore redox homeostasis. Redox regulation of phosphatases and kinases is used to control the activity of select eukaryotic signaling pathways, making reactive oxygen species important second messengers that regulate growth, development and differentiation. In this review we will compare different types of reversible protein thiol modifications, elaborate on their structural and functional consequences and discuss their role in oxidative stress response and ROS adaptation.
1. Reactive Oxygen Species and Oxidative Stress Response
Life in an aerobic environment inevitably leads to the formation of reactive oxygen species (ROS) [1], including hydrogen peroxide (H2O2), superoxide (O2−) and hypochlorous acid (HOCl). These oxidants are endogenously generated as metabolic by-products from processes such as oxidative phosphorylation, or are directly produced by enzymes like NADPH oxidases (i.e., Nox) [2]. Whereas low concentrations of ROS play important roles in cell signaling processes, large amounts of ROS cause irreversible modification and damage to virtually every cellular macromolecule, including lipids, DNA and proteins [3–5]. A number of conserved enzymatic and non-enzymatic systems are present in pro- and eukaryotic organisms that detoxify excess ROS, prevent and repair oxidative damage and maintain redox homeostasis. However, when biological systems are no longer able to deal with ROS, they experience a stress situation commonly known as oxidative stress, which becomes quickly lethal if it is let to run its course.
To protect themselves against oxidative damage, organisms have developed a number of different response systems, whose main function is to sense and rapidly respond to changing levels of specific oxidants. Most of the responses involve transcriptional changes, mediated by oxidative modification of specific transcription factors [6]. The transcriptional activator OxyR, for instance, directly senses increased peroxide levels in E. coli [7]. Upon its activation, OxyR induces the expression of catalase and peroxiredoxin to detoxify hydrogen peroxide and organic peroxides, and also induces the expression thioredoxin and glutaredoxin in order to reduce oxidative thiol modifications and restore redox homeostasis [8]. In yeast, the transcriptional regulator Yap1p takes over this function, sensing the presence of reactive oxygen species and responding to them with the upregulation of antioxidant genes [9]. In addition to these transcriptional changes, organisms also respond to increased oxidant levels with the instantaneous activation of stress-specific chaperones, which protect proteins against oxidative protein aggregation [10, 11]. ROS-mediated change in the function of select metabolic enzymes further serves to redirect metabolic pathways from energy production towards NADPH generation [12]. In concert, these responses provide considerable protection against oxidative damage.
2. Cysteine Oxidation – A Sensitive and (Mostly) Reversible Way to Respond to Oxidants
One of the most commonly employed ways to sense alterations in cellular oxidant levels or the redox environment is through the reversible modification of thiol-containing cysteine side chains in redox-sensitive proteins [6, 13–16]. While most cysteines have pKa values between 8–9 and are therefore fully protonated and largely non-reactive under physiological pH conditions, oxidation-sensitive thiols are often (but not always) characterized by much lower pKa values [17]. These low pKa -values result in the deprotonation of these cysteines under physiological pH conditions. The resulting thiolate anions exhibit much higher reactivity than their protonated thiol counterparts [18]. Reactivity among different thiolate anions also depends on the local protein environment, and currently there is no accurate way to predict thiolate anion reactivity [19]. Upon exposure to oxidants, thiolate anions rapidly form sulfenic acids, which are important intermediates in the thiol oxidation process (Figure 1)[20, 21]. Due to their high reactivity towards nearby thiol groups, sulfenic acids are generally very short-lived [22]. So far, only few redox-regulated proteins, such as the FAD-containing NADH peroxidase from Streptococcus faecalis and matrix metalloproteinase MMP-7 [23, 24] have been shown to use this fully reversible oxidative modification to control their functional activity [20, 25]. Most sulfenic acids, however, rapidly react with other protein thiols to form intra- or intermolecular disulfide bonds, interact with non-protein thiols such as the small redox compound glutathione (GSH) or cysteine to form mixed disulfide bonds (i.e., S-glutathionylation, S-cysteinylation), or react with nearby amino groups to form cyclic sulfenamides (Figure 1). Further oxidation of sulfenic acid or sulfenamides leads to sulfinic acid and sulfonic formation, which are typically irreversible oxidation processes. One exception, is the formation of sulfinic acid at the active site cysteine of select eukaryotic peroxiredoxins [26]. This overoxidation product, which forms part of a functional redox switch, is reduced by sulfiredoxin, a specialized ATP-dependent sulfinic acid reductase [27].
Figure 1. Oxidative thiol modifications commonly found in redox-regulated proteins.

Upon reaction with peroxide (H2O2) or hypochlorous acid (HOCl), redox-sensitive thiol groups (RSH) rapidly form sulfenic acids (RSOH). These sulfenates are highly reactive and tend to quickly react with nearby cysteine thiols to form inter- or intramolecular disulfide bonds (RSSR). Alternatively, they form mixed disulfides with the small tripeptide glutathione (GSH) (RSSG), or undergo cyclic sulfenamide formation (RSNHR). These oxidative thiol modifications are fully reversible, and reduction (RED) is catalyzed by members of the glutaredoxin (Grx) or thioredoxin (Trx) system. Further oxidation of sulfenic acid to sulfinic acid (RSO2H), sulfinamide (RSONHR) or sulfonic acid (RSO3H) is irreversible. One exception is the active site sulfinic acid in peroxiredoxin, whose reduction is mediated by the highly specialized ATP-dependent sulfiredoxin Srx1.
Most oxidative thiol modifications are reduced by the thioredoxin or glutaredoxin systems [28, 29]. At the core of these systems are small, highly conserved oxidoreductases, thioredoxin (e.g., TrxA) and glutaredoxin (e.g., Grx1, Grx3), which use direct thiol-disulfide reactions to reduce oxidized protein thiols. Both oxidoreductases have the characteristic thioredoxin fold, harboring a conserved Cys-X-Y-Cys motif that facilitates the reduction of client proteins [30]. In the initial step, the first Cys of the thioredoxin twin cysteine motif attacks the oxidized thiol of the client protein. In the case of TrxA, this nucleophilic attack leads to the formation of a mixed disulfide bond between TrxA and the client proteins. To resolve this intermolecular disulfide bond and fully reduce the client protein, the second cysteine in TrxA launches another nucleophilic attack at the mixed disulfide bond, forming an intramolecular disulfide bond in TrxA thereby fully reducing the client protein [31]. Reversibility of the thioredoxin system is achieved by thioredoxin reductase (TrxB), which uses the reductive power of NADPH to reduce the disulfide bond in TrxA [28, 32]. Glutaredoxin (Grx) is different from Trx in that it preferentially attacks S-glutathionylated proteins, forming a mixed disulfide with GSH in this process [29, 33]. A second molecule of GSH attacks this intermolecular disulfide bond, reducing Grx and releasing oxidized GSSG, which in turn is reduced by the NADPH-dependent glutathione reductase (GR) [28, 34]. Despite Grx’s preference for S-glutathionylated proteins and Trx’s preference for sulfenic acids, in vivo studies revealed that the two systems can replace each other in the cell [35–37]. This redundancy emphasizes their important roles in maintaining cellular redox homeostasis and preventing higher and typically irreversible thiol modifications.
3. Redox-Regulation – A Powerful Posttranslational Mechanism to Regulate Protein Activity
Select proteins involved in photosynthetic processes have long been known to be inactivated by disulfide bond formation under dark conditions, and activated upon disulfide bond reduction once the organism is photosynthetically active and starts producing NADPH [16, 38, 39]. Moreover, numerous prior studies have reported thiol-containing proteins, which loose their activity during the purification in the absence of reducing agents. Many of these results were historically dismissed as in vitro artifacts, explained by the random and non-specific disulfide bond formation, which alters the structure and hence the function of the proteins. What was more difficult to dismiss as an artifact was the discovery of cytosolic proteins that gain activity when oxidized. One of the first examples of this was OxyR, the peroxide sensitive transcription factor in E. coli. Inactive when reduced, OxyR quickly induced antioxidant gene expression when its conserved cysteine(s) were oxidized by peroxide [7]. Our discovery that a completely unrelated bacterial protein, the chaperone Hsp33, uses a similar type of regulation but with a completely different mechanism [10] provided crucial additional evidence in support of the fact that thiol oxidation of cytosolic proteins is indeed physiologically significant, and that transient oxidation potentially affects many thiol-containing proteins and pathways [40]. With the development of quantitative in vivo thiol trapping methods [41], we now know of many hundreds of different bacterial and eukaryotic proteins, including kinases, phosphatases, transcription factors, and metabolic enzymes that use reversible thiol modifications to rapidly adjust their protein activity to the redox environment of the cell [42–47]. For many of these proteins, the nature of their oxidative thiol modifications is still unknown. While global methods are now available to detect sulfenic acids in proteins [48], cysteines involved in regulatory disulfide bonds still have to be identified by a tedious disulfide mapping process, and sulfenamides are often simply identified by exclusion [49]. However, given the increasing interest in the area of redox regulation, it will only be a matter of time before tools will become available to systematically scrutinize these reversible thiol modifications.
4. Sulfenic Acid – The First Step in the Thiol Oxidation Process
Upon exposure to fast acting oxidants such as HOCl (i.e., bleach), thiolate anions rapidly form sulfenyl chlorides, which subsequently hydrolyze to sulfenic acids (k =106 – 107 M−1s−1) (Figure 1) [50, 51]. This rapid reaction rate likely explains the reversible thiol modifications that many proteins undergo in response to bleach [52]. Hydrogen peroxide, however, although much more oxidizing than HOCl [53], shows ~6 order of magnitude slower reaction rates with most known thiols (k = 5 – 15 M−1 s−1) [54], including many proteins that have been shown to be oxidized by peroxide in vivo. This finding raised the very intriguing question of how peroxide can actually cause the extensive cellular protein thiol oxidation that has been observed in a variety of different model systems [54, 55].
One possible solution to this conundrum came with the discovery that peroxide-mediated oxidation (and activation) of Yap1p, the peroxide-sensitive transcription factor in yeast, is not directly caused by peroxide, but is indirectly mediated by the oxidized form of a small glutathione peroxidase (Gpx/Orp1) [56]. More recently, peroxiredoxins, members of a second family of thiol peroxidases, have also been shown to be involved in the oxidative activation of select thiol-containing proteins [57]. In both peroxidase families, the active site cysteine of thiol peroxidases shows exquisitely high reactivity with peroxide (k = 105 –108 M−1 s−1 [19, 58]), and rapidly undergoes sulfenic acid formation as part of its peroxide detoxification cycle [59]. A cationic environment, which is thought to stabilize the transition state, appears to contribute to this highly unusual reaction rate [60]. Subsequent interaction of this sulfenic acid with the thiol group of other proteins, such as Yap1p causes first the formation of an intermolecular disulfide bond, and subsequently the oxidation of the client protein and the concomitant reduction of the thiol peroxidase [61]. While this discovery explains how non-reactive cysteines can become reversible oxidized upon peroxide treatment in vivo, it is still unclear how many proteins serve as substrates for thiol peroxidase-mediated oxidation. The enormous redundancy of thiol peroxidases in even very simple organisms makes answering this important question challenging.
5. Disulfide Bond Formation as Redox Switch
5.1. Activation of the Oxidative Stress Specific Chaperone Hsp33
Heat shock protein 33 (Hsp33) is a highly conserved cytosolic chaperone, which is present in a large number of different gram positive and negative bacteria as well as in select eukaryotes, including the parasites Leishmania and Trypanosoma, select species of green and red algae and several higher plant species [10]. In bacteria, Hsp33 has been shown to effectively protect cytosolic proteins against oxidative protein aggregation, elicited by reactive chlorine species, such as HOCl, or by a combination of H2O2 and elevated temperatures [10, 11, 62, 63]. Whereas bacteria encounter HOCl, the active ingredient of household bleach, during the innate immune response as well as upon colonization of barrier epithelia in the lung and gut [64], peroxidative heat stress is elicited during inflammation [65]. Presence of Hsp33 significantly increases bacterial resistance towards both of these stress conditions.
Hsp33 is an ATP-independent chaperone, which is posttranslationally activated specifically by these severe oxidative stress conditions to prevent proteins from aggregation [11, 62]. This is crucial for bacterial survival, since under these stress conditions, ATP-dependent bacterial chaperones, such as the DnaK or GroEL-system, which are typically employed to maintain proteostasis under stress conditions, are no longer able to prevent protein aggregation [11]. This is due to the oxidative stress-mediated decrease in cellular ATP levels, which leaves these proteins devoid of their energy source, as well as due to direct oxidative stress-mediated inactivation of some of these chaperones [66, 67].
5.1.1. The Activation Mechanism of Hsp33
How is activation of the chaperone Hsp33 achieved under conditions that cause the oxidative unfolding and aggregation of hundreds of different proteins, including several important chaperones? The answer lies in Hsp33’s unique structural organization, the presence of a highly reactive cysteine-containing redox switch, and the fact that Hsp33 becomes rapidly activated when it gets oxidatively unfolded. Under non-stress conditions, Hsp33 is compactly folded and chaperone inactive (Figure 2). The redox switch resides in Hsp33’s C-terminus. It consists of four cysteines, tetrahedrally arranged in a C-X-C-Xn-C-X-X-C motif. These four cysteines coordinate zinc with very high affinity (Kd of 2.5 × 10−17 at 25°C, pH 7.5) [68]. This zinc coordination appears to lower the pKa of Hsp33’s cysteines, as indicated by the observation that all four cysteines are present in their highly reactive thiolate anion state. The N- and C-terminal domains of Hsp33 are connected by a flexible linker region, which appears to be fully folded when Hsp33 is reduced and zinc-coordinated [69, 70]. Exposure of Hsp33 to oxidizing conditions leads to the formation of two intramolecular disulfide bonds, connecting the next neighbor cysteines [43, 71]. This oxidation leads to the release of zinc, which destabilizes the zinc center and causes the unfolding of the adjacent linker region [63, 72]. Two partially unfolded monomers associate and form the chaperone active dimer (Figure 2) [73]. Disulfide bond formation, a reaction typically considered to be stabilizing and structure forming, converts Hsp33 into a natively unfolded protein [72, 74]. Once partially unfolded, Hsp33 appears to expose binding sites responsible for client binding both in the linker region and the adjacent N-terminus [74, 75]. When reducing conditions are restored, the disulfide bonds are reduced and zinc is re-coordinated [76]. However, to effectively release the client proteins from Hsp33 and return Hsp33 to the chaperone-inactive state, cellular ATP-levels need to be restored as well. Increasing cellular ATP concentrations fuel the ATP-dependent DnaK-chaperone system, which triggers the release of client proteins from Hsp33 and supports their refolding to the native state [74, 76].
Figure 2. Redox cycle of Hsp33.
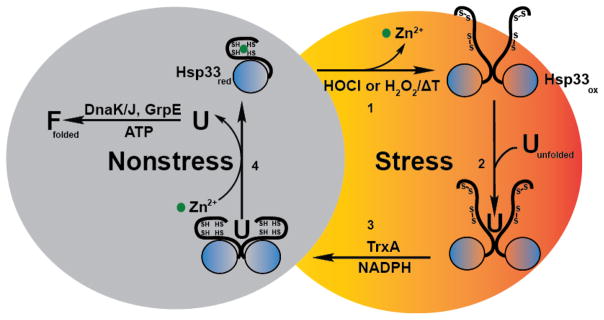
Under non-stress conditions, the heat shock protein Hsp33 is reduced and chaperone-inactive. All four highly conserved cysteines coordinate one zinc ion with high affinity. (1) Upon exposure to bleach (HOCl) or hydrogen peroxide (H2O2) in combination with elevated temperatures, Hsp33’s cysteines form two disulfide bonds, causing the zinc to be released. Loss of zinc binding in combination with disulfide bond formation leads to the partial unfolding of Hsp33, the required step for Hsp33’s activation as a chaperone. Two partially unfolded monomers then associate to form a chaperone-active Hsp33 dimer. (2) Once activated, Hsp33 stabilizes oxidatively unfolding proteins and forms stable client protein – chaperone complexes. (3) Upon return to non-stress conditions, the thioredoxin system reduces Hsp33’s disulfide bonds, generating a chaperone-active reduced dimer – client protein complex. (4) In the last step, the client protein is transferred to the DnaK/DnaJ/GrpE system for productive refolding, and Hsp33 dissociates into its monomeric, inactive state.
The posttranslational activation mechanism of Hsp33 is a new paradigm in chaperone function [77]. Over the past few years, several other chaperones have been discovered which use their own stress-induced protein unfolding as mechanism to activate their chaperone function. One of them is the pH-activated E. coli chaperone HdeA, which rapidly loses structure upon low pH and prevents other chaperones against low pH-mediated aggregation [78–80]. The other ones are members of the small heat shock protein family, which are activated by heat-induced structural rearrangements [81, 82]. By using these activation mechanisms, which do not require time-consuming transcription and translation processes, these specialized chaperones provide immediate protection against rapidly acting protein unfolding stress conditions, such as HOCl treatment or acid stress [77].
Numerous questions still remain in regard to the oxidative activation process of Hsp33 and other proteins with cysteine-containing zinc centers. For instance, how does a tetrahedral cluster of cysteines manage to form disulfide bonds exclusively between the next neighbor cysteines, given that all four cysteines are at an equal distance to each other? How does disulfide bond formation in the far C-terminus of a protein cause unfolding of the protein’s N-terminal regions? Are all cysteine coordinating zinc clusters equally redox sensitive? Studies of the pKa values and reactivities of each of the individual cysteine thiols in Hsp33’s zinc binding cluster, and comparison with other zinc binding proteins may help address some of these questions. A fusion protein consisting of Hsp33’s N-terminus/linker region and the zinc-binding domain of protein kinase C revealed that Hsp33’s chaperone function can be used as read-out for the redox sensitivity of PKC’s zinc centers [83] and potentially other zinc centers. This should help shed light into the mechanism(s) by which zinc-containing cysteine centers gain redox sensitivity or maintain their status as purely structural and redox-inert protein domains.
5.2. Redox-Mediated Activation of Inteins - A Novel Way to Regulate Protein Splicing
In analogy to self-splicing introns, which are nucleotide sequences that reside within the coding regions of DNA (i.e., exon) and catalyze their own excision [84], intervening proteins (i.e., inteins) reside within host proteins (i.e., exteins) and excise themselves upon translation [85, 86]. Exteins become functional once inteins excise and their termini are religated. Depending on their structural organization, inteins can be separated into two basic categories; cis-and trans-splicing inteins [87]. Cis-splicing inteins are single polypeptides embedded into host proteins. Based on their domain structure, they are classified into mini-inteins, which contain the two conserved N-terminal and C-terminal intein fragments IN and IC with protein splicing activity, or maxi-inteins, whose two fragments are separated by a sequence-specific DNA homing endonuclease [88, 89]. In trans-splicing inteins (i.e., split inteins), the N-and C-terminal intein fragments are fused to separate exteins and encoded by two different genes. Association of the two motifs is necessary before autocatalytic cleavage can occur [90]. Inteins are present in organisms of all three domains of life but are found exclusively in unicellular organisms, they are absent in multicellular organisms [87]. The dominant group of proteins with inteins are essential DNA repair and replication enzymes but some metabolic enzymes, proteases and the V-ATPase also contain inteins [91]. Inteins have no known benefit for the host, but these genes have been maintained in genomes over millions of years of evolution as a piece of selfish DNA [88]. Since excision of inteins leads to the activation of the host protein, it is plausible that inteins serve a regulatory role, controlling the activity of these essential proteins [87].
5.2.1. Active Site Cysteines in Inteins – Attractive Targets for Redox Regulation
Inteins are “single turnover enzymes”, which catalyze the splicing reaction without additional factors or energy. Class 1 inteins, which constitute over 90% of all known intervening proteins, use a four-step reaction mechanism to rearrange, excise and re-ligate the host (Figure 3A) [88]. Two active site cysteines (sometimes serines) at the extein-intein boundaries are essential for catalysis; Cys1, found at position 1 of the intein and Cys+1, located at position 1 of the C-terminal extein. In the first step (N-S-Acyl shift), a thioester bond is formed between Cys1 and the adjacent peptide carbonyl group that links N-extein and N-intein fragments. In the second step, a trans-thioesterification reaction occurs in which the ester is transferred onto the sulfur of Cys+1, causing the formation of a new thioester between the N-extein’s carbonyl and the C-extein’s thiol group. These steps are followed by an aminosuccinimide formation and the formation of a new peptide bond between the two extein domains [87]. Class 2 and 3 inteins, which lack the first nucleophile, use variations of this reaction theme [87, 92].
Figure 3. Redox regulation of protein splicing.
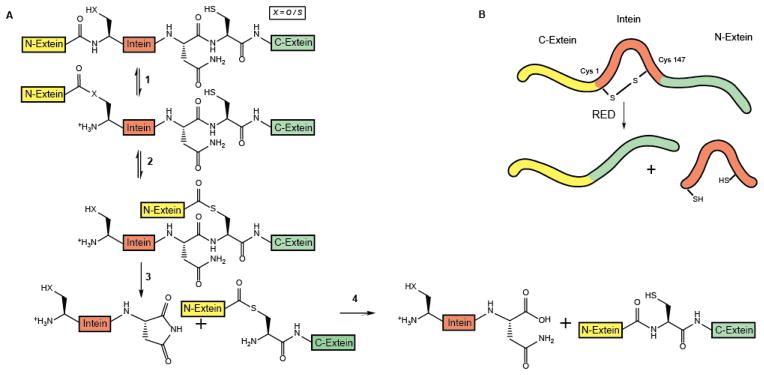
A. Mechanistic scheme of the protein splicing process. The small intervening protein intein (red) is embedded within a host protein (extein), which is inactive until intein is excised and its termini are re-ligated. (1) In the initial reaction, the active site nucleophile XH (cysteine Cys 1 or serine Ser 1) of intein forms a (thio)ester with the adjacent carbonyl carbon of N-extein (yellow). (2) In a subsequent trans-(thio)esterification reaction, the N-extein is transferred onto the side chain of the first C-extein residue (green), which is typically also a cysteine residue (Cys+1). (3) Complete excision of the intein is achieved by the formation of an aminosuccinimide, which involves a highly conserved Asn, located at the very C-terminus of the intein. (4) In the last step, a new peptide bond is formed between the N-extein and C-extein fragments. B. Schematic representation of the intein splicing process of the Mma PolII precursor protein. In the oxidized state, the active site cysteine Cys1 forms an intramolecular disulfide bond with Cys147, which is also located in the intein. This causes the inactivation of intein’s self-splicing function. Incubation with reducing agents (e.g., DTT, TCEP) reduces the disulfide bond, and the intein undergoes the previously described self-splicing process, leading to the activation of the host protein.
Given that nucleophilic cysteines are most often the key players in the intein-mediated splicing process, regulating their redox status appears to be an ideal mechanism to control intein activity. To test this idea, an intein precursor variant was constructed in which the three most C-terminal N-extein residues directly upstream of the active site Cys1 were replaced to generate the oxidoreductase motif of thioredoxin (i.e., Cys-Pro-Gly-Cys1) [93]. Whereas this intein variant showed no substantially altered self-splicing activity when expressed in wild-type E. coli, its activity was drastically lowered upon expression in the E. coli origami mutant strain. This mutant strain lacks both the thioredoxin and glutaredoxin systems and therefore accumulates proteins with non-native disulfide bonds in the cytosol [94]. Importantly, addition of non-nucleophilic reducing agents to the media immediately re-initiated intein splicing, supporting the conclusion that disulfide bond formation reversibly repressed intein’s activity and that this introduced new disulfide bond is redox sensitive [93]. This finding raised the intriguing question whether any naturally occurring inteins might be regulated via reversible disulfide bond formation as well. Indeed, sequence analysis revealed at least one member of the intein family containing a C-X-X-Cys1 motif. This intein was found to be embedded within the molybdate biosynthesis enzyme MoaA and is present in the anaerobic hyperthermophilic archaebacterium Pyrococcus abyssi [93]. Using a splicing reporter construct containing the intein of Pab MoaA flanked by its native N-extein sequence confirmed the previous results and showed that disulfide bond formation occurs within the oxidizing environment of the origami strain, and blocks the self-splicing activity of Pab MoaA’s intein until reducing conditions are restored [93].
A second potentially redox-regulated intein was recently discovered in Pyrococcus abyssi [95]. This intein interrupts the DP2 subunit of DNA polymerase II (Pab PolII), an essential protein involved in DNA replication. Splicing is induced by high temperature, ensuring that the DNA polymerase is only active at the appropriate temperatures. It was therefore surprising when a purified precursor construct, consisting of the Pab PolII intein flanked by a short N-extein and His-tagged C-extein was found to harbor a disulfide bond connecting the two active site cysteines Cys1 and Cys+1 [95]. This disulfide bond effectively prevented the splicing of the intron even at elevated temperatures unless reducing agents were present. Since the two cysteines are 185 aa apart from each other in sequence, it was unclear how disulfide bond formation was achieved. Yet, analysis of the extein sequences directly adjacent to the two cysteines revealed three positively charged amino acids upstream of Cys1 and three negatively charged amino acids immediately downstream of Cys+1. Replacing the three positively charged amino acids with negatively charged amino acids drastically reduced disulfide bond formation, implying that electrostatic interactions might help to bring the two cysteines in close proximity [95]. A third redox-regulated intein was identified in late 2013 the DNA PolII of Methanoculleus marisnigri, another thermophilic and anaerobic archaebacterium [96]. Unlike in Pab PolII, however, only one of the two cysteines involved in the regulatory disulfide bond is an active site cysteine (i.e., Cys1). The other cysteine is located within the intein sequence, at position Cys147, which appears to be also crucial for the splicing process [96] (Figure 3B).
The independent discovery of three redox-sensitive inteins strongly argues that reversible disulfide bond formation might be a general mechanism to regulate intein activity. Since all three inteins have been identified in anaerobic organisms, the authors of these studies speculated that the redox regulation of intein splicing occurs when these anaerobic organisms encounter oxygen. Inactivation of intein splicing in the presence of oxygen would prevent the assembly of the oxidation-sensitive Fe-S-cluster in MoaA, and inhibit DNA replication until non-oxidative stress conditions are restored [93, 95, 96]. At this point it is still unclear, however, if and to what extent disulfide bond formation occurs in vivo and whether oxidative inactivation of intein splicing in archaebacteria is indeed physiologically relevant. Future in vivo thiol trapping will help to provide the answer to this important question. Independent support, however, comes from studies on the Hedgehog (Hh) protein, a protein involved in signaling processes of multicellular organisms [97, 98]. Hedgehog proteins consist of two domains, an N-terminal Hedge domain and a C-terminal, intein-like, Hog domain with self-splicing activity. Like the Cys1 of inteins, Hog domains contain an active site cysteine, which forms a thioester with the carbonyl group of the adjacent Hedge domain (Figure 3A). A cholesterol moiety then displaces the Hog domain, generating a Hedge-cholesterol ester [89], and releasing the Hog protein. Critical for splicing of the Hedgehog protein, which occurs in the endoplasmic reticulum, is the reduction of a disulfide bond, which forms between the active site cysteine, and a second equally highly conserved cysteine [99]. These results suggest that the redox regulation of protein self-splicing activity is a much more widely used control mechanism than initially anticipated.
6. Cyclic Sulfenamide Formation – Another Way to Flip the Redox Switch
Originally identified and characterized as reversible peroxide-mediated modification of the active site cysteine in protein tyrosine phosphatase 1B (PTB1B), formation of cyclic sulfenamides is now a well-accepted mechanism that controls the activity of distinct signaling pathways in eukaryotes and oxidative stress response pathways in bacteria [100, 101]. At the center of this modification is typically a single, highly reactive cysteine, which is oxidized to sulfenic acid upon exposure to peroxide. Depending on the protein and redox environment, this sulfenic acid then either forms a mixed disulfide (e.g. GSH), attacks another nearby cysteine to form a disulfide bond or, as is the case for PTB1B and OhrR, becomes the target of a nucleophilic attack by a nearby backbone amide group, which leads to the formation of a cyclic sulfenamide (Figure 4) [102, 103]. Since the redox-sensitive cysteine plays a critical role in the tyrosine-dephosphorylation reaction, peroxide-mediated thiol modification inactivates PTB1B and leads to major changes in signal transduction pathways, including Ras and integrin signaling [104–106], in response to changes in the local peroxide environment [107].
Figure 4. Mechanism of sulfenamide formation in protein tyrosine phosphatase B1 (PTPB1).
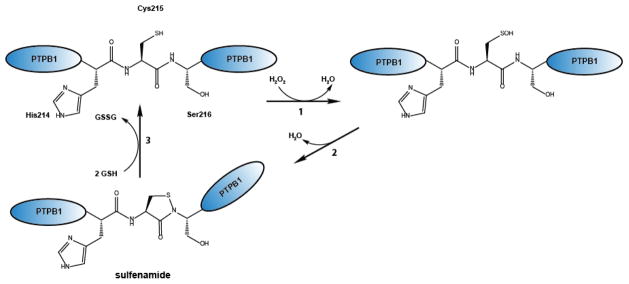
(1) Exposure of the catalytic cysteine 215 (RSH) of PTPB1 to H2O2 leads to the formation of a sulfenic acid (RSOH), which undergoes (2) intramolecular cyclization with a nearby amino group, yielding in sulfenamide (RSNR). (3) Reduction of the sulfenamide is mediated by forming a mixed disulfide formation with glutathione, which is subsequently resolved by glutaredoxin.
In contrast to PTPB1, whose peroxide-mediated oxidation directly inactivates the enzyme, and for which sulfenamide formation appears to serve primarily the purpose of preventing irreversible overoxidation [108, 109], Bacillus subtilis OhrR is a DNA-binding transcriptional repressor, which remains unaltered by the initial oxidation of its cysteine by organic hydroperoxides. Subsequent cyclic sulfenamide formation (or S-thiolation) then induces large conformational changes in OhrR, which are necessary to cause OhrR’s dissociation from the DNA and trigger the expression of select antioxidant enzymes [110, 111]. Reducing conditions reverse sulfenamide formation, reactivate PTP1B and restore the repressor activity of OhrR [112, 113].
It is still unclear how many other redox sensitive protein thiols undergo cyclic sulfenamide formation in vitro or in vivo. The modification causes only a 2 Da loss in mass, which is not always easy to detect and is difficult to distinguish from disulfide bond formation. In contrast to sulfenic acids, which can be specifically derivatized using dimedone and subsequently detected and quantified [114], no global analysis of sulfenamide formation in response to oxidative stress has been reported.
7. Sulfinic Acid Formation in Peroxiredoxin - Functional Changes During Severe Oxidative Stress
Peroxiredoxins, which belong to a highly conserved protein family present in all three domains of life [115], are in charge of detoxifying H2O2, organic hydrogen peroxides and peroxynitrite [116]. They play crucial roles in maintaining redox homeostasis, protecting against oxidative stress conditions, and controlling H2O2-sensitive signaling cascades [116, 117]. Deletion of peroxiredoxin-encoding genes causes severe phenotypes and premature aging in several model organisms [118–120].
The common feature shared by all members of the peroxiredoxin family is their active site cysteine (i.e., peroxidatic cysteine), which, upon reaction with peroxide, forms a sulfenic acid intermediate and reduces the oxidant to harmless water [61, 121] (Figure 5). Regeneration of the active site cysteine in peroxiredoxins depends on the specific type of peroxiredoxins (for excellent recent reviews please see [122]). In 1-Cys peroxiredoxins (e.g., Prx-6) the sulfenic acid is either reduced by ascorbate or low molecular weight thiols, although the physiological reduction mechanisms of peroxiredoxins such as Prx-6 are still not entirely clear. In 2-Cys peroxiredoxins, a second, equally highly conserved cysteine (i.e., resolving cysteine) forms an intra- or intermolecular disulfide bond with the peroxidatic sulfenic acid [123]. This disulfide bond is subsequently reduced by thioredoxin, using a direct thiol-disulfide exchange mechanism [124]. Re-reduction of thioredoxin by thioredoxin reductase and NADPH ultimately closes the catalytic cycle (Figure 5). Alternatively, select 2-Cys peroxiredoxins may interact with and oxidize other redox-sensitive cysteines, getting re-reduced in this process (Figure 5, see section 4).
Figure 5. Peroxiredoxin – An Enzyme with Multiple Personalities.
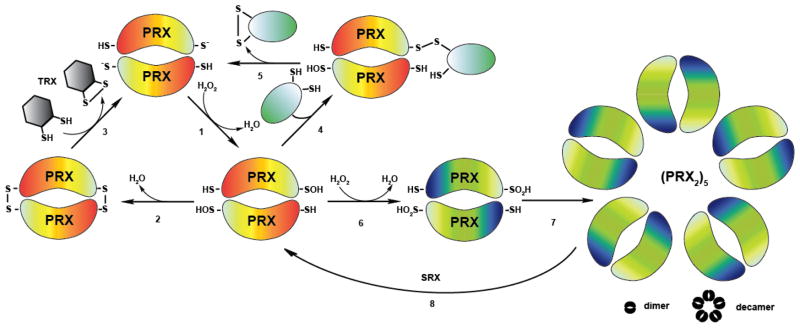
(1) Peroxiredoxins (orange) are responsible for the breakdown of hydrogen peroxide. In this process, their peroxidatic cysteines become oxidized to sulfenic acids. (2) Intermolecular disulfide bond formation with the resolving cysteines in Prx follows. (3) The disulfide bond is subsequently reduced by the Trx system. (4) In an alternative pathway, oxidized peroxiredoxin interacts with the thiol group of a reduced client protein (e.g. Yap1p), forming an intermolecular disulfide bond. (5) This disulfide bond is then resolved by a thiol-disulfide exchange reaction, yielding in an intramolecular disulfide bond within the client protein and reduced Prx. (6) High levels of peroxide lead to the formation of sulfinic acid at the active site cysteine and the inactivation of the peroxidase function. (7) Overoxidation triggers the assembly of peroxiredoxin into higher molecular weight structures, which exert chaperone function in vitro. (8) ATP-dependent sulfiredoxin (SRX) reduces the sulfinic acid in peroxiredoxin.
The active site cysteine in peroxiredoxin is located 13 Å apart from the resolving cysteine. Effective disulfide bond formation requires significant conformational changes the and the local unfolding of the active site [125]. In eukaryotic 2-Cys peroxiredoxins, a C-terminal extension appears to further slow down this unfolding process, making the peroxidatic sulfenic acid prone to react with a second molecule of peroxide to form sulfinic acid [117]. This result puzzled researchers for some time, and raised the question why organisms evolved peroxiredoxins, which lose their peroxidase function under high peroxide conditions. The discovery of sulfiredoxin, an enzyme specifically dedicated to reduce overoxidized peroxiredoxins, however, opened the possibility that this transient inactivation of peroxiredoxin is part of a physiologically relevant, regulatory mechanism [126]. Indeed, a number of recent studies provide supporting evidence that inactivation of peroxiredoxin by overoxidation is indeed beneficial and necessary under conditions of severe oxidative stress. Transient inactivation of peroxiredoxin in mammalian cells, for instance, allows for a localized increase in the amplitude of peroxide, which appears to be necessary for optimal stimulation of signaling cascades (i.e., flood gate hypothesis) [117]. Moreover, recent study in fission yeast revealed that overoxidation of peroxiredoxin frees up the thioredoxin pool, which can then be used to reduce and restore activity of other essential proteins that are prone to oxidation [127]. Rhee and coworkers demonstrated another physiological role for the oxidative inactivation of peroxiredoxin. Prx3, a mitochondrial 2-Cys peroxiredoxin located in the mouse adrenal cortex was found to undergo oxidative inactivation, triggered by Cytochrome P450-mediated H2O2 production during corticosterone synthesis. This inactivation, which followed circadian oscillations, appears to provide an alternative feedback mechanism to regulate steroidogenesis [128]. Finally, overoxidation of peroxiredoxin in yeast and select other model organisms has been shown to cause the formation of very high oligomeric structures, which exert ATP-independent chaperone function similar to the redox regulated chaperone Hsp33 [129] (Figure 5). These studies, which have so far only been conducted in vitro, would explain how eukaryotic cells, which do not contain Hsp33, deal with protein unfolding under ATP-depleted oxidative stress conditions. Oligomeric, chaperone-active peroxiredoxin is thought to expose hydrophobic surfaces capable of binding protein-folding intermediates and prevent their aggregation. So far, however, little is known about the precise mechanism by which chaperone activity is induced, how many proteins rely on peroxiredoxin’s chaperone function in vivo, and what the fate of peroxiredoxin’s client proteins is. It is conceivable that peroxiredoxins use the ATP-dependent reduction by sulfiredoxin to induce client release and support refolding. However, the fact that activation of peroxiredoxin’s chaperone function also occurs in mutant peroxiredoxins that lack the active site cysteine (and hence cannot be overoxidized) [129], or upon incubation in low pH conditions [130], suggests that other factors might be involved in client release. Future in vitro and in vivo studies are necessary to answer these important questions.
8. OUTLOOK
Redox regulation of protein function is a rapidly expanding research area, and it is now clear that many physiological processes in pro- and eukaryotic cells are either directly or indirectly regulated by the cellular redox conditions. With the development of global redox proteomic techniques, we are now in the excellent position to reconstruct these redox-regulated pathways and eventually model and predict how changes in the oxidant levels affect the complete physiology of the organism. Given that oxidative stress has been implicated in countless diseases, ranging from diabetes to heart disease and Parkinson’s disease [131, 132], obtaining a detailed overview of the processes affected and the main players involved, may allow us to design drugs that mitigate the effects of oxidative stress and hence improve the treatment of these diseases.
HIGHLIGHTS.
Reversible cysteine modifications function as redox regulators of protein activity
Disulfide bond formation in Hsp33 activates chaperone function
Disulfide bond formation in select inteins controls protein splicing
Sulfenamide formation in select phosphatases alters signaling processes
Overoxidation of 2-Cys peroxiredoxin switches the peroxidase into a chaperone
Acknowledgments
We thank James Bardwell for critically reading the manuscript. Work in the Jakob lab is supported by the National Institute of Health grants RO1 GM102829 and GM065318.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stadtman ER. Protein oxidation in aging and age-related diseases. Ann N Y Acad Sci. 2001;928:22–38. doi: 10.1111/j.1749-6632.2001.tb05632.x. [DOI] [PubMed] [Google Scholar]
- 2.Nauseef WM. Biological roles for the NOX family NADPH oxidases. Journal of Biological Chemistry. 2008;283(25):16961–5. doi: 10.1074/jbc.R700045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cooke MS, et al. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003;17(10):1195–214. doi: 10.1096/fj.02-0752rev. [DOI] [PubMed] [Google Scholar]
- 4.Bader N, Grune T. Protein oxidation and proteolysis. Biol Chem. 2006;387(10–11):1351–5. doi: 10.1515/BC.2006.169. [DOI] [PubMed] [Google Scholar]
- 5.Reuter S, et al. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic Biol Med. 2010 doi: 10.1016/j.freeradbiomed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antelmann H, Helmann JD. Thiol-based redox switches and gene regulation. Antioxid Redox Signal. 2010 doi: 10.1089/ars.2010.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng M, Aslund F, Storz G. Activation of the OxyR transcription factor by reversible disulfide bond formation. Science. 1998;279(5357):1718–21. doi: 10.1126/science.279.5357.1718. [DOI] [PubMed] [Google Scholar]
- 8.Imlay JA. Cellular defenses against superoxide and hydrogen peroxide. Annu Rev Biochem. 2008;77:755–76. doi: 10.1146/annurev.biochem.77.061606.161055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boronat S, et al. Thiol-based HO signalling in microbial systems. Redox biology. 2014;2:395–399. doi: 10.1016/j.redox.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jakob U, et al. Chaperone activity with a redox switch. Cell. 1999;96(3):341–52. doi: 10.1016/s0092-8674(00)80547-4. [DOI] [PubMed] [Google Scholar]
- 11.Winter J, et al. Severe oxidative stress causes inactivation of DnaK and activation of the redox-regulated chaperone Hsp33. Mol Cell. 2005;17(3):381–92. doi: 10.1016/j.molcel.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 12.Klomsiri C, Karplus PA, Poole LB. Cysteine-Based Redox Switches in Enzymes. Antioxid Redox Signal. 2010 doi: 10.1089/ars.2010.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brandes N, Schmitt S, Jakob U. Thiol-based redox switches in eukaryotic proteins. Antioxid Redox Signal. 2009;11(5):997–1014. doi: 10.1089/ars.2008.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan Z, Banerjee R. Redox remodeling as an immunoregulatory strategy. Biochemistry. 2010;49(6):1059–66. doi: 10.1021/bi902022n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cellular signalling. 2012;24 (5):981–90. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cook KM, Hogg PJ. Post-translational control of protein function by disulfide bond cleavage. Antioxid Redox Signal. 2013;18(15):1987–2015. doi: 10.1089/ars.2012.4807. [DOI] [PubMed] [Google Scholar]
- 17.Roos G, Foloppe N, Messens J. Understanding the pK(a) of redox cysteines: the key role of hydrogen bonding. Antioxid Redox Signal. 2013;18(1):94–127. doi: 10.1089/ars.2012.4521. [DOI] [PubMed] [Google Scholar]
- 18.Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45(5):549–61. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 19.Randall LM, Ferrer-Sueta G, Denicola A. Peroxiredoxins as preferential targets in H2O2-induced signaling. Methods in Enzymology. 2013;527:41–63. doi: 10.1016/B978-0-12-405882-8.00003-9. [DOI] [PubMed] [Google Scholar]
- 20.Lo Conte M, Carroll KS. The redox biochemistry of protein sulfenylation and sulfinylation. J Biol Chem. 2013;288(37):26480–8. doi: 10.1074/jbc.R113.467738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kettenhofen NJ, Wood MJ. Formation, Reactivity, and Detection of Protein Sulfenic Acids. Chem Res Toxicol. 2010 doi: 10.1021/tx100237w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poole LB, Karplus PA, Claiborne A. Protein sulfenic acids in redox signaling. Annu Rev Pharmacol Toxicol. 2004;44:325–47. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- 23.Claiborne A, et al. Protein-sulfenic acid stabilization and function in enzyme catalysis and gene regulation. FASEB J. 1993;7(15):1483–90. doi: 10.1096/fasebj.7.15.8262333. [DOI] [PubMed] [Google Scholar]
- 24.Shetty V, Spellman DS, Neubert TA. Characterization by tandem mass spectrometry of stable cysteine sulfenic acid in a cysteine switch peptide of matrix metalloproteinases. J Am Soc Mass Spectrom. 2007;18(8):1544–51. doi: 10.1016/j.jasms.2007.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kettenhofen NJ, Wood MJ. Formation, reactivity, and detection of protein sulfenic acids. Chem Res Toxicol. 2010;23(11):1633–46. doi: 10.1021/tx100237w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rhee SG, et al. Sulfiredoxin, the cysteine sulfinic acid reductase specific to 2-Cys peroxiredoxin: its discovery, mechanism of action, and biological significance. Kidney Int Suppl. 2007;(106):S3–8. doi: 10.1038/sj.ki.5002380. [DOI] [PubMed] [Google Scholar]
- 27.Moon JC, et al. Reversal of 2-Cys peroxiredoxin oligomerization by sulfiredoxin. Biochem Biophys Res Commun. 2013;432(2):291–5. doi: 10.1016/j.bbrc.2013.01.114. [DOI] [PubMed] [Google Scholar]
- 28.Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med. 2013 doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 29.Berndt C, Lillig CH, Holmgren A. Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: implications for diseases in the cardiovascular system. Am J Physiol Heart Circ Physiol. 2007;292(3):H1227–36. doi: 10.1152/ajpheart.01162.2006. [DOI] [PubMed] [Google Scholar]
- 30.Martin JL. Thioredoxin--a fold for all reasons. Structure. 1995;3(3):245–50. doi: 10.1016/s0969-2126(01)00154-x. [DOI] [PubMed] [Google Scholar]
- 31.Roos G, et al. How thioredoxin dissociates its mixed disulfide. PLoS Comput Biol. 2009;5(8):e1000461. doi: 10.1371/journal.pcbi.1000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williams CH., Jr Mechanism and structure of thioredoxin reductase from Escherichia coli. FASEB J. 1995;9(13):1267–76. doi: 10.1096/fasebj.9.13.7557016. [DOI] [PubMed] [Google Scholar]
- 33.Gallogly MM, Mieyal JJ. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr Opin Pharmacol. 2007;7(4):381–91. doi: 10.1016/j.coph.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 34.Holmgren A, Aslund F. Glutaredoxin. Methods Enzymol. 1995;252:283–92. doi: 10.1016/0076-6879(95)52031-7. [DOI] [PubMed] [Google Scholar]
- 35.Aslund F, Beckwith J. The thioredoxin superfamily: redundancy, specificity, and gray-area genomics. Journal of bacteriology. 1999;181(5):1375–9. doi: 10.1128/jb.181.5.1375-1379.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meyer Y, et al. Thioredoxin and glutaredoxin systems in plants: molecular mechanisms, crosstalks, and functional significance. Antioxidants & Redox Signaling. 2012;17(8):1124–60. doi: 10.1089/ars.2011.4327. [DOI] [PubMed] [Google Scholar]
- 37.Du Y, et al. Thioredoxin 1 is inactivated due to oxidation induced by peroxiredoxin under oxidative stress and reactivated by the glutaredoxin system. The Journal of biological chemistry. 2013;288(45):32241–7. doi: 10.1074/jbc.M113.495150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Masuda S, et al. Repression of photosynthesis gene expression by formation of a disulfide bond in CrtJ. Proc Natl Acad Sci U S A. 2002;99(10):7078–83. doi: 10.1073/pnas.102013099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Portis AR, Jr, et al. Regulation of Rubisco activase and its interaction with Rubisco. J Exp Bot. 2008;59(7):1597–604. doi: 10.1093/jxb/erm240. [DOI] [PubMed] [Google Scholar]
- 40.Aslund F, et al. Regulation of the OxyR transcription factor by hydrogen peroxide and the cellular thiol-disulfide status. Proc Natl Acad Sci U S A. 1999;96 (11):6161–5. doi: 10.1073/pnas.96.11.6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang CS, et al. Chemical approaches for trapping protein thiols and their oxidative modification. Yao Xue Xue Bao. 2012;47(3):280–90. [PubMed] [Google Scholar]
- 42.Sethuraman M, et al. Isotope-coded affinity tag (ICAT) approach to redox proteomics: identification and quantitation of oxidant-sensitive cysteine thiols in complex protein mixtures. J Proteome Res. 2004;3(6):1228–33. doi: 10.1021/pr049887e. [DOI] [PubMed] [Google Scholar]
- 43.Leichert LI, et al. Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc Natl Acad Sci U S A. 2008;105:8197–202. doi: 10.1073/pnas.0707723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gianazza E, Eberini I, Ghezzi P. Detection of protein glutathionylation. Methods Mol Biol. 2009;519:397–415. doi: 10.1007/978-1-59745-281-6_26. [DOI] [PubMed] [Google Scholar]
- 45.Brandes N, et al. Using Quantitative Redox Proteomics to Dissect the Yeast Redoxome. Journal of Biological Chemistry. 2011;286(48):41893–41903. doi: 10.1074/jbc.M111.296236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Knoefler D, et al. Quantitative in vivo redox sensors uncover oxidative stress as an early event in life. Molecular Cell. 2012;47(5):767–76. doi: 10.1016/j.molcel.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar V, et al. Redox proteomics of thiol proteins in mouse heart during ischemia/reperfusion using ICAT reagents and mass spectrometry. Free radical biology & medicine. 2013;58:109–17. doi: 10.1016/j.freeradbiomed.2013.01.021. [DOI] [PubMed] [Google Scholar]
- 48.Nelson KJ, et al. Use of dimedone-based chemical probes for sulfenic acid detection methods to visualize and identify labeled proteins. Methods Enzymol. 2010;473:95–115. doi: 10.1016/S0076-6879(10)73004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jones DP, Go YM. Mapping the cysteine proteome: analysis of redox-sensing thiols. Curr Opin Chem Biol. 2011;15(1):103–12. doi: 10.1016/j.cbpa.2010.12.014. [DOI] [PubMed] [Google Scholar]
- 50.Gray MJ, Wholey WY, Jakob U. Bacterial responses to reactive chlorine species. Annu Rev Microbiol. 2013;67:141–60. doi: 10.1146/annurev-micro-102912-142520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pattison DI, Davies MJ, Hawkins CL. Reactions and reactivity of myeloperoxidase-derived oxidants: differential biological effects of hypochlorous and hypothiocyanous acids. Free radical research. 2012;46(8):975–95. doi: 10.3109/10715762.2012.667566. [DOI] [PubMed] [Google Scholar]
- 52.Leichert LI, et al. Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc Natl Acad Sci U S A. 2008;105(24):8197–202. doi: 10.1073/pnas.0707723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ferrer-Sueta G, et al. Factors affecting protein thiol reactivity and specificity in peroxide reduction. Chem Res Toxicol. 2011;24(4):434–50. doi: 10.1021/tx100413v. [DOI] [PubMed] [Google Scholar]
- 54.Winterbourn CC. The biological chemistry of hydrogen peroxide. Methods in Enzymology. 2013;528:3–25. doi: 10.1016/B978-0-12-405881-1.00001-X. [DOI] [PubMed] [Google Scholar]
- 55.Winterbourn CC, Metodiewa D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic Biol Med. 1999;27 (3–4):322–8. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- 56.Delaunay A, et al. A thiol peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell. 2002;111(4):471–81. doi: 10.1016/s0092-8674(02)01048-6. [DOI] [PubMed] [Google Scholar]
- 57.Jarvis RM, Hughes SM, Ledgerwood EC. Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radic Biol Med. 2012;53(7):1522–30. doi: 10.1016/j.freeradbiomed.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 58.Peskin AV, et al. The high reactivity of peroxiredoxin 2 with H(2)O(2) is not reflected in its reaction with other oxidants and thiol reagents. The Journal of biological chemistry. 2007;282(16):11885–92. doi: 10.1074/jbc.M700339200. [DOI] [PubMed] [Google Scholar]
- 59.Nagy P, et al. Model for the exceptional reactivity of peroxiredoxins 2 and 3 with hydrogen peroxide: a kinetic and computational study. The Journal of biological chemistry. 2011;286(20):18048–55. doi: 10.1074/jbc.M111.232355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hall A, et al. Structural evidence that peroxiredoxin catalytic power is based on transition-state stabilization. Journal of Molecular Biology. 2010;402(1):194–209. doi: 10.1016/j.jmb.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Poole LB. The catalytic mechanism of peroxiredoxins. Subcell Biochem. 2007;44:61–81. doi: 10.1007/978-1-4020-6051-9_4. [DOI] [PubMed] [Google Scholar]
- 62.Winter J, et al. Bleach activates a redox-regulated chaperone by oxidative protein unfolding. Cell. 2008;135(4):691–701. doi: 10.1016/j.cell.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ilbert M, et al. The redox-switch domain of Hsp33 functions as dual stress sensor. Nat Struct Mol Biol. 2007 doi: 10.1038/nsmb1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ryu JH, Ha EM, Lee WJ. Innate immunity and gut-microbe mutualism in Drosophila. Dev Comp Immunol. 2010;34(4):369–76. doi: 10.1016/j.dci.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 65.Jacquier-Sarlin MR, et al. Protective effects of hsp70 in inflammation. Experientia. 1994;50(11–12):1031–8. doi: 10.1007/BF01923458. [DOI] [PubMed] [Google Scholar]
- 66.Tamarit J, Cabiscol E, Ros J. Identification of the major oxidatively damaged proteins in Escherichia coli cells exposed to oxidative stress. J Biol Chem. 1998;273(5):3027–32. doi: 10.1074/jbc.273.5.3027. [DOI] [PubMed] [Google Scholar]
- 67.Khor HK, Fisher MT, Schoneich C. Potential role of methionine sulfoxide in the inactivation of the chaperone GroEL by hypochlorous acid (HOCl) and peroxynitrite (ONOO−) The Journal of biological chemistry. 2004;279(19):19486–93. doi: 10.1074/jbc.M310045200. [DOI] [PubMed] [Google Scholar]
- 68.Jakob U, Eser M, Bardwell JC. Redox switch of hsp33 has a novel zinc-binding motif. J Biol Chem. 2000;275(49):38302–10. doi: 10.1074/jbc.M005957200. [DOI] [PubMed] [Google Scholar]
- 69.Janda I, et al. The crystal structure of the reduced, Zn2+-bound form of the B. subtilis Hsp33 chaperone and its implications for the activation mechanism. Structure (Camb) 2004;12(10):1901–7. doi: 10.1016/j.str.2004.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vijayalakshmi J, et al. The 2.2 A crystal structure of Hsp33: a heat shock protein with redox-regulated chaperone activity. Structure. 2001;9(5):367–75. doi: 10.1016/s0969-2126(01)00597-4. [DOI] [PubMed] [Google Scholar]
- 71.Barbirz S, Jakob U, Glocker MO. Mass spectrometry unravels disulfide bond formation as the mechanism that activates a molecular chaperone. J Biol Chem. 2000;275(25):18759–66. doi: 10.1074/jbc.M001089200. [DOI] [PubMed] [Google Scholar]
- 72.Graf PC, et al. Activation of the redox-regulated chaperone Hsp33 by domain unfolding. J Biol Chem. 2004;279(19):20529–38. doi: 10.1074/jbc.M401764200. [DOI] [PubMed] [Google Scholar]
- 73.Graumann J, et al. Activation of the redox-regulated molecular chaperone Hsp33--a two-step mechanism. Structure. 2001;9(5):377–87. doi: 10.1016/s0969-2126(01)00599-8. [DOI] [PubMed] [Google Scholar]
- 74.Reichmann D, et al. Order out of disorder: working cycle of an intrinsically unfolded chaperone. Cell. 2012;148(5):947–57. doi: 10.1016/j.cell.2012.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cremers CM, et al. Unfolding of metastable linker region is at the core of Hsp33 activation as a redox-regulated chaperone. J Biol Chem. 2010;285(15):11243–51. doi: 10.1074/jbc.M109.084350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hoffmann JH, et al. Identification of a redox-regulated chaperone network. EMBO J. 2004;23(1):160–8. doi: 10.1038/sj.emboj.7600016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bardwell JC, Jakob U. Conditional disorder in chaperone action. Trends Biochem Sci. 2012;37(12):517–25. doi: 10.1016/j.tibs.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tapley TL, et al. Structural plasticity of an acid-activated chaperone allows promiscuous substrate binding. Proc Natl Acad Sci U S A. 2009;106(14):5557–62. doi: 10.1073/pnas.0811811106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Foit L, et al. Chaperone activation by unfolding. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(14):E1254–62. doi: 10.1073/pnas.1222458110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tapley TL, et al. Protein refolding by pH-triggered chaperone binding and release. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(3):1071–6. doi: 10.1073/pnas.0911610107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Haslbeck M, et al. A domain in the N-terminal part of Hsp26 is essential for chaperone function and oligomerization. J Mol Biol. 2004;343(2):445–55. doi: 10.1016/j.jmb.2004.08.048. [DOI] [PubMed] [Google Scholar]
- 82.Peschek J, et al. Regulated structural transitions unleash the chaperone activity of alphaB-crystallin. Proc Natl Acad Sci U S A. 2013;110(40):E3780–9. doi: 10.1073/pnas.1308898110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhao F, et al. Are zinc-finger domains of protein kinase C dynamic structures that unfold by lipid or redox activation? Antioxid Redox Signal. 2011;14(5):757–66. doi: 10.1089/ars.2010.3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cech TR. Self-splicing of group I introns. Annu Rev Biochem. 1990;59:543–68. doi: 10.1146/annurev.bi.59.070190.002551. [DOI] [PubMed] [Google Scholar]
- 85.Cooper AA, Stevens TH. Protein splicing: self-splicing of genetically mobile elements at the protein level. Trends Biochem Sci. 1995;20(9):351–6. doi: 10.1016/s0968-0004(00)89075-1. [DOI] [PubMed] [Google Scholar]
- 86.Perler FB, et al. Protein splicing elements: inteins and exteins--a definition of terms and recommended nomenclature. Nucleic Acids Res. 1994;22(7):1125–7. doi: 10.1093/nar/22.7.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Volkmann G, Mootz HD. Recent progress in intein research: from mechanism to directed evolution and applications. Cell Mol Life Sci. 2013;70(7):1185–206. doi: 10.1007/s00018-012-1120-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gogarten JP, et al. Inteins: structure, function, and evolution. Annu Rev Microbiol. 2002;56:263–87. doi: 10.1146/annurev.micro.56.012302.160741. [DOI] [PubMed] [Google Scholar]
- 89.Perler FB. Protein splicing of inteins and hedgehog autoproteolysis: structure, function, and evolution. Cell. 1998;92(1):1–4. doi: 10.1016/s0092-8674(00)80892-2. [DOI] [PubMed] [Google Scholar]
- 90.Shah NH, et al. Naturally Split Inteins Assemble through a “Capture and Collapse” Mechanism. Journal of the American Chemical Society. 2013;135(49):18673–81. doi: 10.1021/ja4104364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu XQ. Protein-splicing intein: Genetic mobility, origin, and evolution. Annu Rev Genet. 2000;34:61–76. doi: 10.1146/annurev.genet.34.1.61. [DOI] [PubMed] [Google Scholar]
- 92.Southworth MW, Benner J, Perler FB. An alternative protein splicing mechanism for inteins lacking an N-terminal nucleophile. EMBO J. 2000;19(18):5019–26. doi: 10.1093/emboj/19.18.5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Callahan BP, et al. Structure of catalytically competent intein caught in a redox trap with functional and evolutionary implications. Nat Struct Mol Biol. 2011;18 (5):630–3. doi: 10.1038/nsmb.2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Prinz WA, et al. The role of the thioredoxin and glutaredoxin pathways in reducing protein disulfide bonds in the Escherichia coli cytoplasm. The Journal of biological chemistry. 1997;272(25):15661–7. doi: 10.1074/jbc.272.25.15661. [DOI] [PubMed] [Google Scholar]
- 95.Chen W, et al. Intramolecular disulfide bond between catalytic cysteines in an intein precursor. Journal of the American Chemical Society. 2012;134(5):2500–3. doi: 10.1021/ja211010g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nicastri MC, et al. Internal disulfide bond acts as a switch for intein activity. Biochemistry. 2013;52(34):5920–7. doi: 10.1021/bi400736c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pietrokovski S. Intein spread and extinction in evolution. Trends Genet. 2001;17 (8):465–72. doi: 10.1016/s0168-9525(01)02365-4. [DOI] [PubMed] [Google Scholar]
- 98.Xie J, et al. H, C, and N NMR assignments of a Drosophila Hedgehog autoprocessing domain. Biomol NMR Assign. 2013 doi: 10.1007/s12104-013-9500-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chen X, et al. Processing and turnover of the Hedgehog protein in the endoplasmic reticulum. J Cell Biol. 2011;192(5):825–38. doi: 10.1083/jcb.201008090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Salmeen A, Barford D. Methods for preparing crystals of reversibly oxidized proteins: crystallization of protein tyrosine phosphatase 1B as an example. Methods Mol Biol. 2008;476:101–16. doi: 10.1007/978-1-59745-129-1_8. [DOI] [PubMed] [Google Scholar]
- 101.Yang J, et al. Reversible oxidation of the membrane distal domain of receptor PTPalpha is mediated by a cyclic sulfenamide. Biochemistry. 2007;46(3):709–19. doi: 10.1021/bi061546m. [DOI] [PubMed] [Google Scholar]
- 102.Meng FG, Zhang ZY. Redox regulation of protein tyrosine phosphatase activity by hydroxyl radical. Biochim Biophys Acta. 2013;1834(1):464–9. doi: 10.1016/j.bbapap.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ostman A, et al. Regulation of protein tyrosine phosphatases by reversible oxidation. Journal of biochemistry. 2011;150(4):345–56. doi: 10.1093/jb/mvr104. [DOI] [PubMed] [Google Scholar]
- 104.Liang F, et al. The role of protein-tyrosine phosphatase 1B in integrin signaling. J Biol Chem. 2005;280(26):24857–63. doi: 10.1074/jbc.M502780200. [DOI] [PubMed] [Google Scholar]
- 105.Dube N, Cheng A, Tremblay ML. The role of protein tyrosine phosphatase 1B in Ras signaling. Proc Natl Acad Sci U S A. 2004;101(7):1834–9. doi: 10.1073/pnas.0304242101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Salmeen A, Barford D. Functions and mechanisms of redox regulation of cysteine-based phosphatases. Antioxidants & Redox Signaling. 2005;7(5–6):560–77. doi: 10.1089/ars.2005.7.560. [DOI] [PubMed] [Google Scholar]
- 107.Bae YS, et al. Regulation of reactive oxygen species generation in cell signaling. Molecules and cells. 2011;32(6):491–509. doi: 10.1007/s10059-011-0276-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.van Montfort RL, et al. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature. 2003;423(6941):773–7. doi: 10.1038/nature01681. [DOI] [PubMed] [Google Scholar]
- 109.Salmeen A, et al. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423(6941):769–73. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 110.Eiamphungporn W, et al. Oxidation of a single active site suffices for the functional inactivation of the dimeric Bacillus subtilis OhrR repressor in vitro. Nucleic acids research. 2009;37(4):1174–81. doi: 10.1093/nar/gkn1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lee JW, Soonsanga S, Helmann JD. A complex thiolate switch regulates the Bacillus subtilis organic peroxide sensor OhrR. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(21):8743–8. doi: 10.1073/pnas.0702081104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dubbs JM, Mongkolsuk S. Peroxide-sensing transcriptional regulators in bacteria. J Bacteriol. 2012;194(20):5495–503. doi: 10.1128/JB.00304-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pan J, Carroll KS. Chemical biology approaches to study protein cysteine sulfenylation. Biopolymers. 2013 doi: 10.1002/bip.22255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Seo YH, Carroll KS. Quantification of protein sulfenic acid modifications using isotope-coded dimedone and iododimedone. Angew Chem Int Ed Engl. 2011;50(6):1342–5. doi: 10.1002/anie.201007175. [DOI] [PubMed] [Google Scholar]
- 115.Hofmann B, Hecht HJ, Flohe L. Peroxiredoxins. Biol Chem. 2002;383(3–4):347–64. doi: 10.1515/BC.2002.040. [DOI] [PubMed] [Google Scholar]
- 116.Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38(12):1543–52. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 117.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300(5619):650–3. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 118.Lee TH, et al. Peroxiredoxin II is essential for sustaining life span of erythrocytes in mice. Blood. 2003;101(12):5033–5038. doi: 10.1182/blood-2002-08-2548. [DOI] [PubMed] [Google Scholar]
- 119.Neumann CA, et al. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature. 2003;424(6948):561–565. doi: 10.1038/nature01819. [DOI] [PubMed] [Google Scholar]
- 120.Kumsta C, Thamsen M, Jakob U. Effects of Oxidative Stress on Behavior, Physiology, and the Redox Thiol Proteome of Caenorhabditis elegans. Antioxidants & Redox Signaling. 2011;14(6):1023–1037. doi: 10.1089/ars.2010.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hall A, et al. Structure-based insights into the catalytic power and conformational dexterity of peroxiredoxins. Antioxidants & redox signaling. 2011;15(3):795–815. doi: 10.1089/ars.2010.3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Poynton RA, Hampton MB. Peroxiredoxins as biomarkers of oxidative stress. Biochimica et biophysica acta. 2014;1840(2):906–12. doi: 10.1016/j.bbagen.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 123.Knoops B, Loumaye E, Van Der Eecken V. Evolution of the peroxiredoxins. Subcell Biochem. 2007;44:27–40. doi: 10.1007/978-1-4020-6051-9_2. [DOI] [PubMed] [Google Scholar]
- 124.Du Y, et al. Thioredoxin 1 Is Inactivated Due to Oxidation Induced by Peroxiredoxin under Oxidative Stress and Reactivated by the Glutaredoxin System. J Biol Chem. 2013;288(45):32241–7. doi: 10.1074/jbc.M113.495150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schroder E, et al. Crystal structure of decameric 2-Cys peroxiredoxin from human erythrocytes at 1.7 A resolution. Structure. 2000;8(6):605–15. doi: 10.1016/s0969-2126(00)00147-7. [DOI] [PubMed] [Google Scholar]
- 126.Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425(6961):980–4. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 127.Day AM, et al. Inactivation of a peroxiredoxin by hydrogen peroxide is critical for thioredoxin-mediated repair of oxidized proteins and cell survival. Molecular Cell. 2012;45(3):398–408. doi: 10.1016/j.molcel.2011.11.027. [DOI] [PubMed] [Google Scholar]
- 128.Kil IS, et al. Feedback control of adrenal steroidogenesis via H2O2-dependent, reversible inactivation of peroxiredoxin III in mitochondria. Molecular cell. 2012;46(5):584–94. doi: 10.1016/j.molcel.2012.05.030. [DOI] [PubMed] [Google Scholar]
- 129.Jang HH, et al. Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell. 2004;117(5):625–35. doi: 10.1016/j.cell.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 130.Saccoccia F, et al. Moonlighting by different stressors: crystal structure of the chaperone species of a 2-Cys peroxiredoxin. Structure. 2012;20(3):429–39. doi: 10.1016/j.str.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Aliev G, et al. The role of oxidative stress in the pathophysiology of cerebrovascular lesions in Alzheimer’s disease. Brain Pathol. 2002;12(1):21–35. doi: 10.1111/j.1750-3639.2002.tb00419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Valko M, et al. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39(1):44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]