

Abstract
Oxygen homeostasis plays a critical role in angiogenesis, erythropoiesis and cell metabolism. Oxygen homeostasis is set by the hypoxia inducible factor-1α (HIF-1α) pathway, which is controlled by Factor Inhibiting HIF-1α (FIH). FIH is a non-heme Fe(II), α-ketoglutarate dependent dioxygenase that inhibits HIF-1α by hydroxylating the C-terminal transactivation domain (CTAD) of HIF-1α at HIF-Asn803. A tight coupling between CTAD binding and O2-activation is essential for hypoxia sensing, making changes in the coordination geometry of Fe(II) upon CTAD encounter a crucial feature of this enzyme. Although the consensus chemical mechanism for FIH proposes that CTAD binding triggers O2-activation by causing the Fe(II) cofactor to release an aquo ligand, experimental evidence of this point has been absent. More broadly, this proposed coordination change at Fe(II) has not been observed during steady-state turnover in any αKG oxygenase to date. In this manuscript, solvent isotope effects (SIEs) were used as a direct mechanistic probe of substrate triggered aquo release in FIH, as inverse SIEs (SIE < 1) are signatures for pre-equilibrium aquo release from metal ions. Our mechanistic studies of FIH have revealed inverse solvent isotope effects in the steady-state rate constants at limiting concentrations of CTAD or αKG: D2Okcat/KM(CTAD) = 0.40 ± 0.07, D2Okcat/KM(αKG) = 0.32 ± 0.08, providing direct evidence for aquo release during steady-state turnover. Furthermore, the SIE at saturating concentrations of CTAD and αKG was inverse, D2Okcat = 0.51 ± 0.07, indicating that aquo release occurs after CTAD binds. The inverse kinetic SIEs observed in the steady state for FIH can be explained by a strong Fe-OH2 bond. The stable Fe-OH2 bond plays an important part in FIH’s regulatory role over O2 homeostasis in humans, and points toward a strategy for tightly coupling O2-activation with CTAD hydroxylation that relies on substrate triggering.
Introduction
Oxygen homeostasis is essential for proper cellular function in humans and is tightly controlled by a small group of non-heme Fe(II), alpha-ketoglutarate (αKG)-dependent dioxygenases. Oxygen-dependent regulation of the transcriptional coactivator hypoxia inducible factor-1α (HIF-1α) maintains O2 homeostasis as HIF-1α controls more than 100 genes, directing processes such as angiogenesis, glycolysis and erythropoeisis.(1–3) The Fe(II)/αKG-dependent dioxygenase ‘factor inhibiting HIF-1α’ (FIH-1 or FIH) inhibits HIF-1α in the presence of sufficient [O2] by hydroxylating the C-terminal activation domain (CTAD) of HIF-1α at HIF-Asn803.(4–7) This hydroxylation prevents recruitment of the CREB binding protein, effectively down regulating HIF-1α dependent gene expression in response to normal or elevated [O2].
The consensus mechanism for Fe(II)/αKG-dependent dioxygenases proposes that binding of primary substrate triggers the Fe(II) to bind O2, due to aquo release opening a coordination site for O2 (Scheme 1). Substrate triggering is a central tenet of the consensus mechanism, as it can explain the relative O2-reactivity of αKG oxygenases. (11)(49) Subsequent O2 activation allows for oxidative decarboxylation of αKG and the formation of a highly reactive ferryl intermediate. H-atom abstraction and ensuing •OH rebound effectively hydroxylates the primary substrate. As sensing [O2] requires that O2-activation only occur when HIF-1α is bound, substrate triggering is the key to O2-sensing by FIH.
Scheme 1.
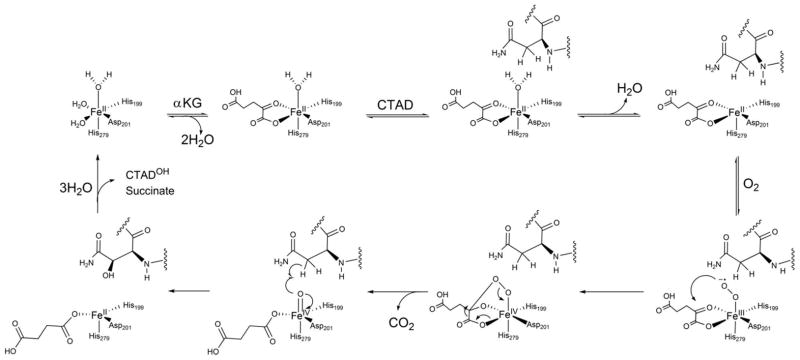
Consensus chemical mechanism for FIH.
Various experimental techniques have been used to test the consensus mechanism of the Fe(II)/αKG-dependent dioxygenases. Extensive spectroscopic studies on the Fe(II)/αKG-dependent dioxygenase clavaminate synthase (CAS) have provided insight to the coordination change due to aquo release induced by primary substrate. MCD and CD studies of (Fe + αKG)CAS indicate the Fe(II) is predominantly 6 coordinate. Upon addition of primary substrate (S) the active site iron of (Fe + αKG + S)CAS adopts a 5 coordinate square-pyramidal geometry (8–10) due to release of the aquo ligand. Stopped-flow and freeze-quench techniques have been used to study the Fe(II)/αKG-dependent dioxygenase, taurine dioxygenase (TauD). Loss of the aquo ligand was observed under single-turnover conditions, and these studies identified a Fe(IV)=O intermediate as the active oxidant in TauD.(11–13) However these studies did not address aquo release under multiple turnover conditions leaving open the possibility that aquo release is not a recurring step in the steady-state.
To date, neither in depth spectroscopic studies of the coordination changes proposed upon substrate binding, nor mechanistic probes for substrate triggering have been reported for FIH, making substrate triggering in this enzyme largely speculative. The X-ray crystallographic data for FIH bound to various substrates is ambiguous with respect to the coordination geometry of Fe(II), making it difficult to draw conclusions about the link between substrate binding and aquo release. Structures of (Fe + αKG)FIH are reported as containing either 5-coordinate(15) or 6-coordinate(16) Fe(II), however the crystal structure of (Fe + αKG + CTAD)FIH shows a 5-coordinate Fe(II),(15) suggesting that aquo release may partially occur prior to CTAD binding. EPR spectroscopy of Co(II)-substituted (Co + αKG)FIH revealed a mixture of 5-coordinate and 6-coordinate Co(II), further suggesting that aquo release may occur prior to CTAD binding.(17, 18) Crystal structures of both (Fe + αKG + Notch)FIH and (Fe + αKG + TNKS2)FIH were refined with an axial aquo ligand present, suggesting that aquo release occurs after these substrates bind.(19, 20) Direct mechanistic probes of aquo release during turnover would provide a crucial view into the mechanism of this enzyme, as well as providing insight into substrate triggering by this broad class of enzymes.
We propose that the extensive H-bonding network surrounding the active site of FIH constitutes a second coordination sphere, which transduces CTAD binding into aquo release. Steady-state kinetics of FIH point mutants indicate that removal of hydrogen-bonds from several residues surrounding the Fe(II) impairs catalytic efficiency(14), suggesting that substrate binding causes alterations to those hydrogen bonds. FIH has been shown to have a tightly coupled oxidative and reductive half reaction, avoiding the formation of reactive oxygen species during turnover.(21) Point mutations that perturb this H-bonding network significantly decrease FIH activity and lead to uncoupled αKG consumption.(14) Based on FIH crystal structures and mechanistic data for point mutants, we hypothesized that aquo release occurs on every turnover during the steady state.
To test this hypothesis, we applied mechanistic probes that are sensitive to those steps preceding O2-activation. Specifically, we focused on the steps involving αKG and CTAD binding, as these steps lead to substrate triggering: the aquo release and priming of the Fe(II) for O2 binding/activation. Steady-state kinetic assays were performed to identify the preferred binding order for CTAD and αKG. Solvent isotope effects (SIEs) on steady-state rate constants were measured to test for the release of aquo ligands from the Fe(II) cofactor. When the rate constants are larger in D2O-containing buffers than for H2O-containing buffer, the kinetics SIEs are ‘inverse’ and are diagnostic for pre-equilibrium release of aquo ligands from metal cofactors. We observed inverse SIEs on steady-state rate constants, providing direct evidence for the pre-equilibrium release of 1, 2 and 3 aquo ligands from the Fe(II) under different conditions. Our results establish two very important points. First, CTAD-binding to (Fe + αKG)FIH leads to release of 1 aquo ligand, suggesting that the overall strategy to control hydroxylation in FIH relies on substrate triggered changes in hydrogen-bonding to release the aquo ligand. Second, the rate-limiting step in the steady-state precedes or coincides with the first irreversible step – this is likely to be decarboxylation (Scheme 1), meaning that FIH does not accumulate oxidized intermediates. Both observations point to an overall strategy for O2-activation that relies on careful control over substrate triggering in FIH.
Experimental Procedures
Materials
All buffers and reagents were purchased from commercial vendors and were not further purified, with the exception of the CTAD peptide. The CTAD peptide corresponding to the C-terminal activation domain of human HIF-1α, (HIF-1α788–826) contained a Cys800 → Ala point mutation(21): DESGLPQLTSYDAEVNAPIQGSRNLLQGEELLRALDQVN. The CTAD peptide was purchased as a desalted peptide from EZBiolab (Carmel, IN, USA) with free N and C-termini. RP-HPLC utilizing a gradient from 30% acetonitrile/0.1% trifluoroacetic acid (TFA) to 95% acetonitrile/0.1% TFA was used to obtain CTAD of >95% purity.
Protein Expression and Purification
FIH was overexpressed in E. coli with an N-terminal His6 tag and purified as previously described.(18) Briefly, His6-FIH was separated from cell lysate via Ni-NTA column chromatography and the affinity tag was then cleaved with thrombin. Three additional residues, from the fusion protein (NH2-GlySerHis-) remained on the N-terminus following thrombin cleavage. Cleaved FIH was collected as flow through from a Ni-NTA column and then incubated with EDTA to removed metal. Dimeric FIH was obtained with size-exclusion chromatography using Sephadex G-75 resin and 50 mM NaCl 50 mM Tris pH 8.00 as the running buffer. Purified FIH was buffer exchanged into 50 mM HEPES pH 7.00. The molecular weight was confirmed by QSTAR TOF-MS (expected, 40.566 kDa; observed, 40.574 kDa), while the purity (>95%) was assessed by SDS-PAGE.
Steady-state kinetics assays
All assays were performed at 37.0°C with saturating concentrations of FeSO4 (50 μM) and ascorbate (2 mM), and ambient O2 concentration. Assays in which CTAD was the varied substrate (15 μM – 250 μM) utilized saturating αKG (500 μM) unless specified otherwise. Assays with αKG as the varied substrate (5 μM – 200 μM) utilized a fixed CTAD concentration of either 39 μM (~1/2KM(CTAD)) or 306 μM (~4xKM(CTAD)). Assay reagents were mixed and incubated for two minutes at 37.0°C in microcentrigue tubes. Then the reaction was initiated with the addition of enzyme ([E]T = 0.5 μM). Reaction aliquots (5 μL) were quenched with 75% acetonitrile/0.2% TFA (20 μL) saturated with 3,5-dimethoxy-4-hydroxycinnamic acid and analyzed for peptide hydroxylation using a Bruker Daltonics Omniflex MALDI-TOF-MS. The mole fraction of product, χCTAD-OH, was determined by the relative intensities of hydroxylated CTAD (CTADOH, 4271 m/z) and unhydroxylated CTAD (4255 m/z). Initial rates were determined from 5–7 quenched timepoints. The steady-state rate constants, kcat and kcat/KM, were obtained by nonlinear least-squares fitting of initial rate data (0 to ~15% fractional conversion) to the Michaelis-Menten equation.
Viscosity Assays
Steady-state assays were performed in 50 mM HEPES pH 7.00, using sucrose as the viscosogen to test for the rate limitation of diffusional steps. Initial rates were measured as described above, using buffers containing 10% (ηrel=1.3), 18% (ηrel=1.8) and 25% (ηrel=2.8) sucrose solutions (w/w).(22)
Solvent Isotope Assays
Deuterium oxide (D, 99.9%) was purchased from Cambridge Isotope Laboratories (Andover, MA, USA) and used as received. The pD was determined by pre-soaking the pH meter in D2O for ten minutes and then adding 0.4 to the meter reading of the D2O solution of interest (pD = pHread + 0.4).(23) Steady-state assays were performed as described above. All reagent stocks used in the steady-state assays in D2O, were prepared using D2O. Working FIH stock solutions were made by diluting high concentration stocks from H2O into D2O containing 50mM HEPES pD 7.00. Assays were performed in 50 mM HEPES, pD 7.00, with a final D2O percentage of 97%. SIEs were calculated from the ratio of rate constants observed in buffers containing H2O or D2O; e.g. D2Okcat = kcat(H2O)/kcat(D2O).
Coupling Ratio
The extent of coupling between FIH’s two half reactions in D2O was determined from monitoring the succinate and CTADOH concentrations throughout a reaction. Reactions containing αKG (500 μM), FeSO4 (50 μM), CTAD (200 μM) and FIH (5 μM) were performed in 50 mM Tris pD 7.00, and analyzed similarly to previously reported procedures.(14, 24) A Hamilton PRP-X300 anion exclusion column was used to separate the succinate produced from the quenched reactions. UV detection at 210 nm was used to determine the succinate concentration. Using aliquots from the same quenched assay, a Bruker Daltonics Omniflex MALDI-TOF-MS was used to determine the CTADOH concentration. The coupling ratio (C) was determined by fitting [succinate] as a linear function of [CTADOH] for multiple quench points, where C = [succinate]/[CTADOH].
Results
Solvent isotope effects
Solvent isotope effects (SIEs) were measured to test for aquo release from the Fe(II) as part of substrate triggering during turnover in FIH. By varying the degree of saturation with respect to αKG and CTAD, the SIE on kinetic constants reporting on different microscopic steps were accessed. SIEs arising from the pre-equilibirum release of 1, 2, and 3 aquo ligands were predicted from the consensus mechanism, depending on the assay conditions (Scheme 1). As the limiting SIEs are multiples of the proton fractionation factor (Φ) for the O-L bond (L = H or D)(25), they are quite distinct for the number of aquo ligands released.
The fractionation factor for L2O (where L = H or D) is inverse when water is bonded to another Lewis acid, leading to a tendency for D2O to accumulate in bulk solvent. For example, ΦO-L = 0.69 for L3O+ (26–28) due to zero-point energy differences. The fractionation factor, ΦO-L, for aquo release from Co(II) in two carbonic anhydrase isozymes has been found to be between 0.72 and 0.90,(25, 29) agreeing closely with the fractionation factor for in solution, (ΦO-L = 0.73).(30) As the fractionation factor for the Fe2+-OH2 in FIH is unknown, we estimate Φ = 0.70 as is commonly done for fractionation of water from other M-OH2 ⇌ M + OH2 equilibria.(30) As Φ is reported on a per-bond basis, and are multiplicative, D2OKeq for the release of aquo ligands are diagnostic for the numbers of H2O released from Fe: one (D2OKeq = Φ2 = 0.49), two (D2OKeq = Φ4 = 0.24) and three (D2OKeq = Φ6 = 0.12).
The equilibrium fractionation factors will lead to inverse kinetic SIEs on different kinetic constants provided that the aquo release is in a pre-equilibrium prior to a subsequent rate-limiting step. As shown in the Appendix, kcat and kcat/KM(CTAD) can report on one aquo release – leading to the prediction that the limiting values for D2Okcat ~ D2O(kcat/KM(CTAD)) = 0.49. In contrast, the limiting values for D2O(kcat/KM(αKG)) will report on the release of 2 or 3 aquo ligands depending on the degree to which FIH is saturated with CTAD. It is important to note that our assays used ambient [O2] (220μM), which is approximately 2xKM(O2),(31) making our kinetic constants apparent.
The rate constants obtained under conditions of saturating αKG, ambient [O2] and varied [CTAD] are kcat and kcat/KM(CTAD). Under these conditions, kcat reports on all steps after CTAD binding up through product release, and is predicted to involve the release of one aquo ligand.(Scheme 1) The initial rate assays in D2O exhibited increased rates when compared to similar assays in H2O, indicating an inverse SIE on both kcat and kcat/KM(CTAD). We observed D2Okcat = 0.51 ± 0.07, in good agreement with the limiting SIE (0.49) for the pre-equilibrium release of one aquo ligand (Figure 1). Similarly, we observed D2O(kcat/KM(CTAD)) = 0.40 ± 0.07, which is also in good agreement with one aquo release prior to a rate-limiting step.
Fig. 1.
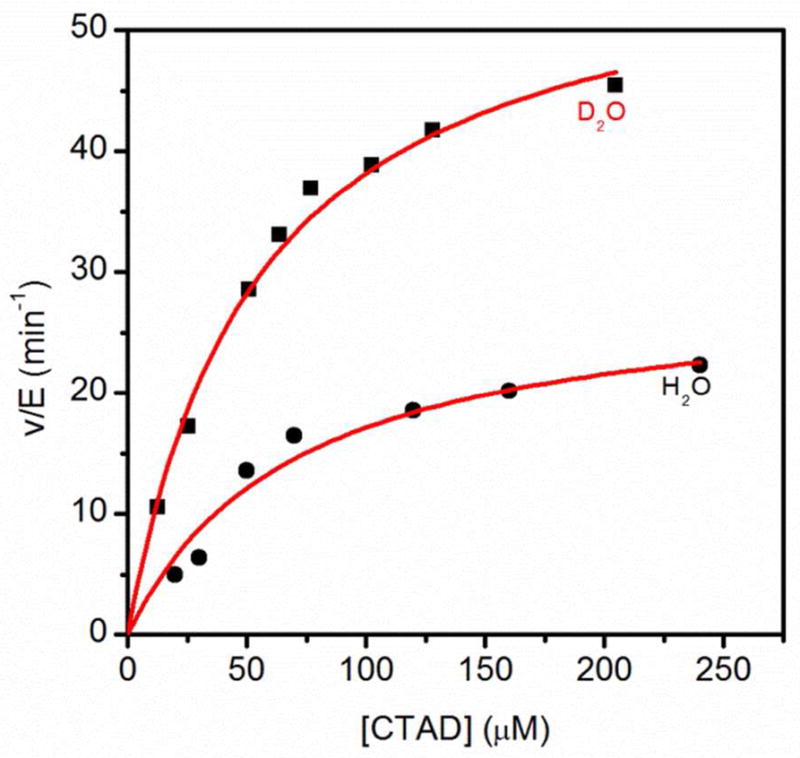
Steady-state kinetics of FIH in H2O (●) and 97% D2O (■) buffers. FIH (0.5 μM), ascorbate (2 mM), αKG (500 μM), FeSO4 (50 μM) and CTAD (0–250 μM) were in 50mM HEPES pL 7.00.
Due to the sequential ordered mechanism (see below), the apparent kcat/KM(αKG) encompasses distinct steps depending on the fixed concentration of CTAD. Since kcat/KM(αKG) reports on steps from the encounter with αKG up through the first irreversible step, high or low fixed concentrations of CTAD were used to isolate different microscopic steps. CTAD binding is kinetically irreversible at high CTAD concentrations, meaning that kcat/KM(αKG)High[CTAD] reports on only those steps between αKG encounter and CTAD binding, encompassing the release of two aquo ligands. The observed SIE of 0.32 ± 0.08 on kcat/KM(αKG)High[CTAD] was in reasonable agreement with the limiting value (0.24) expected for the release of two aquo ligands. In contrast, CTAD binding is kinetically reversible at sub-saturating CTAD concentrations, making a subsequent step (thought to be O2 activation) the first irreversible step. Thus, kcat/KM(αKG)Low[CTAD] reports on all steps between αKG binding and O2 activation, which encompasses the release of three aquo ligands in the consensus mechanism. The observed SIE of 0.11 ± 0.03 for kcat/KM(αKG)Low[CTAD] agreed closely with the limiting value (0.12) expected for the release of three aquo ligands prior to a rate-limiting step (Figure 2).
Fig. 2.
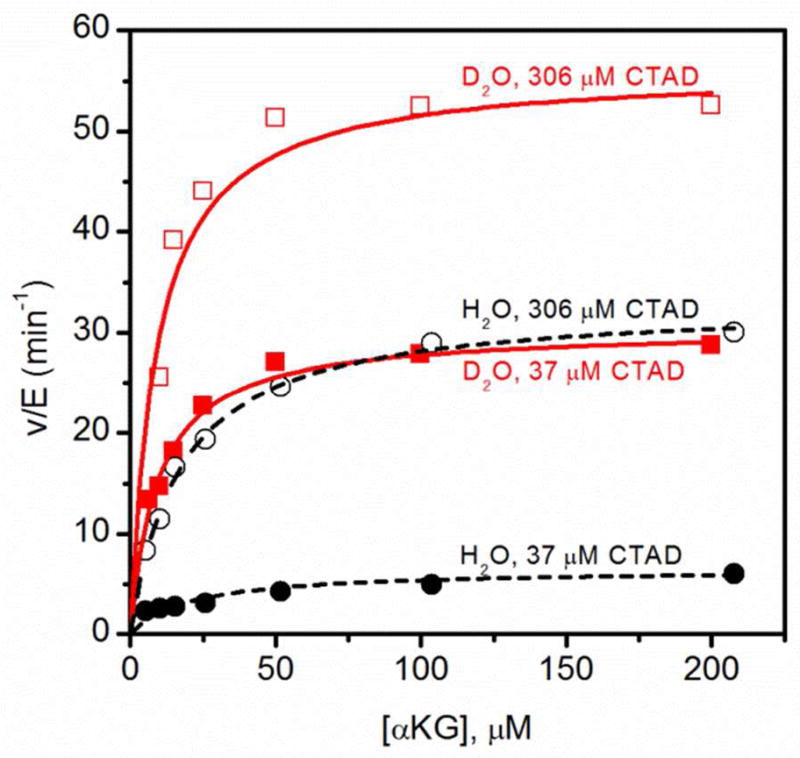
Steady-state kinetics of FIH for αKG at 37μM CTAD and 306μM CTAD, in H2O (●,■) and D2O(○,□)at 37.0°C. FIH (0.5μM), ascorbate (2 mM), αKG (0 – 210 μM), FeSO4 (50 μM) and CTAD (37 μM and 306 μM) were in 50 mM HEPES pL 7.00.
Validation of Observed SIEs
A series of control experiments were completed to ensure that the SIEs arose from aquo release. Control assays showed the steady-state rate constants, kcat and kcat/KM(CTAD), were pH independent between pH 6.50 and pH 8.00 at fixed ionic strength (I = 120 mM). Furthermore, steady-state assays at pH 7.00 showed that kcat and kcat/KM(CTAD) were independent of ionic strength (Figure S1). Viscosity experiments were completed as a control for the increased relative viscosity of D2O, as solvent viscosity can affect the rate of diffusional steps, or of conformational changes. The binding order of αKG and CTAD was also investigated to define the chemical steps reported on by kcat/KM(CTAD) and kcat/KM(αKG), as these rate constants proved different microscopic steps within the chemical mechanism reporting the release of up to three aquo ligands.
FIH’s activity increased approximately two fold in D2O, prompting us to check the coupling between the oxidative and reductive half-reactions in D2O. As the oxidative half reaction produces succinate, and the reductive half-reaction produces CTADOH, the [succinate]/[CTADOH] ratio is equal to the coupling between these half reactions (C=[succinate]/[CTADOH]). Previously we showed tight coupling between succinate production and CTAD hydroxylation in H2O.(14) Here we observed that FIH’s two half reactions remained tightly coupled in D2O, with C equal to unity within experimental uncertainty(C = 1.1 ± 0.1) (Table 2).
Table 2.
Coupling of [Succinate] and [CTADOH] for FIH. 50 mM Tris pL 7.00, at 37.0°C.
Reactions contained αKG (500 μM), FeSO4 (50 μM), CTAD (200 μM) and FIH (5 μM) in 50 mM Tris pD 7.00
Reference (14)
Diffusional steps are not rate-limiting
Steady-state rate constants were measured as a function of relative viscosity to test for diffusional steps that might contribute to the observed rate constants and SIEs, as the viscosity of D2O is greater than that of H2O. Initial rate data were collected in 50 mM HEPES, pH 7.00, with sucrose as the viscosogen, and fitted to the Michaelis-Menten equation. Analysis of kcat, kcat/KM(CTAD) and kcat/KM(αKG) as a function of relative viscosity indicated that each of the steady-state rate constants were independent of solvent viscosity (Figure S2).
The normalized regression plot showed that relative viscosity had no effect on any rate constant. Thus, neither diffusional encounter, relevant for kcat/KM, nor product release, relevant for kcat, are rate-limiting. If diffusional encounter were rate limiting, kcat/KM would decrease as the relative solvent viscosity increased.(32, 33) Furthermore, the lack of a viscosity effect indicates FIH does not undergo a viscosity-sensitive conformational change, which has been shown to obscure SIEs for some enzymes.(34, 35)
Sequential Order: αKG binds before CTAD
The consensus mechanism for αKG oxygenases is a sequential ordered model that, when applied to FIH, predicts αKG binds prior to CTAD. Although steady-state analyses of several other αKG-dependent oxygenases have been shown to follow this binding order,(36–38) we tested this sequential binding model for FIH due to the interesting SIEs that we observed under varied [CTAD]. As the binding order of αKG and CTAD affects the microscopic steps defined by the steady-state kinetic parameters, this is critical to our interpretations of the SIEs.
Four different fixed CTAD concentrations, ranging from ½KM(CTAD) to 4xKM(CTAD) were chosen for steady state kinetic assays where the αKG concentration was the varied substrate. In close agreement with previously reported values,(14, 31) the KM(αKG) remained constant at 20 ± 2μM for all CTAD concentrations. The regression plot of kcat/KM(αKG) as a function of [CTAD] passed through the origin, as expected for ordered sequential binding of αKG prior to CTAD (Figure 3).(39)
Fig. 3.
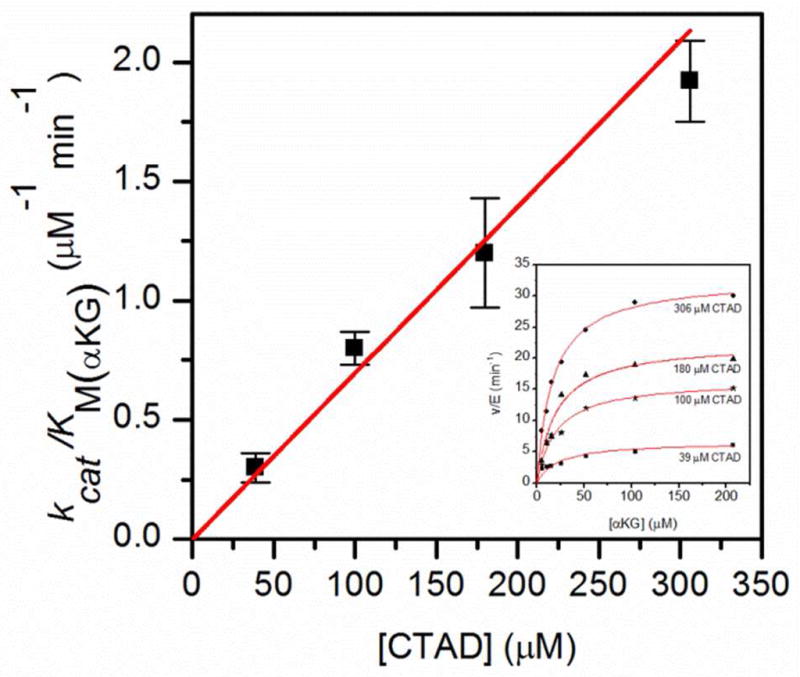
Regression plot showing kcat/KM(αKG) as a function of CTAD concentration. Inset: Steady-state kinetics of FIH with αKG as varied substrate, at different fixed CTAD concentrations at 37.0°C. FIH (0.5 μM), ascorbate (2 mM), αKG (0–210 μM), FeSO4 (50 μM) and CTAD (39 – 306 μM) were in 50mM HEPES pH 7.00.
Discussion
O2 sensing by FIH is critical to cellular growth and development making close consonance between [O2] and the FIH-catalyzed hydroxylation of CTAD central to O2-homeostasis. An additional benefit is that controlled O2 activation by FIH would avoid ROS production (21), preventing anomalous oxidations. Although it has been shown that decarboxylation of αKG and hydroxylation of CTAD by FIH is tightly coupled,(14) the mechanistic strategy used by FIH to ensure that O2 activation leads to CTAD hydroxylation with high fidelity remains unclear. Our overall hypothesis is the hydrogen bonding from the second coordination sphere ensures that the Fe(II) in (Fe + αKG)FIH remains 6-coordinate, and that CTAD binding alters these hydrogen bonds leading to O2 activation – the substrate triggering model. CTAD binding could trigger FIH by affecting any one of the microscopic steps in the overall chemical mechanism, so long as the rate of O2-activation were increased upon CTAD binding. Our observation of inverse SIEs on kcat/KM(CTAD) and kcat/KM(αKG) indicate that the rate-limiting step in the steady state follows aquo release, and is either coincident with or precedes decarboxylation (Scheme 1). This points towards a strategy for tightly coupling O2-activation with CTAD hydroxylation that relies on substrate triggering.
O2-activation is rate-limiting in FIH
Inverse kinetic SIEs (SIE < 1) are diagnostic for aquo release from metal ions, as the vast majority of SIEs are normal (SIE > 1).(29) Two other causes of inverse kinetic SIEs, the involvement of a CysS- nucleophile in catalysis, and a conformational change, were ruled out for FIH. In the former case, there are no active site CysSH residues; in the latter case, we verified that viscosity did not affect the rate of turnover.
Inverse kinetic SIEs have been reported for several metalloenzymes, including a number of hydrolases, carbonic anhydrase, and a point mutant of SLO.(29, 40–43) In each case, the inverse kinetic SIEs indicated that aquo release was a pre-equilibrium step prior to a subsequent rate limiting step. Our observation of inverse kinetic SIEs corresponding to multiple aquo release steps is the first such report, to the best of our knowledge. The inverse SIEs arise from an unfavorable equilibrium for aquo release, combined with a slow O2 activation step.
The consensus mechanism predicts that both kcat and kcat/KM(CTAD) encompass one aquo release for a limiting predicted SIE = 0.49. The experimentally observed kinetic SIEs correlate nicely with the theoretical value expected for the equilibrium release of one aquo ligand, as D2Okcat = 0.51 ± 0.07 and D2Okcat/KM(CTAD) = 0.40 ± 0.07.(Table 1) A kinetic model with separate microscopic steps for αKG, CTAD and O2 binding, as well as the final aquo release, was constructed from the consensus chemical mechanism and used to analyze the observed SIEs (Scheme 2). As kcat encompasses all steps with the exception of diffusional encounter with substrates, a separate, reversible microscopic step for aquo release was needed, as the D2Okcat < 1 indicated that aquo release must be separate from CTAD encounter.
Table 1.
Solvent Isotope Effects for FIH. 50 mM HEPES pL 7.00, at 37.0°C.
| H2O | D2O | Experimental SIE | Theoretical SIE | # of Aquo Ligands | |
|---|---|---|---|---|---|
| kcat (min−1)a | 30 ± 2.5 | 59 ± 2.0 | 0.51 ± 0.07 | 0.49 | 1 |
| kcat/KMCTAD (μM−1 min−1)a | 0.43 ± 0.10 | 1.09 ± 0.11 | 0.40 ± 0.07 | 0.49 | 1 |
| kcat/KMαKG(High[CTAD]) (μM−1 min−1)b | 1.94 ± 0.13 | 6.05 ± 1.37 | 0.32 ± 0.08 | 0.24 | 2 |
| kcat/KMαKG(Low[CTAD]) (μM−1 min−1)c | 0.29 ± 0.07 | 2.60 ± 0.34 | 0.11 ± 0.03 | 0.12 | 3 |
CTAD as the varied substrate; FIH (0.5μM), ascorbate (2 mM), αKG (500 μM), FeSO4 (50 μM), and CTAD (0 – 250 μM)
αKG as the varied substrate; FIH (0.5μM), ascorbate (2 mM), αKG (5 – 200 μM), FeSO4 (50 μM), and CTAD (306 μM)
αKG as the varied substrate;FIH (0.5μM), ascorbate (2 mM), αKG (5 – 200 μM), FeSO4 (50 μM), and CTAD (37 μM)
Scheme 2.

Proposed kinetic mechanism for FIH (S = CTAD).
The similarity in the SIEs for kcat and kcat/KM(CTAD) strongly suggests that the overall rate-limiting step in FIH turnover is common to both rate constants. As kcat/KM(CTAD) encompasses steps between CTAD encounter and the first irreversible step, the common step has to be the O2-activation step (Scheme1). This step is typically viewed as the initial attack on the 2-oxo group of αKG which leads to oxidative decarboxylation. This is noteworthy as that this predicts that FIH will not accumulate the ferryl intermediate during turnover, in contrast to TauD.(12) Slow O2-activation can account for the tight coupling between decarboxylation and hydroxylation seen for FIH(14) as hydroxylation must be very fast relative to the decarboxylation step. Thus, we conclude that FIH likely adopts a strategy to ensure tight coupling between O2-binding and -activation in which O2-activation is rate-limiting.
We analyzed the observed kinetic SIEs using language predominately developed by Northrop and Cleland (44, 45), in which observed kinetic SIEs are a function of “commitments to catalysis” and the equilibrium and kinetic isotope effects (D2OKqq and D2Ok5) on the aquo release step (k5). The commitments to catalysis, Cf and Cr, are ratios of microscopic rate constants that describe the tendency of an enzyme to go forward or backward through the isotopically sensitive step. Full expressions are provided in the Appendix. In the case of D2Okcat/KM(CTAD), this expression takes the form below. The primary virtue of such a presentation is that it permits us to focus on specific segments of the kinetic mechanism. As equilibrium isotope effects reflect fractionation factors, the equilibrium SIE on aquo release is predicted to be ~ 0.5 (D2OK5 = 0.49).
| (1) |
It is clear that the only way for D2Okcat/KM(CTAD) = D2OKeq is for the reverse commitment to be very large (Cr ≫ Cf). Reverse commitment (Cr) is the kinetic competition between H2O and O2 for the triggered form of enzyme – when Cr is large, it means that H2O is the preferred ligand (Appendix – Eqn. A10). Furthermore, the Cf/Cr ratio is, to a first approximation, the equilibrium for aquo release – when Cf/Cr ≪1, it means that K5 is much less than one and that the position of this equilibrium favors the aquo-on state. Similar analysis of the commitment factors for D2Okcat further supports the conclusion that aquo release is disfavored thermodynamically, despite a slightly different SIE expression and forward commitment on kcat (Cvf) (Appendix).
Substrate triggering is faster than O2-activation
Steps involved in substrate triggering were accessed by measuring the kinetic SIE with sub-saturating [αKG]. As kcat/KM(αKG) includes steps between diffusional encounter of αKG with FIH, up through and including the subsequent irreversible step, CTAD binding and substrate triggering may be observable on this kinetic constant. Under conditions of high [CTAD], only the two aquo ligands released upon αKG binding may be observed, as saturating FIH with CTAD makes CTAD binding kinetically irreversible for kcat/KM(αKG)High[CTAD]. In contrast, subsaturating [CTAD] makes it possible to access release of all three aquo ligands on kcat/KM(αKG)Low[CTAD], provided that a later step be rate-limiting.
The observed SIE for kcat/KM(αKG)High[CTAD] (SIE = 0.32 ± 0.08) correlates well with the pre-equilibrium release of two aquo ligands (Table 1), as expected for kinetically irreversible CTAD binding. The key observation supporting rapid substrate triggering was the SIE on kcat/KM(αKG)Low[CTAD] (SIE = 0.11 ± 0.03), as this indicated the pre-equilibrium release of all three aquo ligands prior to a subsequent rate-limiting step. This establishes that steps associated with CTAD binding and substrate triggering are not slow under these conditions, and further points to O2-activation as the rate-limiting step.
Hydrogen bonding from the second coordination sphere
Turnover in FIH and other αKG oxygenases depends on substrate triggering – minimally the release of the aquo ligand from the (Fe + αKG + Substr) enzyme form – prior to O2 binding. What is striking about the kinetics of FIH are the inverse kinetic SIEs, implicating slow O2-activation in this enzyme. In the case of FIH, substrate triggering leads to rate-limiting O2-activation, meaning that the intermediate which is expected to accumulate in the steady-state is a relatively innocuous Fe2+ center. In contrast, TauD exhibits SIEs of unity(46), and the partially rate-limiting steps include product release and H-atom transfer by the [FeIVO]2+ intermediate.(11) For TauD, substrate triggering leads to rapid O2-activation, and the partial accumulation of a powerful oxidant, [FeIVO]2+.(12) It is as if TauD were built for speed, but FIH built for fidelity. As the ligands to the Fe(II) are identical in these two enzymes, the reasons for such disparate strategies for the oxidation reaction must lie beyond the primary coordination sphere.
We attribute the difference in oxidation strategies between these enzymes to their second coordination spheres. The most striking difference between their second coordination spheres is in the hydrogen bonding between the facial triad Asp201 ligand and the axial aquo ligand. In FIH, the remote O-atom of Asp201 forms a 2.8 Å hydrogen bond to the bound aquo ligand in (Fe + αKG)FIH (Figure 4) – once CTAD binds, two new hydrogen bonds form to the Asp201 which appear to serve to partially stimulate aquo release.(16, 17) The extensive hydrogen bonding to this aquo ligand likely serves to stabilize the Fe-OH2 bond, which would explain the large reverse commitments to catalysis (Cr) and the inverse SIEs. Such high affinity for the aquo ligand could serve to throttle back the oxidation reaction such that O2-activation only occurs when CTAD is present. We note a certain similarity to the hydrogen bonding network found in PHD2, which leads to an inverse D2Okcat (47) and the absence of any accumulating intermediates in pre-steady state experiments.(24)
Fig. 4.
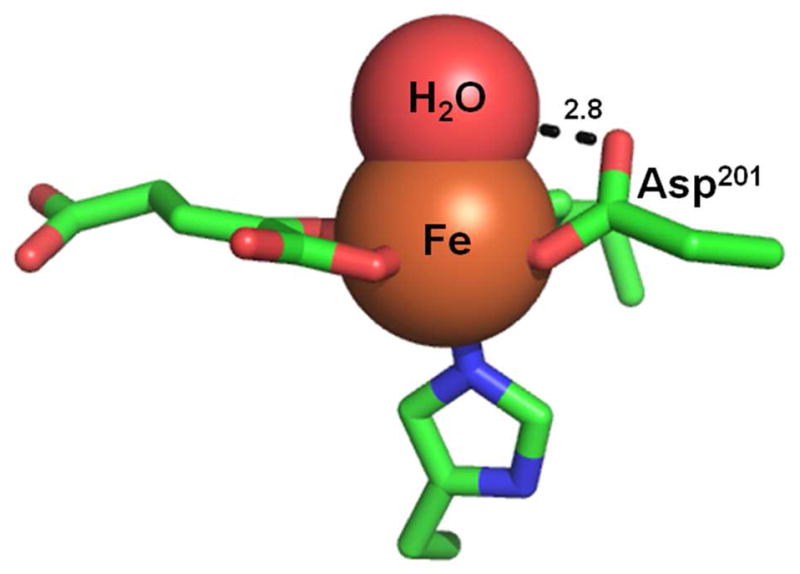
FIH active site with the 2.8 Å Asp201-aquo hydrogen bond. (PDBID: 3P3P)
TauD is a significant contrast to FIH, structurally and kinetically. The facial triad Asp of TauD is rotated away from the aquo ligand, and cannot form a hydrogen bond to the bound aquo ligand. This leads to a weak Fe2+-OH2 bond strength(48), and TauD readily activates O2 in the absence of the substrate taurine.(49, 50) The second coordination sphere of TauD favors aquo-release, which can lead to rapid O2-activation – unfortunately, this leads to a reduction in fidelity.
Conclusion
Inverse kinetic SIEs on kcat/KM and kcat limit the possible rate-limiting step for FIH to a step between aquo release and decarboxylation (Scheme 1). This points to a step very early in the catalytic cycle, such as O2 activation, as the likely rate-limiting step. Such a strategy would aid in FIH’s regulatory role over O2 homeostasis in humans, as it would lead to direct transduction of intracellular O2 levels into a readable signal, hydroxylated HIF-Asn803. This tight control over O2-activation strongly suggests that oxidized intermediates will not accumulate in FIH under normal turnover conditions, and may reflect the demands of a regulatory function in O2-sensing by tightly correlating O2-activation with substrate hydroxylation.
Supplementary Material
Acknowledgments
We thank the NIH for funding (R01-GM077413).
Abbreviations
- HIF
hypoxia inducible factor-1α
- FIH
factor-inhibiting HIF
- αKG
α-ketoglutarate
- CTAD
C-terminal transactivation domain
- CREB
cAMP response element-binding protein
- CAS
clavaminate synthase
- MCD
magnetic circular dichroism
- CD
circular dichroism
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- SIE
solvent isotope effect
- ROS
reactive oxygen species
- PHD2
prolyl hydroxylase dioxygenase 2
- TauD
taurine dioxygenase
- SLO
soybean lipoxygenase
Appendix
As there are multiple isotope sensitive steps for the kinetic mechanism of FIH (Scheme 2), algebraic expressions for each kinetic parameter were derived using the net rate method of Tian.(51) The expression for D2Okcat takes the form:
| (A1) |
| (A2) |
| (A3) |
The parameters CVf and Cr are the forward and reverse commitments to catalysis. D2Ok5 is the kinetic SIE for water release, D2OK5 is the equilibrium SIE on aquo release and D2Ok15 the kinetic SIE on water re-binding. The equations derived for the SIEs for kcat/KM(CTAD), kcat/KM(αKG)High[CTAD], kcat/KM(αKG)Low[CTAD] take similar form and are shown below.
| (A4) |
| (A5) |
| (A6) |
| (A7) |
| (A8) |
| (A9) |
| (A10) |
Footnotes
Supporting Information Available: Control experiments showing the effect of ionic strength and viscosity on the steady state kinetics of FIH. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hausinger RP. FeII/alpha-ketoglutarate-dependent hydroxylases and related enzymes. Crit Rev Biochem Mol Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 2.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, Zhou J. Geometric and electronic structure/function correlations in non-heme iron enzymes. Chem Rev. 2000;100:235–350. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 3.Costas M, Mehn MP, Jensen MP, Que L., Jr Dioxygen activation at mononuclear nonheme iron active sites: enzymes, models, and intermediates. Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 4.Hewitson KS, McNeill LA, Riordan MV, Tian YM, Bullock AN, Welford RW, Elkins JM, Oldham NJ, Bhattacharya S, Gleadle JM, Ratcliffe PJ, Pugh CW, Schofield CJ. Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J Biol Chem. 2002;277:26351–26355. doi: 10.1074/jbc.C200273200. [DOI] [PubMed] [Google Scholar]
- 5.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 6.Lando D, Peet DJ, Gorman JJ, Whelan Da, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Devel. 2002;16:1466–1471.1. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McNeill LA, Hewitson KS, Claridge TD, Seibel JF, Horsfall LE, Schofield CJ. Hypoxia-inducible factor asparaginyl hydroxylase (FIH-1) catalyses hydroxylation at the beta-carbon of asparagine-803. Biochem J. 2002;367:571–575. doi: 10.1042/BJ20021162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou J, Gunsior M, Bachmann BO, Townsend CA, Solomon EI. Substrate Binding to the α-Ketoglutarate-Dependent Non-Heme Iron Enzyme Clavaminate Synthase 2: Coupling Mechanism of Oxidative Decarboxylation and Hydroxylation. J Am Chem Soc. 1998;120:13539–13540. [Google Scholar]
- 9.Zhou J, Kelly WL, Bachmann BO, Gunsior M, Townsend CA, Solomon EI. Spectroscopic studies of substrate interactions with clavaminate synthase 2, a multifunctional alpha-KG-dependent non-heme iron enzyme: Correlation with mechanisms and reactivities. J Am Chem Soc. 2001;123:7388–7398. doi: 10.1021/ja004025+. [DOI] [PubMed] [Google Scholar]
- 10.Pavel EG, Zhou J, Busby RW, Gunsior M, Townsend CA, Solomon EI. Circular dichroism and magnetic circular dichroism spectroscopic studies of the non-heme ferrous active site in clavaminate synthase and its interaction with alpha-ketoglutarate cosubstrate. J Am Chem Soc. 1998;120:743–753. [Google Scholar]
- 11.Price JC, Barr EW, Hoffart LM, Krebs C, Bollinger JM. Kinetic dissection of the catalytic mechanism of taurine: alpha-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry. 2005;44:8138–8147. doi: 10.1021/bi050227c. [DOI] [PubMed] [Google Scholar]
- 12.Price JC, Barr EW, Tirupati B, Bollinger JM, Jr, Krebs C. The first direct characterization of a high-valent iron intermediate in the reaction of an alpha-ketoglutarate-dependent dioxygenase: a high-spin FeIV complex in taurine/alpha-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 13.Price JC, Barr EW, Glass TE, Krebs C, Bollinger JM., Jr Evidence for hydrogen abstraction from C1 of taurine by the high-spin Fe(IV) intermediate detected during oxygen activation by taurine:alpha-ketoglutarate dioxygenase (TauD) J Am Chem Soc. 2003;125:13008–13009. doi: 10.1021/ja037400h. [DOI] [PubMed] [Google Scholar]
- 14.Saban E, Chen YH, Hangasky JA, Taabazuing CY, Holmes BE, Knapp MJ. The Second Coordination Sphere of FIH Controls Hydroxylation. Biochemistry. 2011;50:4733–4740. doi: 10.1021/bi102042t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elkins JM, Hewitson KS, McNeill LA, Seibel JF, Schlemminger I, Pugh CW, Ratcliffe PJ, Schofield CJ. Structure of factor-inhibiting hypoxia-inducible factor (HIF) reveals mechanism of oxidative modification of HIF-1 alpha. J Biol Chem. 2003;278:1802–1806. doi: 10.1074/jbc.C200644200. [DOI] [PubMed] [Google Scholar]
- 16.Dann CE, Bruick RK, Deisenhofer J. Structure of factor-inhibiting hypoxia-inducible factor 1: An asparaginyl hydroxylase involved in the hypoxic response pathway. Proc Natl Acad Sci USA. 2002;99:15351–15356. doi: 10.1073/pnas.202614999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y-H, Comeaux LM, Eyles SJ, Knapp MJ. Auto-hydroxylation of FIH-1, an Fe(II), α-ketoglutarate dependent human hypoxia sensor. Chem Commun. 2008:4768–4770. doi: 10.1039/b809099h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen YH, Comeaux LM, Herbst RW, Saban E, Kennedy DC, Maroney MJ, Knapp MJ. Coordination changes and auto-hydroxylation of FIH-1: Uncoupled O-2-activation in a human hypoxia sensor. J Inorg Biochem. 2008;102:2120–2129. doi: 10.1016/j.jinorgbio.2008.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang M, Chowdhury R, Ge W, Hamed RB, McDonough MA, Claridge TDW, Kessler BM, Cockman ME, Ratcliffe PJ, Schofield CJ. Factor-inhibiting hypoxia-inducible factor (FIH) catalyses the post-translational hydroxylation of histidinyl residues within ankyrin repeat domains. FEBS J. 2011;278:1086–1097. doi: 10.1111/j.1742-4658.2011.08022.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coleman ML, McDonough MA, Hewitson KS, Coles C, Mecinovic J, Edelmann M, Cook KM, Cockman ME, Lancaster DE, Kessler BM, Oldham NJ, Ratcliffe PJ, Schofield CJ. Asparaginyl hydroxylation of the notch ankyrin repeat domain by factor inhibiting hypoxia-inducible factor. J Biol Chem. 2007;282:24027–24038. doi: 10.1074/jbc.M704102200. [DOI] [PubMed] [Google Scholar]
- 21.Saban E, Flagg SC, Knapp MJ. Uncoupled O2-activation in the human HIF-asparaginyl hydroxylase, FIH, does not produce reactive oxygen species. J Inorg Biochem. 2011;105:630–636. doi: 10.1016/j.jinorgbio.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.CRC Handbook of Chemistry and Physics. 61. CRC Press; Boca Raton: 1981. [Google Scholar]
- 23.Quinn DM, Sutton LD. Theoretical Basis and Mechanistic Utility of Solvent Isotope Effects. In: Cook PF, editor. Enzyme Mechanism from Isotope Effects. CRC Press; Boca Raton: 1991. pp. 73–126. [Google Scholar]
- 24.Flashman E, Hoffart LM, Hamed RB, Bollinger JM, Krebs C, Schofield CJ. Evidence for the slow reaction of hypoxia-inducible factor prolyl hydroxylase 2 with oxygen. FEBS J. 2010;277:4089–4099. doi: 10.1111/j.1742-4658.2010.07804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Enzyme Mechanism from Isotope Effects. CRC Press; 1991. [Google Scholar]
- 26.McFarland J, Bernhard SA. Catalytic Steps During Single-Turnover Reduction of Aldehydes by Alcohol-Dehydrogenase. Biochemistry. 1972;11:1486. doi: 10.1021/bi00758a025. [DOI] [PubMed] [Google Scholar]
- 27.Lee KM, Dahlhauser KF, Plapp BV. Reactivity of Horse Liver Alcohol-Dehydrogenase with 3-Methylcyclohexanols. Biochemistry. 1988;27:3528–3532. doi: 10.1021/bi00409a060. [DOI] [PubMed] [Google Scholar]
- 28.Dunn MF, Hutchison JS. Roles of zinc ion and reduced coenzyme in the formation of a transient chemical intermediate during the equine liver alcohol dehydrogenase catalyzed reduction of an aromatic aldehyde. Biochemistry. 1973;12:4882–4892. doi: 10.1021/bi00748a012. [DOI] [PubMed] [Google Scholar]
- 29.Kassebaum JW, Silverman DN. Hydrogen/deuterium fractionation factors of the aqueous ligand of cobalt in Co(H2O)62+ and Co(II)-substituted carbonic anhydrase. J Amer Chem Soc. 1989;111:2691–2696. [Google Scholar]
- 30.Cook PF. Enzyme Mechanism from Isotope Effects. CRC Press; Boca Raton: 1991. Kinetic and regulatory mechanisms of enzymes from isotope effects; pp. 203–230. [Google Scholar]
- 31.Koivunen P, Hirsilä M, Günzler V, Kivirikko KI, Myllyharju J. Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J Biol Chem. 2004;279:9899–9904. doi: 10.1074/jbc.M312254200. [DOI] [PubMed] [Google Scholar]
- 32.Blacklow SC, Raines RT, Lim Wa, Zamore PD, Knowles JR. Triosephosphate isomerase catalysis is diffusion controlled. Biochemistry. 1988;27:1158–1167. doi: 10.1021/bi00404a013. [DOI] [PubMed] [Google Scholar]
- 33.Brouwer AC, Kirsch JF. Investigation of diffusion-limited rates of chymotrypsin reactions by viscosity variation. Biochemistry. 1982;21:1302–1307. doi: 10.1021/bi00535a030. [DOI] [PubMed] [Google Scholar]
- 34.Raber ML, Freeman MF, Townsend CA. Dissection of the Stepwise Mechanism to β-Lactam Formation and Elucidation of a Rate-determining Conformational Change in β-Lactam Synthetase. J Biol Chem. 2009;284:207–217. doi: 10.1074/jbc.M805390200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karsten WE, Lai CJ, Cook PF. Inverse Solvent Isotope Effects in the NAD-Malic Enzyme Reaction Are the Result of the Viscosity Difference between D2O and H2O: Implications for Solvent Isotope Effect Studies. J Amer Chem Soc. 1995;117:5914–5918. [Google Scholar]
- 36.Holme E. Kinetic study of thymine 7-hydroxylase from Neurospora crassa. Biochemistry. 1975;14:4999–5003. doi: 10.1021/bi00693a033. [DOI] [PubMed] [Google Scholar]
- 37.Rundgren M. Steady state kinetics of 4-hydroxyphenylpyruvate dioxygenase from human liver (III) J Biol Chem. 1977;252:5094–5099. [PubMed] [Google Scholar]
- 38.De Carolis E, De Luca V. Purification, characterization, and kinetic analysis of a 2-oxoglutarate-dependent dioxygenase involved in vindoline biosynthesis from Catharanthus roseus. J Biol Chem. 1993;268:5504–5511. [PubMed] [Google Scholar]
- 39.Cook PF, Cleland WW. Enzyme Kinetics and Mechanism. Garland Science; New York: 2007. [Google Scholar]
- 40.Izquierdo MC, Stein RL. Mechanistic studies of thermolysin. J Amer Chem Soc. 1990;112:6054–6062. [Google Scholar]
- 41.Born TL, Zheng R, Blanchard JS. Hydrolysis of N-succinyl-L,L-diaminopimelic acid by the Haemophilus influenzae dapE-encoded desuccinylase: metal activation, solvent isotope effects, and kinetic mechanism. Biochemistry. 1998;37:10478–10487. doi: 10.1021/bi9806807. [DOI] [PubMed] [Google Scholar]
- 42.Harrison RK, Chang B, Niedzwiecki L, Stein RL. Mechanistic studies on the human matrix metalloproteinase stromelysin. Biochemistry. 1992;31:10757–10762. doi: 10.1021/bi00159a016. [DOI] [PubMed] [Google Scholar]
- 43.Tomchick DR, Phan P, Cymborowski M, Minor W, Holman TR. Structural and Functional Characterization of Second-Coordination Sphere Mutants of Soybean Lipoxygenase-1. Biochemistry. 2001;40:7509–7517. doi: 10.1021/bi002893d. [DOI] [PubMed] [Google Scholar]
- 44.Cook PF, Cleland WW. Mechanistic deductions from isotope effects in multireactant enzyme mechanisms. Biochemistry. 1981;20:1790–1796. doi: 10.1021/bi00510a013. [DOI] [PubMed] [Google Scholar]
- 45.Northrop DB. Isotope Effects on Enzyme-Catalyzed Reactions. University Park Press; Baltimore, MD: 1977. [Google Scholar]
- 46.Grzyska PK, Ryle MJ, Monterosso GR, Liu J, Ballou DP, Hausinger RP. Steady-State and Transient Kinetic Analyses of Taurine/alpha-Ketoglutarate Dioxygenase: Effects of Oxygen Concentration, Alternative Sulfonates, and Active-Site Variants on the FeIV-oxo Intermediate. Biochemistry. 2005;44:3845–3855. doi: 10.1021/bi048746n. [DOI] [PubMed] [Google Scholar]
- 47.Flagg SC, Giri N, Pektas S, Maroney MJ, Knapp MJ. Inverse Solvent Isotope Effects Demonstrate Slow Aquo Release from Hypoxia Inducible Factor-Prolyl Hydroxylase (PHD2) Biochemistry. 2012;51:6654–6666. doi: 10.1021/bi300229y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neidig ML, Brown CD, Light KM, Fujimori DG, Nolan EM, Price JC, Barr EW, Bollinger JM, Krebs C, Walsh CT, Solomon EI. CD and MCD of CytC3 and taurine dioxygenase: role of the facial triad in alpha-KG-dependent oxygenases. J Amer Chem Soc. 2007;129:14224–14231. doi: 10.1021/ja074557r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ryle MJ, Liu A, Muthukumaran RB, Ho RYN, Koehntop KD, McCracken J, Que L, Hausinger RP. O2- and alpha-Ketoglutarate-Dependent Tyrosyl Radical Formation in TauD, an alpha-Keto Acid-Dependent Non-Heme Iron Dioxygenase. Biochemistry. 2003;42:1854–1862. doi: 10.1021/bi026832m. [DOI] [PubMed] [Google Scholar]
- 50.Koehntop KD, Marimanikkuppam S, Ryle MJ, Hausinger RP. Self-hydroxylation of taurine/a-ketoglutarate dioxygenase: evidence for more than one oxygen activation mechanism. J Biol Inorg Chem. 2006:63–72. doi: 10.1007/s00775-005-0059-4. [DOI] [PubMed] [Google Scholar]
- 51.Tian GC. Effective Rate Constants and General Isotope Effect Equations for Steady-State Enzymatic-Reactions with Multiple Isotope-Sensitive Steps. Bioorg Chem. 1992;20:95–106. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.