

Abstract
The pleiotropic pro-inflammatory cytokine interleukin (IL)-1β has co-evolved with a competitive inhibitor, IL-1 receptor antagonist (IL-1Ra). IL-1β initiates cell signaling by binding the IL-1 receptor (IL-1R) whereas IL-1Ra acts as an antagonist, blocking receptor signaling. The current paradigm for agonist/antagonist functions for these two proteins is based on the receptor–ligand interaction observed in the crystal structures of the receptor–ligand complexes. While IL-1Ra and IL-1β are structurally homologous, IL-1Ra engages only two of the three extracellular domains of the receptor, whereas IL-1β engages all three. We find that an allosteric functional switch exists within a highly conserved pocket of residues, residues 111–120. This region is maintained across all IL-1 family members and serves as a hydrophobic mini-core for IL-1β folding. A key difference across species is a conserved aromatic residue at position 117 in IL-1β, versus a conserved cysteine in IL-1Ra at the analogous position, 116. We find that the replacement of C116 with a phenylalanine switches the protein from an antagonist to an agonist despite the distant location of C116 relative to receptor interaction sites. These results suggest new ways to develop designer cytokine activity into the β-trefoil fold and may be of general use in regulation of this large family of signaling proteins.
Keywords: structural plasticity, barrel-cap interface, allostery
Introduction
Cytokines are one of the tightly regulated currencies of cellular communication.1-3 Within the cytokine family, the interleukin (IL)-1 family of proteins is the only group to have identified natural antagonist proteins.4 IL-1β has co-evolved with a competitive inhibitor, IL-1 receptor antagonist (IL-1Ra). IL-1β binds the IL-1 receptor (IL-1RI) and triggers a signal cascade whereas IL-1Ra competes for the same site and inhibits signaling.5-7 The IL-1R belongs to the immunoglobulin (Ig) superfamily characterized by three extracellular Ig domains (I, II, and III), a single trans-membrane spanning sequence, and a globular intracellular domain.8-11 In addition, a soluble form of the receptor (sIL-1R), arising from alternative splicing and comprising residues 1–311 of the IL-1R, is found circulating in the bloodstream, inhibiting IL-1β signaling.5,12 The structures of the complexes of sIL-1R with IL-1β and IL-1Ra, respectively, have been solved and reveal a novel interaction motif for a cytokine–receptor complex where the three Ig domains wrap around the trefoil-folded proteins with a 1:1 stoichiometry.8,9,13 IL-1β has two known binding sites for the IL-1R, designated A and B (Fig. 1). Site A binds the first two Ig domains on the receptor while site B interacts with the third domain. Similarly, IL-1Ra maintains the interactions between site A and the IL-1R but differs in its interaction with Ig domain III. Comparison of the structures of the IL-1β/receptor and IL-1Ra/ receptor complexes has led to the proposal that binding to the B-site of IL-1β induces a conformational change in the receptor, triggering the signal cascade8,9,13 (Fig. 1a). The IL-1β/IL-1RI complex recruits a co-receptor, IL-1R accessory protein (IL-1RAcP), and this heterotrimeric complex is responsible for initialing cell signaling (Fig. 1b). The Nf-κB pathwayis the major target for IL-1 signaling in various cell types, but especially in monocytes and macrophages.
Fig. 1.

(a) Comparison of IL-1β (left) and IL-1Ra (right) bound to the IL-1RI receptor. Ig domains I, II, and III of sIL-1RI are labeled on the complex on the left for clarity. IL-1β is thought to induce a conformational change (helical structure in the linker between domains II and III and a 20° rotation of Ig domain III with respect to that observed in the IL-1RI complex of IL-1Ra) and binds the third domain of the receptor. The binding sites in IL-1β are highlighted in red (binding site A) and cyan (binding site B), respectively. When bound to IL-1β, contacts in binding site B result in a closed conformation of the receptor domains while the receptor adopts an inactive open conformation when bound to IL-1Ra. (b) The heterotrimeric complex of IL-1RI (brown) and co-receptor, IL-1RAcP (magenta), modeled with IL-1Ra (blue). The C116F mutation (yellow) is highlighted to illustrate the location of the change with respect to the receptor as well as the barrel core of the protein.
The allosteric mechanism of IL-1RI activation by IL-1β binding is apparent when comparing the crystal structures of IL-1β and IL-1Ra bound to the cytoplasmic domain of the IL-1RI receptor protein.8,9,13 When bound to IL-1β, contacts in binding site B result in a “closed” conformation of the receptor domains while the receptor adopts an inactive “open” conformation when bound to IL-1Ra (Fig. 1a). Functionally, proteins can act as allosteric effectors or inhibitors for other proteins, as is the case for IL-1Ra and IL-1β regulating the multidomain IL-1R. While allosteric regulation of activation/inhibition is unusual in single domain proteins that do not undergo obvious structural reorganization, a hallmark feature appears to be communication between distal residues. Recent studies reveal the occurrence of hot spot residues,14 where allosteric signal propagation primarily involves conserved amino acids.15-19 Additionally, there is emerging evidence for allosteric disulfide bonds in proteins that can control function by triggering a functional event when it breaks and/or forms.20
IL-1Ra and IL-1β share the same single-domain tertiary structure, the β-trefoil fold, despite having only 30% sequence identity. While IL-1Ra and IL-1β are structurally homologous, each makes a noticeably distinct number and type of contacts at the protein–receptor interface. Key structural differences are in loop regions, including the β-bulge “trigger loop” between β-strands 4 and 5 in IL-1β and the large loop between β-strands 11 and 12 in IL-1Ra. Interestingly, the most conserved sequence region, with the exception of a handful of residues, is from residue 111 to 121 in both proteins. These residues compose a hydrophobic “mini-core” important for folding in IL-1β21 and maintaining the stability of the barrel–cap interface.22 This stable mini-core bridges the barrel and cap and is located diametrically opposed to the receptor A and B (IL-1β) or A (IL-1Ra) interfaces (Fig. 1a) and the accessory protein binding site (Fig. 1b). Key differences between the agonist (IL-1β) and the antagonist (IL-1Ra) protein include the replacement of an aromatic residue at the structurally homologous position in agonist IL-1β (Phe117) with a cysteine at position 116 in IL-1Ra. Furthermore, this region bridges the early and late stages of folding23 as cavity-changing mutations in the barrel core of IL-1β confirmed that core packing is rate limiting in folding.24,25
Transmission of information occurs through the hydrogen-bonding network and is significant for receptor binding and signaling for IL-1β.26 The resistance to overt structural changes24,26-28 despite potential changes in function suggests that the β-trefoil fold may provide an effective scaffold for functional variation. Is this structure/function plasticity a general feature of the cytokine β-trefoil fold? We explored this question by replacing the C116 in IL-1Ra with the agonist’s phenylalanine. The replacement of C116 and analysis of the thermodynamics, folding, and functional properties with respect to wild-type (WT) IL-1Ra were performed. Strikingly, our results reveal that mutation of this site in IL-1Ra, which is diametrically opposed to the receptor binding site, converts IL-1Ra from an antagonist into an agonist. Taken together, our findings reveal the potential for and the design of new internal allosteric regulation strategies targeting inflammation control.
Results
Replacement of the cysteine residue with phenylalanine yields a more stable mutant protein compared to WT IL-1Ra
Equilibrium unfolding studies were undertaken to determine the effect of mutation on the thermodynamic stability between WT IL-1Ra and the C116F mutant proteins. IL-1Ra has two tryptophan residues, W16 and W120, which are useful probes for fluorescence studies of equilibrium unfolding.29 The equilibrium fluorescence-detected unfolding curves for WT (cyan) and C116F mutant (yellow) IL-1Ra are given in Fig. 2a. The normalized fluorescence intensities were fit to a two-state model (Fig. 2a, colored lines) as previously described for IL-1β26-28 to determine the free energy of stabilization (ΔG) and cooperativity (m-value) of the folding transition for each protein (Table 1). Replacing Cys116 with a phenylalanine results in an increase of 1.3 ± 0.5 in the ΔΔG as evident in the shift of the observed transition curve to higher denaturant concentrations than that observed for WT IL-1Ra, with no measurable change in the cooperativity of the transition (Table 1). Similar experiments conducted with WT IL-1Ra under reducing conditions reveal that the observed change in stability is not a result of disrupting the disulfide bond (Fig. S1). The observed stabilization in the C116F mutant protein suggests that increasing the hydrophobicity of the loop and altering the cavity interaction at the interface of the barrel core and hairpin cap of the protein are favorable interactions. Additionally, the apparent loss of disulfide interaction as a result of the mutation has little effect on the stability of the mutant protein molecule. In order to assess site-specific changes to the native state of the C116F mutant protein, NMR experiments were performed, including chemical shift analysis and native-state hydrogen–deuterium exchange (HDX) on the WT and mutant proteins.
Fig. 2.

Comparison of the thermodynamic stability of mutant and WT proteins. Fit of the fraction of unfolded protein, Fapp, as a function of denaturant concentration for the equilibrium unfolding of IL-1Ra and C116F (cyan and yellow, respectively). The stability for C116F is greater than that observed for WT IL-1Ra as indicated by the shift of the observed transition curve to higher denaturant (right) concentrations.
Table 1.
The thermodynamic parameter comparison for WT IL-1Ra and the C116F construct
| IL-1Ra | ΔGN–U (kcal mol−1)a,b | ΔΔGN–U (kcal mol–1)a,b | m-valueN–U (kcal mol–1 M–1) | Cw (M)c |
|---|---|---|---|---|
| WT IL-1Ra | 8.1 ± 0.2 | 0 | 5.7 ± 0.4 | 1.4 |
| C116F | 9.3 ± 0.5 | 1.3 ± 0.5 | 5.6 ± 0.4 | 1.7 |
Changes in folding parameters upon mutation of IL-1Ra. The equilibrium data were fit using MATLAB in order to obtain equilibrium parameters for folding, ΔGN–U. Changes in ΔGN–U (ΔΔGN–U) were obtained using WT as a reference m-value indicates changes in the accessible surface area upon folding and indicate cooperativity of folding.
Equilibrium transition data were evaluated using a two-state folding model.
Data were obtained by calculating the average wavelength and calculating the relative average wavelength in terms of Fw as a function of [Gdn·HCl].
Cw values were taken from dividing the ΔGN–U by the m-value.
NMR studies confirm the conservation of the native fold while revealing new connectivities between the binding sites
In order to determine whether the mutation at the interface of the hairpin cap and β-barrel disrupts the native structure of C116F IL-1Ra mutant, we used 1H–15N NMR spectroscopy to assess the global fold of the mutant relative to WT. The 1H–15N cross-peaks of the IL-1Ra backbone resonances in a heteronuclear single quantum coherence (HSQC) spectrum have chemical shift dispersion indicative of specific secondary and tertiary interactions, and a pattern unique to the protein structure.30 Hetero-nuclear 1H–15N HSQC spectra were collected for both mutant and WT IL-1Ra proteins, shown in Fig. 3a. The spectra overlay of C116F (Fig. 3a, yellow) indicates chemical shift patterns and dispersion quite similar to that observed for the WT IL-1Ra (Fig. 3a, cyan). Further comparison of the global folds by assessing change in 1H–15N chemical shifts for the C116F mutant versus WT IL-1Ra are highlighted in Fig. 3b and mapped onto the molecule in Fig. 3c. Several residues local to the C116 mutation site show changes compared to WT 1H–15N chemical shifts. The majority of chemical shift changes are concentrated within the central trefoil unit of the molecule across from the receptor binding interface, and, more specifically, in the hairpin cap extending from the site of mutation (Fig. 3c). These observed changes are consistent with altering the packing of the local amino acid side chains and disruption of the disulfide bond interaction as a result of adding the aromatic side chain via mutation, changing the molecular environment of the local region adjacent to the mutation.
Fig. 3.

Characterization of the effects of C116F mutation on the protein structure with NMR. (a) An overlay of the HSQC spectra for WT (cyan) and mutant (yellow) IL-1Ra. The global pattern of chemical shifts and dispersion of the backbone resonances in the 1H–15N HSQC spectra of WT IL-1Ra and C116F indicate a similar overall global fold. (b) Graphic of the combined weighted proton/nitrogen chemical shift changes in IL-1Ra as a result of the C116F mutation. The changes greater than 0.1 are as indicated above the grey shaded area. (c) Significant chemical shift differences observed in the free protein upon mutation are indicated by red spheres and mapped back onto the molecule. The yellow sphere indicates the site of mutation.
To further assess the effect of mutation on the native state of IL-1Ra, we performed HDX via NMR on both mutant and WT proteins, as described for IL-1β.27,28 Comparison of the change in the amide proton signal over time after introduction into deuterated buffer (Fig. 4a) reveals that many residues maintain the same protection from solvent exchange as that observed for WT IL-1Ra. The main differences in amide proton exchange were observed for amide backbone probes located in turns and hinge points throughout the mutant molecule (Fig. 4b, red spheres), where the rate of exchange for the mutant protein resulted in loss of the amide backbone probe within the first time point (exchange was faster than WT). These changes are located at the interface of the cap and barrel and within the receptor–ligand interaction surfaces (Fig. 4b). However, when bound to the receptor, additional significant destabilization in HDX is observed in β-barrel strands 1, 8, 9, and 12 in the C116F mutant protein (see below). Interestingly, the stabilizing mutation also resulted in probes within the area of the mutation demonstrating a protection from exchange (Fig. 4b, blue spheres) compared to WT. These changes are consistent with stabilization of the hydrophobic mini-core and hairpin cap–barrel interface upon introduction of the aromatic side chain.
Fig. 4.

Summary of the observed native HDX results for WT and C116F mutant proteins. (a) Representative comparisons of the change in amide proton signals as a function of time after introduction into deuterated buffer for WT IL-1Ra ( ) and C116F (
) and C116F ( ). The upper trace (residue T23) displays amide protons that are slow exchanging in both the WT and mutant protein. The lower traces (residue R15 and F149) are representative of those observable probes that exhibit intermediate (middle) and fast (bottom) exchange in both proteins. All three probes are examples of HDX kinetics that are similar for both mutant and WT proteins. (b) Representation of amide probes that are destabilized to exchange in C116F such that they are not observed in the first time point (red spheres). These backbone amide probes are located at topologically equivalent turns or hinge points of the mutant protein. Three backbone probes exhibit increased protection from amide exchange in C116F and are indicated by blue spheres.
). The upper trace (residue T23) displays amide protons that are slow exchanging in both the WT and mutant protein. The lower traces (residue R15 and F149) are representative of those observable probes that exhibit intermediate (middle) and fast (bottom) exchange in both proteins. All three probes are examples of HDX kinetics that are similar for both mutant and WT proteins. (b) Representation of amide probes that are destabilized to exchange in C116F such that they are not observed in the first time point (red spheres). These backbone amide probes are located at topologically equivalent turns or hinge points of the mutant protein. Three backbone probes exhibit increased protection from amide exchange in C116F and are indicated by blue spheres.
A preliminary examination of how the mutation affects binding sites A and B were undertaken using hydrogen–deuterium exchange mass spectrometry (DXMS) with the heterotrimeric complex of WT or C116F IL-1Ra, IL-1RI, and IL-1RAcP. Comparison of HDX of the WT and mutant protein bound to the receptor demonstrates that when bound to the receptor, significant destabilization in HDX is observed in β-barrel strands 1, 8, 9, and 12 in the C116F mutant protein (Fig. 7, pink strands). These results indicate that the mutant protein adjusts upon binding such that it can now engage both sites A and B (Fig. 1) as it becomes signaling competent. This long-range control of a binding interface is distinct from the classic allosteric mechanism.
Fig. 7.
Schematic of proposed interaction of the C116F mutant with respect to receptor binding and subsequent signaling as a result of mutation. (a) The agonist-like behavior from the introduction of a phenylalanine at position C116 creates a subtle rearrangement of the residues around area of mutation (yellow lock), disrupting interactions related to a potential intramolecular disulfide. This unlocks the pinned hairpin cap from the β-barrel, allowing for engagement of the B-site at the opposite end of the protein, eliciting a signal response. (b) Molecular representation of both the chemical shift NMR results on the free ligand (red spheres) and DXMS (pink strands) of the receptor complex. When bound to the receptor, significant destabilization in HDX is observed in β-barrel strands 1, 8, 9, and 12 in the C116F mutant protein (pink strands). These results indicate that the mutant protein adjusts upon binding such that it can now engage both sites A and B (Fig. 1) as it becomes signaling competent. This long-range control of a binding interface is distinct from the classic allosteric mechanism.
Fluorescence-detected folding studies indicate conservation of mechanism with an increased rate of refolding consistent with the change in stability and increased buried hydrophobic surface area in the mutant protein
A series of time-dependent fluorescence-detected stopped-flow and manual-mixing techniques were performed to determine if the C116F point mutation alters the folding mechanism of the protein compared to WT. The observed change in fluorescence intensity as a function of time (Fig. 5a) for each denaturant concentration was collected and fit as described previously.26-28 A plot of the naturallog of kobs for refolding and unfolding of WT IL-1Ra and C116F mutant proteins as a function of final denaturant concentration is presented in Fig. 5b. The WT IL-1Ra data are represented in cyan, while those for the C116F mutant are represented in yellow. The overall refolding and unfolding kinetics are similar; however, the refolding of the C116F mutant exhibits a stronger denaturant dependence (Fig. 5b) consistent with a slightly faster collapse to the native state by altering the loop hydrophobicity and alternate packing of the hairpin cap to the barrel core of the protein.
Fig. 5.
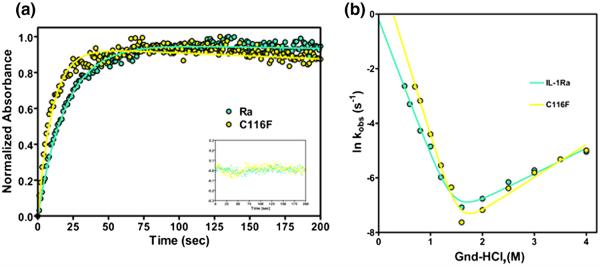
Representative traces of the folding kinetics and chevron plot of the relaxation rates indicating the effects of altered chain connectivity. (a) WT IL-1Ra is in cyan and C116F is in yellow. The traces are of the manual mixing kinetic refolding jumps from 2.2 M guanidine–HCl to 0.4 M guanidine–HCl. The residuals of the fit, following the same coloring scheme, are represented in the inset. (b) Plot of the natural log of kobs obtained by both stopped-flow and manual-mixing refolding and unfolding experiments for both protein variants as a function of final denaturant concentration. The data points (left and right, circles) and fits depict the rate of formation of the native protein.
Assessment of the biological activity of the C116F mutant protein reveals an allosteric switch to agonist activity
IL-1 proteins are multifunctional cytokines that mediate various inflammation and cellular immune responses. In many cases, the transcriptional regulatory properties of IL-1 proteins are mediated by their abilities to activate Nf-κB.31 IL-1 activity can be assayed for activity through cell stimulation and monitoring the resulting import of Nf-κB into the nucleus. Typically, upon stimulation with IL-1β, the signal for Nf-κB first increases and then decreases over time, as the IL-1RI receptor complex is down-regulated. In the identical assay, the C116F mutant was tested for agonist activity by direct stimulation of 3T3 mouse embryonic fibroblast (MEF) cells and compared with WT IL-1Ra and IL-1β followed by an electrophoretic mobility shift assay (EMSA) measuring the amount of Nf-κB imported into the nuclei of the stimulated cells.32 A plot of the normalized band intensities from the full EMSA time courses for controls IL-1β (black, positive), WT IL-1Ra (cyan, negative), and the C116F mutant (yellow) is presented in Fig. 6. The C116F point mutation shows a significant change in intensity over time compared to WT IL-1Ra (Fig. 6), indicating a switch to agonist cytokine activity for the mutant protein, despite the alteration occurring distal to the binding regions of the molecule.
Fig. 6.

Plot of the nuclear import of Nf-κB upon stimulation over time for IL-1 activity. The normalized band intensities from the full EMSA time courses for mutant (yellow), WT IL-1Ra (cyan), and IL-1β (black), plotted over time. The time course indicates stimulation interaction where human IL-1β (positive control) and C116F display signaling as a result of Nf-κB importation compared to IL-1Ra (negative control), which shows negligible activity.
Discussion
Structural relationship of competing receptor-binding cytokines
The location of C116 in IL-1Ra (Fig. 1, yellow) is in the analogous position to F117 in IL-1β, across from the potential disulfide partner C69. For both proteins, this is in a tight turn between β-strand 9 of the barrel and cap β-strand 10, which is hydrophobic in nature. The addition of the conserved phenylalanine aromatic side chain to IL-1Ra increases the hydrophobicity of this turn, resulting in an overall stabilizing effect compared to WT (Fig. 2a; Table 1). For many proteins, changes within the hydrophobic core are oftentimes accompanied by destabilization of the native fold, unless a particular variant fills a solvated cavity within the interior of the protein.25 For the C116F mutant, adding the aromatic phenylalanine ring between β-strand 7 in the hairpin cap, and the hydrophobic loop between barrel β-strand 9 and cap β-strand 10, stabilizes the protein. The increase in stabilization is most likely due to increasing favorable hydrophobic packing interactions of the phenylalanine side chain with the surrounding residues’ side chains. In IL-1β, F117 packs in between residues V72, P78, Q116, and W120. The analogous region for C116 in IL-1Ra shows side-chain interactions with V70, K71, T76, R97, A115, P117, and W120. This area of the sequence between IL-1Ra and IL-1β is highly conserved within the IL-1 family. For IL-1Ra, the replacement of C116 with the residue with a conserved familial residue in the same position in IL-1β does not destabilize the protein; rather, it stabilizes the protein further. This increase in stability is an unusual result for a disulfide interaction. Indeed, while disulfides are often employed by proteins for adding to their stability,33,34 in the case of IL-1Ra, our results indicate that the role of the disulfide bridge is not related to the stability of the protein.
Non-uniform changes to the β-trefoil by single point mutation
Conserved aromatic residues line the central cavity of β-trefoil proteins35 and appear to be essential for both stability and folding.24 The structural “hinge point” between the hairpin cap and β-barrel in both IL-1β and IL-1Ra proteins, adjacent to one of the conserved topologically symmetric phenylalanine residues (Phe101), is a conserved region in both proteins. Both C116 in IL-1Ra and F117 in IL-1β reside within this conserved region. Structural comparison of the C116F mutant with WT IL-1Ra (Fig. 3a) reveals similarities in the overall fold, although changes are observed within the region of mutation where chemical shift deviations are propagated (Fig. 3b and c). More specifically, the introduction of the aromatic residue appears to have the greatest effect on the chemical shifts of the conserved side-chain interactions at the hairpin cap–β-barrel interface (Fig. 3b), as the point mutation appears to reflect changes in “pinning” the cap on the β-barrel (Fig. 3c). While this disruption is evident in the chemical shift analysis, many of the residues with observed chemical shift changes (Fig. 3) maintain similarities in HDX protection in the free ligand compared to WT protein. Differences in HDX resulting in complete loss of backbone amide probe/protection (Fig. 4b, red spheres) are observed at topological turns and hinge points throughout the protein, consistent with previous β-trefoil protein behavior.22 For a small subset of probes (Fig. 4b, blue spheres), while there are clear differences in chemical shifts, backbone amide protection increases compared to WT IL-1Ra and are in close proximity to the site of mutation and likely result from increased stability in the hydrophobic mini-core. Interestingly, the observed changes as a result of the mutation appear to conserve the overall shape of the molecule, maintaining the integrity of the cap and barrel of the protein and the ligand-binding interactions with the receptor.
Similar folding behavior for mutant protein observed despite changes in stability and dynamics
Geometrically, the conserved, stable mini-core region is significant in the folding of IL-1β and biologically bridges two important functional regions, the interface between the A and B receptor binding sites (Fig. 1a). While the structures between the agonist (IL-1β) and antagonist (IL-1Ra) are conserved, a key difference aside from the β-bulge trigger loop is the potential C116–C69 disulfide interaction. The comparative analysis of the folding of the C116F mutant with WT IL-Ra reveals that the observed increase in stability of the C116F mutant protein is reflected in an increase in the denaturant dependence of the refolding reaction (Fig. 5). It is well established that disulfide bonds can either increase or decrease the stability and/or rate of folding, depending on their location with respect to the folding nucleus.36 Taken together, our results indicate that the disulfide is not important for folding but hydrophobic packing at this site leads to stabilization of the fold.
Increased structural plasticity and allosteric regulation of agonist activity observed by single point mutation in IL-1Ra
In response to the mutation at C116, there are local structural rearrangements observed near the site of mutation (Fig. 3). However, most residues retain WT stability in the free ligand except where noted (Fig. 4a), with residues local to the mutation showing the greatest effects in stability. However, when bound to the receptor, significant destabilization in HDX is observed in β-barrel strands 1, 8, 9, and 12 in the C116F mutant protein (Fig. 7). These results indicate that the mutant protein is unlocked upon binding such that it can now engage both sites A and B and is signaling competent. More significantly, the C116F mutant data reveal the capacity to bind IL-1RI and activate Nf-κB importation into the nucleus as detected by EMSA (Fig. 6). The concomitant effects of stabilizing the native state via the mini-core cause increasing structural plasticity in order to accommodate the aromatic side chain that effectively produced an IL-1Ra agonist protein. The agonist-like behavior from the introduction of a phenylalanine at position C116 creates a subtle rearrangement of the residues around that area, disrupting the interactions related to an intramolecular disulfide. This “unlocks” the opposite side of the protein, allowing for engagement of the B-site, eliciting a signal response (Fig. 7). This long-range control of a binding interface is distinct from the classic allosteric mechanism and is unusual in a single domain protein that does not undergo obvious structural reorganization.
Conclusion
The consequence of substituting the conserved antagonist residue (C116) with the conserved aromatic residue (phenylalanine) from the agonist into IL-1Ra illustrates the subtle interplay between residue stability, protein structure, dynamics, and function. Similar to other familial structural homo-logues, the structural plasticity evident in the IL-1Ra cytokine is revealed through converting antagonist activity into agonist via mutation of a single site far removed from the receptor interaction surface and offers new insight into avenues for the development of designer cytokines.
Materials and Methods
WT IL-1Ra and C116F expression and purification
The cDNA for human IL-1Ra isoform 1 was obtained from Open Biosystems (GenBank number NM_173842). The DNA corresponding to the mature secreted form (residues 26–177 of the precursor protein) was amplified by PCR, restriction digested, and ligated into the pET-24a expression vector (Novagen). The ligated DNA was transformed into Escherichia coli DH5α competent cells (Novagen) and insert-containing DNA purified from isolated colonies verified by sequencing (Eton Bioscience). The insert-containing vector was transformed into E. coli BL21 (DE3) competent cells (Invitrogen). An LB culture was inoculated with these cells and grown at 37 °C until they reached an OD600 of 0.6. Protein expression was induced by adding 1 mM IPTG (final concentration) and the temperature was reduced to 30 °C. The cells were harvested after 4 h by centrifuging at 6238g for 10 min. The media were carefully decanted, and the resulting pellets were suspended in a total volume of 80 ml of 25 mM ammonium acetate, pH 5.2, 1 mM ethylenediaminetetraacetic acid (EDTA), and 1 mM PMSF. The resulting supernatant was extensively dialyzed into 25 mM ammonium acetate, pH 5.2, and 1 mM EDTA. The dialyzed protein was spun again at 20,201g for 20 min, and the supernatant was filtered by a 0.2-μm filter. The filtered protein was injected onto a HiTrap-S HP cation-exchange column (GE Healthcare) equilibrated with Buffer A. The protein eluted as a single peak between a gradient of 30–70% B over 100 ml (Buffer B is 750 mM ammonium acetate, pH 5.2, and 1 mM EDTA). The purity of the protein was assessed by SDS-PAGE. The C116F IL-1Ra point mutation was prepared following the same procedure. Uniformly 15N-enriched samples of WT and mutant IL-1Ra were grown in M9 minimal media salts, with 99% [15N] ammonium sulfate (Isotec) at 2 g/L (Isotec) and purified as previously described. Protein concentrations were determined by measuring the A280 on a UV–Vis spectrometer (Bio-Rad).−The extinction coefficient for WT IL-1Ra is 15,460 L cm−1 mol−1, calculated as previously described.37 All proteins were exchanged into 10 mM 2-(N-morpholino)ethanesulfonic acid, pH 6.5, 90 mM NaCl, and 1 mM EDTA buffer (1× 4-morpholineethanesulfonic acid) unless noted otherwise.
Equilibrium and kinetic assays
Equilibrium unfolding titrations were measured using average fluorescence wavelength as previously described for IL-1β.26-28 Experiments in reducing conditions were performed in the presence of 10 mM DTT. The data were fit to a two-state model as previously described38 using in-house software. Stopped-flow fluorescence-detected refolding and manual-mixing fluorescence-detected refolding/unfolding kinetics studies were carried out as previously described for IL-1β.26-28 Manual-mixing and stopped-flow fluorescence kinetic data were fit as previously described.26-28
Heteronuclear NMR and HDX experiments
NMR 1H–15N HSQC experiments were performed similar to previous experiments on IL-1β27 with modifications. WT and mutant IL-1Ra were collected on a Varian DMX spectrometer with an operating frequency of 500 MHz equipped with a triple-resonance probe. Samples in the range of 250 μM to 1 mM were exchanged into buffer containing either 25 mM NaPO4, 100 mM NaCl, pH 6.5, or 100 mM d3-NaOAc, pH 5.4, and 10% final volume of D2O. HSQC spectra of the non-deuterated samples were recorded with a spectral width of 12.0 ppm in the 1H dimension (centered at 4.63 ppm) and 32.0 ppm in the 15N dimension (centered at 121.0 ppm). The number of points acquired was 1024, with 64 increments in the 15N dimension. The HDX time course was performed similar to previous experiments.27 All spectra were recorded at 308 K. All NMR experiments were processed as previously described.39
Chemical shift analysis was performed using the following equation:
| (1) |
The combined weighted average was plotted. Changes greater than 0.1 were deemed significant.
MEF cellular stimulation time course and nuclear extract preparation and EMSA
3T3 immortalized MEFs were generated and maintained as described elsewhere.32 Cells were grown to approximately 100% confluence and then starved for 12–16 h in 0.5% bovine calf serum/1 × Dulbecco’s modified Eagle’s medium before stimulation with the appropriate concentration of cytokine. Cells were stimulated with WT and mutant IL-1Ra following previously described protocol.32 Normalized samples and standards were incubated in binding buffer and 32P-labeled probe (specific for Nf-κB) for 15 min at room temperature before loading onto a 5% non-denaturing polyacyrlamide gel. After 2 h at 200 V, the gels were dried and then placed in a storage phosphor cassette before visualizing on a Typhoon 9410 Variable Mode Imager (GE Healthcare). The bands were normalized based on the total amount of protein added, and the bands were quantified in ImageQuant (GE Healthcare). Similar experiments in reducing conditions were not amenable.
Hydrogen–deuterium exchange mass spectrometry
Briefly, the heterotrimeric complex of WT or C116F IL-1Ra, IL-1RI (Sigma-Aldrich), and IL-1RAcP (Sigma-Aldrich) in equimolar amounts was exchanged into 1 × phosphate-buffered saline and equilibrated at room temperature. A deuterium exchange time course was initiated by adding phosphate-buffered saline at pD 7.40 to 66.7% final deuterium, and aliquots were removed and quenched with 0.5% formic acid, 16.6% glycerol, and 3.2 M guanidine–HCl (quench buffer) at 0 °C and then immediately frozen on dry ice and stored at −80 °C until analysis. Back-exchange and fully deuterated controls were run as before.40 All samples were subsequently thawed at 4 °C and passed over an AL-20-pepsin column [16 ml bed volume, 30 mg/ml porcine pepsin (Sigma)], at a flow rate of 20 ml/min. The resulting peptides were collected on a C18 trap (Michrom MAGIC C18AQ 0.262) and separated using a C18 reversed-phase column (Michrom MAGIC C18AQ 0.2650 3 mm 200 A) running a linear gradient of 0.046% (v/v) trifluoroacetic acid, 6.4% (v/v) acetonitrile to 0.03% (v/v) trifluoroacetic acid, 38.4% (v/v) acetonitrile over 30 min with column effluent directed into an LCQ mass spectrometer (Thermo-Finnigan LCQ Classic). Data were acquired in both data-dependent MS1:MS2 mode and MS1 profile mode. SEQUEST software (Thermo Finnigan Inc.) was used to identify the sequence of the peptide ions. DXMS Explorer (Sierra Analytics Inc., Modesto, CA) was used for the analysis of the mass spectra.
Supplementary Material
Acknowledgements
This work was supported by National Institutes of Health grants R01 GM101467 and R01 GM054038 (P.A.J.). We would like to thank members of the Jennings laboratory for discussion and review of this work. We would also like to thank Alex Hoffmann for the generous gift of the WT-2 3T3 MEF cells, Dr. Shannon Werner for help with the initial biological assay development and training, and Dr. Ellen O’Dea and Kim Ngo for their advice and help. We would like to thank Dr. Sheng Li for running the DXMS samples (NIH grants R01 GM093325 and S10 RR029388).
Abbreviations
- IL
interleukin
- IL-1R
IL-1 receptor
- IL-1Ra
IL-1 receptor antagonist
- IL-1RAcP
IL-1R accessory protein
- WT
wild type
- HDX
hydrogen–deuterium exchange
- HSQC
heteronuclear single quantum coherence
- MEF
mouse embryonic fibroblast
- EMSA
electrophoretic mobility shift assay
- EDTA
ethylenediaminetetraacetic acid
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jmb.2013.03.016
References
- 1.Dinarello CA. Interleukin-1beta. Crit. Care Med. 2005;33:S460–S462. doi: 10.1097/01.ccm.0000185500.11080.91. [DOI] [PubMed] [Google Scholar]
- 2.Netea MG, Nold-Petry CA, Nold MF, Joosten LA, Opitz B, van der Meer JH, et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood. 2009;113:2324–2335. doi: 10.1182/blood-2008-03-146720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siders WM, Mizel SB. Interleukin-1 beta secretion. A possible multistep process that is regulated in a cell type-specific manner. J. Biol. Chem. 1995;270:16258–16264. doi: 10.1074/jbc.270.27.16258. [DOI] [PubMed] [Google Scholar]
- 4.Arend WP, Malyak M, Guthridge CJ, Gabay C. Interleukin-1 receptor antagonist: role in biology. Annu. Rev. Immunol. 1998;16:27–55. doi: 10.1146/annurev.immunol.16.1.27. [DOI] [PubMed] [Google Scholar]
- 5.Arend WP, Malyak M, Smith MF, Jr, Whisenand TD, Slack JL, Sims JE, et al. Binding of IL-1 alpha, IL-1 beta, and IL-1 receptor antagonist by soluble IL-1 receptors and levels of soluble IL-1 receptors in synovial fluids. J. Immunol. 1994;153:4766–4774. [PubMed] [Google Scholar]
- 6.Baumann JB, Christen E, Gamboni G, Joss U, van Oostrum J, Girard J, Eberle AN. Receptor binding and biological activity of IL-1 alpha, IL-1 beta, IL-1 beta analogues and an IL-1 antagonist in A375 human melanoma cells. J. Recept. Res. 1993;13:245–262. doi: 10.3109/10799899309073658. [DOI] [PubMed] [Google Scholar]
- 7.Evans RJ, Bray J, Childs JD, Vigers GP, Brandhuber BJ, Skalicky JJ, et al. Mapping receptor binding sites in interleukin (IL)-1 receptor antagonist and IL-1 beta by site-directed mutagenesis. Identification of a single site in IL-1ra and two sites in IL-1 beta. J. Biol. Chem. 1995;270:11477–11483. doi: 10.1074/jbc.270.19.11477. [DOI] [PubMed] [Google Scholar]
- 8.Schreuder H, Tardif C, Trump-Kallmeyer S, Soffientini A, Sarubbi E, Akeson A, et al. A new cytokine-receptor binding mode revealed by the Allosteric Point Mutation in IL-1Ra 2391 crystal structure of the IL-1 receptor with an antagonist. Nature. 1997;386:194–200. doi: 10.1038/386194a0. [DOI] [PubMed] [Google Scholar]
- 9.Vigers GP, Anderson LJ, Caffes P, Brandhuber BJ. Crystal structure of the type-I interleukin-1 receptor complexed with interleukin-1beta. Nature. 1997;386:190–194. doi: 10.1038/386190a0. [DOI] [PubMed] [Google Scholar]
- 10.O’Neill LA, Dinarello CA. The IL-1 receptor/toll-like receptor superfamily: crucial receptors for inflammation and host defense. Immunol. Today. 2000;21:206–209. doi: 10.1016/s0167-5699(00)01611-x. [DOI] [PubMed] [Google Scholar]
- 11.O’Neill LA. The interleukin-1 receptor/Tolllike receptor superfamily: signal transduction during inflammation and host defense. Sci. STKE. 2000;2000:re1. doi: 10.1126/stke.442000re1. [DOI] [PubMed] [Google Scholar]
- 12.Lang D, Knop J, Wesche H, Raffetseder U, Kurrle R, Boraschi D, Martin MU. The type II IL-1 receptor interacts with the IL-1 receptor accessory protein: a novel mechanism of regulation of IL-1 responsiveness. J. Immunol. 1998;161:6871–6877. [PubMed] [Google Scholar]
- 13.Wang D, Zhang S, Li L, Liu X, Mei K, Wang X. Structural insights into the assembly and activation of IL-1beta with its receptors. Nat. Immunol. 2010;11:905–911. doi: 10.1038/ni.1925. [DOI] [PubMed] [Google Scholar]
- 14.Yu H, Ma L, Yang Y, Cui Q. Mechanochemical coupling in the myosin motor domain. II. Analysis of critical residues. PLoS Comput. Biol. 2007;3:e23. doi: 10.1371/journal.pcbi.0030023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chennubhotla C, Bahar I. Markov propagation of allosteric effects in biomolecular systems: application to GroEL–GroES. Mol. Syst. Biol. 2006;2:36. doi: 10.1038/msb4100075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lockless SW, Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science. 1999;286:295–299. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- 17.Ranson NA, Clare DK, Farr GW, Houldershaw D, Horwich AL, Saibil HR. Allosteric signaling of ATP hydrolysis in GroEL–GroES complexes. Nat. Struct. Mol. Biol. 2006;13:147–152. doi: 10.1038/nsmb1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Russ WP, Lowery DM, Mishra P, Yaffe MB, Ranganathan R. Natural-like function in artificial WW domains. Nature. 2005;437:579–583. doi: 10.1038/nature03990. [DOI] [PubMed] [Google Scholar]
- 19.Ota N, Agard DA. Intramolecular signaling pathways revealed by modeling anisotropic thermal diffusion. J. Mol. Biol. 2005;351:345–354. doi: 10.1016/j.jmb.2005.05.043. [DOI] [PubMed] [Google Scholar]
- 20.Schmidt B, Ho L, Hogg PJ. Allosteric disulfide bonds. Biochemistry. 2006;45:7429–7433. doi: 10.1021/bi0603064. [DOI] [PubMed] [Google Scholar]
- 21.Clementi C, Jennings PA, Onuchic JN. How native-state topology affects the folding of dihydrofolate reductase and interleukin-1beta. Proc. Natl Acad. Sci. USA. 2000;97:5871–5876. doi: 10.1073/pnas.100547897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roy M, Chavez LL, Finke JM, Heidary DK, Onuchic JN, Jennings PA. The native energy landscape for interleukin-1beta. Modulation of the population ensemble through native-state topology. J. Mol. Biol. 2005;348:335–347. doi: 10.1016/j.jmb.2005.02.059. [DOI] [PubMed] [Google Scholar]
- 23.Varley P, Gronenborn AM, Christensen H, Wingfield PT, Pain RH, Clore GM. Kinetics of folding of the all beta-sheet protein interleukin-1-beta. Science. 1993;260:1110–1113. doi: 10.1126/science.8493553. [DOI] [PubMed] [Google Scholar]
- 24.Heidary DK, Jennings PA. Three topologically equivalent core residues affect the transition state ensemble in a protein folding reaction. J. Mol. Biol. 2002;316:789–798. doi: 10.1006/jmbi.2001.5270. [DOI] [PubMed] [Google Scholar]
- 25.Capraro DT, Lammert H, Heidary DK, Roy M, Gross LA, Onuchic JN, Jennings PA. Cavity volume and altered water interactions contribute to route switching during the folding of interleukin-1β (IL-1β) Biophys J. 2012 doi: 10.1016/j.bpj.2013.06.019. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heidary DK, Roy M, Daumy GO, Cong Y, Jennings PA. Long-range coupling between separate docking sites in interleukin-1beta. J. Mol. Biol. 2005;353:1187–1198. doi: 10.1016/j.jmb.2005.08.072. [DOI] [PubMed] [Google Scholar]
- 27.Capraro DT, Gosavi S, Roy M, Onuchic JN, Jennings PA. Folding circular permutants of IL-1beta: route selection driven by functional frustration. PLoS One. 2012;7:e38512. doi: 10.1371/journal.pone.0038512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Capraro DT, Roy M, Onuchic JN, Gosavi S, Jennings PA. beta-Bulge triggers routeswitching on the functional landscape of interleukin-1beta. Proc. Natl Acad. Sci. USA. 2012;109:1490–1493. doi: 10.1073/pnas.1114430109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Latypov RF, Harvey TS, Liu D, Bondarenko PV, Kohno T, Fachini RA, 2nd, et al. Biophysical characterization of structural properties and folding of interleukin-1 receptor antagonist. J. Mol. Biol. 2007;368:1187–1201. doi: 10.1016/j.jmb.2007.02.031. [DOI] [PubMed] [Google Scholar]
- 30.Stockman BJ, Scahill TA, Roy M, Ulrich EL, Strakalaitis NA, Brunner DP, et al. Secondary structure and topology of interleukin-1 receptor antagonist protein determined by heteronuclear three-dimensional NMR spectroscopy. Biochemistry. 1992;31:5237–5245. doi: 10.1021/bi00138a001. [DOI] [PubMed] [Google Scholar]
- 31.Hayden MS, Ghosh S. Signaling to NFkappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 32.Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 2002;298:1241–1245. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- 33.Darby N, Creighton TE. Disulfide bonds in protein folding and stability. Methods Mol. Biol. 1995;40:219–252. doi: 10.1385/0-89603-301-5:219. [DOI] [PubMed] [Google Scholar]
- 34.Villafranca JE, Howell EE, Oatley SJ, Xuong NH, Kraut J. An engineered disulfide bond in dihydrofolate reductase. Biochemistry. 1987;26:2182–2189. doi: 10.1021/bi00382a017. [DOI] [PubMed] [Google Scholar]
- 35.Adamek DH, Guerrero L, Blaber M, Caspar DL. Structural and energetic consequences of mutations in a solvated hydrophobic cavity. J. Mol. Biol. 2005;346:307–318. doi: 10.1016/j.jmb.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 36.Mamathambika BS, Bardwell JC. Disulfide-linked protein folding pathways. Annu. Rev. Cell Dev. Biol. 2008;24:211–235. doi: 10.1146/annurev.cellbio.24.110707.175333. [DOI] [PubMed] [Google Scholar]
- 37.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 38.Chrunyk BA, Wetzel R. Breakdown in the relationship between thermal and thermodynamic stability in an interleukin-1 beta point mutant modified in a surface loop. Protein Eng. 1993;6:733–738. doi: 10.1093/protein/6.7.733. [DOI] [PubMed] [Google Scholar]
- 39.Capraro DT, Roy M, Onuchic JN, Jennings PA. Backtracking on the folding landscape of the beta-trefoil protein interleukin-1beta? Proc. Natl Acad. Sci. USA. 2008;105:14844–14848. doi: 10.1073/pnas.0807812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hailey KL, Li S, Andersen MD, Roy M, Woods VL, Jr, Jennings PA. Prointerleukin (IL)-1beta shares a core region of stability as compared with mature IL-1beta while maintaining a distinctly different configurational landscape: a comparative hydrogen/deuterium exchange mass spectrometry study. J. Biol. Chem. 2009;284:26137–26148. doi: 10.1074/jbc.M109.027375. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.