

Abstract
Next-generation sequencing results from bead-based aptamer libraries have demonstrated that traditional DNA/RNA alignment software is insufficient. This is particularly true for X-aptamers containing specialty bases (W, X, Y, Z, ...) that are identified by special encoding. Thus, we sought an automated program that uses the inherent design scheme of bead-based X-aptamers to create a hypothetical reference library and Markov modeling techniques to provide improved alignments. Aptaligner provides this feature as well as length error and noise level cutoff features, is parallelized to run on multiple central processing units (cores), and sorts sequences from a single chip into projects and subprojects.
DNA aptamers are quickly maturing as research and therapeutic tools because of their ability to bind specific proteins with high affinity.1−3 Using solution methods,4 DNA aptamers are generally determined by serial selection rounds. However, during bead-based aptamer selection,5,6 one collects beads in a single round or uses serial magnet heights. Traditionally, subcloned and sequenced aptamers were aligned using methods such as ClustalW,7 and more recently methods such as Tallymer,8 using k-mer analysis to find transposable elements, and structural alignments9 have been reported. X-aptamers10 are created by a bead-based pseudorandom process incorporating encoded specialty 5-X-dU bases (labeled as W, X, Y, or Z). Traditional alignment algorithms ignore the inherent X-aptamer bead-based, encoded design and fail to properly align and decode the sequences. Thus, we sought to create mapping software for X-aptamers, similar to BLAST11 or BLAT,12 but one using a Markov model based on the X-aptamer library designs.
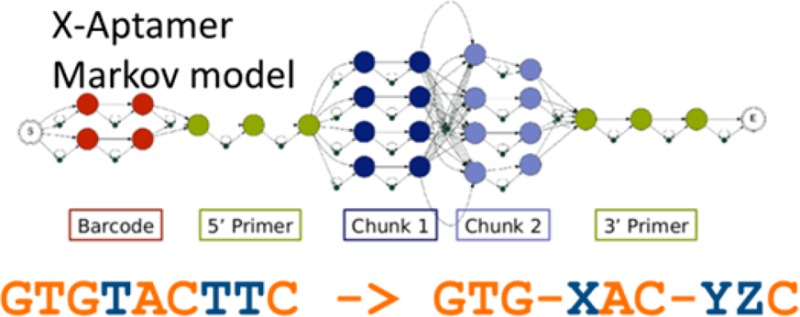
Traditional aptamers use two primers flanking a random region. However, X-aptamers10 use a pseudorandom process wherein the variable region is built in 10 or mores stages, using a split and pool method5,6,10 in which only a small number of DNA fragments are allowed at any one stage (Table S1 of the Supporting Information). Some fragments contain functionalized deoxyuracil bases (5-X-dU) that are sequenced as a T.
Although next-generation sequencing (NGS) provides copious data, analyzing a single aptamer or X-aptamer project per chip is not cost-effective. For optimal utility, NGS aptamer alignment software should be capable of separating NGS data into multiple projects (Figure S2 of the Supporting Information) and subprojects when using bar coding. Aptaligner was designed to accomplish such analyses for X-aptamers and other aptamers.
Logistically, Aptaligner performs the following steps: (1) determines the number of projects, (2) reads and error-checks library design files, (3) reads FastQ NGS data and reduces it to an enumerated unique list, (4) removes sequences outside of a user-defined length range, (5) removes rare sequences with a user-defined noise level, (6) asks for central processing units (CPUs), (7) builds Markov models for each library and finds the optimal alignment, (8) assigns each sequence to a project, (9) decodes positions W–Z, and (10) conducts statistical analysis. These steps are explained in more detail below and are explicitly detailed in the program user guide.
In the first two steps, Aptaligner requests the number of projects having data in the FastQ file. For each project, graphical user interfaces (GUIs) ask for a project name and library design file (.csv format). The design file describes aptamer library construction from the 5′-end to the 3′-end. To sequence multiple aptamer selection rounds at once, bar codes (tags) may be added during the final polymerase chain reaction step, and they may be located at either end of 5′- or 3′-primer. For each line, two flags indicate whether the line contains primer information or bar code information (Table S1 of the Supporting Information). As a precaution, the code checks each library design file for typical errors and aborts if needed.
In step 3, the FastQ file is reduced to a set of enumerated unique sequences. In our experience, using a 5 million sequence NGS chip, this step typically reduces the list from more than 5 million nonunique sequences to ∼3.5 million unique sequences. This step is used to avoid reanalyzing identical sequences thousands of times.
In step 4, the user supplies a length error cutoff value to remove sequences that are excessively short or long. The GUI shows the number of unique sequences and how many of them will be retained using length cutoff values of 0 (∼20%), 5 (∼50%), 10 (∼55%), 15 (∼60%), and the current choice. The default value is five bases, and the user may accept or change this value. With a change, the window will recalculate the retained sequences.
In step 5, a noise level cutoff removes sequences that occur “noise-level” or fewer times. The dialogue box indicates unique sequences retained in step 4 (length cutoff) and how many will be retained after application of the noise filter using noise levels from 0 to 5. A noise level of 1 often reduces the sequences by 90%, greatly reducing the computation time.
After the user (step 6) inputs the number of CPUs (cores) to use for the calculation, Aptaligner (step 7) performs the alignments using a Markov model. The topology of the Markov model (Figure S1 of the Supporting Information) is derived individually from each library design file (Table S1 of the Supporting Information). Each base is represented as a node, with transitions linking each base to the next. Alternative sequences (alternate bar codes, alternate DNA fragments) are modeled as parallel tracks, with no transitions between them. Insertion nodes link each base to the next, allowing bases to be inserted because of technical errors. Deletions are modeled by transitions from each node to every possible node that occurs later in the library. The deletion penalty is based on the number of bases skipped. However, we do have special transitions that allow whole fragments to be deleted. Given the Markov model, and a specification of base match, mismatch, insertion, and deletion probabilities, we align an observed sequence and recover the highest-scoring alignment using the Viterbi algorithm.13 A theoretical X-aptamer library might contain up to 100 billion perfect sequences.
For each sequence, the alignment scores are used to choose the winning project. Good scores typically range from −11 to −60 (Figure S2 of the Supporting Information) but are slightly project-dependent. A list of sequences, scores, and their winning projects is retained in the main directory, and project-specific sequences are recorded into project subdirectories.
Once sequences are assigned to a project, the specialty 5-X-dU bases are decoded (step 9) from T back to W, X, Y, or Z. A fragment substitution is performed for perfect fragments. For imperfect fragments, the code calculates all possible Levenshtein edit distances14 between the fragment and theoretical choices. If, and only if, a unique smallest distance is found with a T/X alignment, the T is converted to X. All letters except B and J, and A, C, G, T, and U, can denote specialty bases, but additional mapping (“ --lib2seq X:T”) is needed.
The final step calculates statistics for each bar code or random fragment, clusters the top sequences (Figure S3 of the Supporting Information), and, for the top 50 sequences, finds all close sequences (edit distance of <3) in the top 5000 sequences (Table S2 of the Supporting Information).
In addition to providing superior alignment (Table 1), by ignoring small primer errors Aptaligner finds 40–100% more counts per random region total compared to exact primer matches (Exact Match). Examples are listed in Table 2.
Table 1. Top X-Aptamer Sequences Aligned against a Library (not each other) Based on a Markov Model.
| T1 | T2 | T3 | T4 | T5 | T6 | T7 | T8 | T9 | T10 | T11 | T12 | all | R1 | R2 | R3 | R4 | R5 | R6 | R7 | R8 | R9 | R10 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 0 | 0 | 3426 | 0 | 28 | 0 | 2 | 6 | 0 | 2 | 3467 | GC | GTG | GT | GX | -- | TTC | GTG | --- | TX | GCC |
| 0 | 0 | 0 | 0 | 1391 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1392 | XT | TGG | -- | GG | AT | TTC | GTG | GTG | GT | GGC |
| 1466 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1469 | XT | GCG | XC | AC | AT | GCC | GTT | CCG | TG | GCC |
| 7 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 1150 | 0 | 0 | 1159 | XT | TGG | XC | GG | CC | --- | GTT | --- | -- | GCC |
| 9 | 2 | 0 | 0 | 102 | 0 | 0 | 0 | 972 | 59 | 0 | 0 | 1144 | GC | TX- | TA | AC | CC | TTC | GTG | GTG | GT | GCG |
| 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 1080 | 3 | 0 | 0 | 1085 | TG | TXG | TG | GX | AT | TGC | GTT | GTG | -- | GXG |
| 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 1 | 1001 | 0 | 0 | 1004 | TC | TXG | GT | GX | AT | TGC | GTT | --- | -- | GCC |
| 0 | 0 | 0 | 0 | 2 | 1 | 2 | 0 | 0 | 982 | 0 | 0 | 987 | TC | TXG | GT | GX | -- | TTC | GTG | CCG | TX | GCC |
| 2 | 1 | 0 | 0 | 0 | 0 | 3 | 0 | 1 | 959 | 0 | 0 | 966 | GC | GTG | -- | GG | AT | TGC | GTT | GTG | TG | GXG |
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 877 | 1 | 0 | 0 | 878 | -- | --- | TA | -- | CC | TGC | GTT | GAG | -- | GXG |
| 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 824 | 0 | 0 | 826 | TC | GTG | XC | GX | GT | XAC | GTG | GTG | GT | GCC |
| 722 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 774 | TG | GTG | TG | GG | CC | TTC | GTT | GXC | GT | GCC |
Table 2. Sequence Frequencies Determined by Aptaligner and Exact Matcha.
| sequence | Exact Match | Aptaligner | |
|---|---|---|---|
| 1 | GCGTGGTGTTTCGTGTTGCC | 5328 | 7602 |
| 2 | TTTGGGGATTTCGTGGTGGTGGC | 2664 | 3662 |
| 3 | GCGTGTGGTATTGCGTTGTCTTGGC | 2260 | 3616 |
| 4 | TTTGGTCGGCCGTTGCC | 2099 | 2686 |
| 5 | TTGCGTCACATGCCGTTCCGTGGCC | 2080 | 3245 |
| 6 | GCTTTAACCCTTCGTGGTGGTGCG | 1921 | 3793 |
| 7 | TGTTGTGGTATTGCGTTGTGGTG | 1837 | 2819 |
| 8 | TCTTGGTGTATTGCGTTGCC | 1785 | 2487 |
| 9 | TTTGGTCGTATTGCGTAGTCGTGGC | 1615 | 2718 |
| 10 | GCGGGATCCGTTGTG | 1615 | 2965 |
| 11 | GCGTGGGATTGCGTTGTGTGGTG | 1593 | 2836 |
| 12 | TCTTGGTGTTTCGTGCCGTTGCC | 1566 | 2298 |
Confidential sequences were scrambled postanalysis. There was an increase in sequence frequency for Aptaligner compared to Exact Match. These are the same sequences listed in Table 1, but they were scrambled after alignment.
The software and test files (library design, FastQ, and output) are free to noncommercial users at www.uth.edu/nbme/Aptaligner.htm or bioinformatics.uth.tmc.edu. It requires linux (tested on RedHat and Fedora) and the following: Python (www.python.org), including xlrd, argeparse, and openpyxl; Biopython (www.biopython.org); R,15 including R libraries MiscPsycho,16 stringr,17 dendroextras,18 and statmod;19 numpy (http://www.numpy.org/); and C.
The software was tested on a laptop (Intel Core2 Duo, CPU P8400 @2.27 GHz, 4GB RAM, 64-bit Operating System, RHEL 6.5), a Dell Precision T7500N computer workstation with dual quad core Intel Xeon processor E5603 (1.6 GHz) with 12 GB RAM, with a Redhat Linux (RHEL 6.5) operating system, and a server with 48 cores (Dell PowerEdge R815; 2× AMD Opteron 6174 2.2 GHz processors (4 total); 8× 32Gb 1333 MHz Dual Ranked RDIMM RAM; No OS; RAID 5; PERC H700, 512 Mb Cache; 6 × 300Gb 10k RPM SCSI HD; 1100w power supply). It was also run using a Fedora 20 virtual machine. Aptaligner’s complexity is O(NMM), similar to pairwise alignments, where N equals sequence length and M equals the library length. Accordingly, the software requires significant memory (3G) and up to 12GB free disk space per NGS chip. Insufficient resources elicit a run-time warning.
Acknowledgments
We thank Drs. A. Annapragada (Texas Children’s Hospital); N. Vigneswaran, D. Gorenstein, and V. Thiviyanathan (University of Texas Health Science Center); Haifa Shen (Methodist Hospital Research Institute); and Mark Shumbera (AM Biotechnologies) for providing confidential data sets for software testing.
Supporting Information Available
Supplemental figures and tables, including a library design input file. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∇ M.-A.E.-R.: Centro Universitario Contra el Cáncer, Hospital Universitario “Dr. José Eleuterio González”, Universidad Autónoma de Nuevo León, Monterrey, N.L. México.
Author Contributions
E.L. and M.-A.E.-R. contributed equally to this work.
Supported by NIH/NCATS (UL1 TR000371), NCI (CA151668), Welch Foundation (AU-1296), NIAID (HHSN272200800048C and AI054827), and NHLBI (HHSN268201000037C) grants.
The authors declare the following financial interests. The authors and the University of Texas Health Science Center would share any future licensing fees.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Jayasena S. D. (1999) Clin. Chem. 45, 1628–1650. [PubMed] [Google Scholar]
- Keefe A. D.; Pai S.; Ellington A. (2010) Nat. Rev. Drug Discovery 9, 537–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiviyanathan V.; Somasunderam A.; Volk D. E.; Klostergaard J.; Gorenstein D. G. (2012) Proceedings of the Institute of Chemistry of Ceylon 204–209(June). [Google Scholar]
- Tuerk C.; Gold L. (1990) Science 249, 505–510. [DOI] [PubMed] [Google Scholar]
- Yang X.; Bassett S. E.; Li X.; Luxon B. A.; Herzog N. K.; Shope R. E.; Aronson J.; Prow T. W.; Leary J. F.; Kirby R.; Ellington A. D.; Gorenstein D. G. (2002) Nucleic Acids Res. 30, e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.; Li X.; Prow T. W.; Reece L. M.; Bassett S. E.; Luxon B. A.; Herzog N. K.; Aronson J.; Shope R. E.; Leary J. F.; Gorenstein D. G. (2003) Nucleic Acids Res. 31, e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenna R.; Suqawara H.; Koike T.; Lopez R.; Gibson T. J.; Higgins D. G.; Thompson J. D. (2003) Nucleic Acids Res. 31(13), 3497–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S.; Narechania A.; Stein J. C.; Ware D. (2008) BMC Genomics 9, 517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theil W. H.; Bair T.; Peek A. S.; Liu X.; Dassie J.; Stockdate K. R.; Behike M. A.; Milleer F. J. Jr.; Giangrande P. H. (2012) PLoS One 7(9), e43836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W.; Elizondo-Riojas M.-A.; Li X.; Lokesh G. L. R.; Somasunderam A.; Thiviyanathan V.; Volk D. E.; Durland R. H.; Engelhardt J.; Cavasotto C. N.; Gorenstein D. G. (2012) Biochemistry 51(42), 8321–8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F.; Gish W.; Miller W.; Myers E. W.; Lipman D. J. (1990) J. Mol. Biol. 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Kent W. J. (2002) Genome Res. 12, 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning C., and Schütze H. (1999) Foundations of Statistical Natural Language Processing, MIT Press, Cambridge, MA. [Google Scholar]
- Levenshtein V. I. (1966) Phys.-Dokl. 10, 707–710. [Google Scholar]
- R Core Team (2013) R: A language and environment for statistical computing, R Foundation for Statistical Computing, Vienna. [Google Scholar]
- Doran H. C. (2010) MiscPsycho: Miscellaneous Psychometric Analyses. R, version 1.6 (http://CRAN.R-project.org/package=MiscPsycho). [Google Scholar]
- Wickham H. (2012) stringr: Make it easier to work with strings. R, version 0.6.2 (http://CRAN.R-project.org/package=stringr). [Google Scholar]
- Jefferis G. (2013) dendroextras: Extra functions to cut, label and colour dendrogram clusters. R, version 0.1–2 (http://CRAN.R-project.org/package=dendroextras). [Google Scholar]
- Smyth G. with contributions from Yifang Hu, Peter Dunn, and Belinda Phipson (2013) statmod: Statistical Modeling. R, version 1.4.17 (http://CRAN.R-project.org/package=statmod). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.