

Summary
Establishing lifelong infection and periodically shedding infectious progeny is a successful strategy employed by several persistent pathogens. In this issue of Cell Host & Microbe, Pan et al. demonstrate that a cell-type-specific host microRNA can restrict gene expression and pathogenicity of Herpes Simplex Virus-1, thereby promoting long-term infection.
In any prolonged war, détente: a long term relaxation of strained relations or tensions between two nations represents a desirable outcome. This also applies for the ongoing war between host and pathogen, and particularly so for those pathogens that have long co-evolved with their hosts. For a virus, establishing lifelong infection maximizes the opportunity to spread to more hosts, thereby providing an evolutionary advantage. Viruses have evolved a number of strategies for persistent infection and arguably the most elegant is latency. Latent viruses can undergo a lytic growth phase, involving replication of the viral genome, production of progeny virus, lysis of cells, infection of neighboring cells, and spread to other organisms. During this phase, the virus also infects cells within the host where it is able to establish latency. Latency results in drastically reduced expression of most viral genes and eliminates the production of infectious progeny, allowing the virus to evade the immune response. Periodically, the virus reactivates from this quiescent state in response to a variety of environmental cues and re-enters the lytic phase of its life cycle.
Members of the Herpesviridae family could be considered the champions of latent infection. There are several human pathogens in this family, including Epstein-Barr virus, human cytomegalovirus (HCMV), varicella-zoster virus, and herpes simplex virus type I (HSV-1). HSV-1 replicates primarily in epithelial cells but establishes latency in neural ganglia. During latency HSV-1 limits gene expression to a single locus, the non-protein-coding latency associated transcript (LAT), and coordinately reduces lytic gene expression to near-undetectable levels. Both viral and host factors are believed to be involved in establishing and maintaining latency in neuronal cells, but the mechanisms are still poorly understood.
In one sense, the latent phase of the viral life cycle is simply the repression of the lytic phase. In herpesviruses, the immediate early (IE) genes drive initiation of the lytic phase. In HSV-1, the IE viral protein, ICP0 is required for reactivation(Boutell and Everett, 2013) and for controlling its own expression as well as other IE genes that are critical to maintaining latency. MicroRNA (miRNA)-mediated silencing has garnered considerable attention as a potential mechanism for this regulation during latency(Cullen, 2011). Consistent with this hypothesis, members of both the Herpesviridae and Polyomaviridae families encode autoregulatory miRNAs that control expression of their lytic genes. For example, the LAT locus of HSV-1 encodes several miRNAs (Umbach et al., 2008; Jurak et al., 2011), which have been linked to down-regulation of the IE genes ICP0 and ICP 34.5 (Umbach et al., 2008). Thus, it is firmly established that diverse viruses can use miRNAs to regulate the expression of their own genes – a potential factor in both establishing and maintaining latent infections.
miRNA regulation is prevalent in multicellular eukaryotes and, directly or indirectly, likely regulates all cellular pathways. miRNAs are central components of the RNA induced silencing complex (RISC), targeting it to messenger RNAs (mRNAs)via base pairing. Numerous miRNAs have cell-type-specific expression, making them attractive candidates as host factors involved in circumstantial regulation of lytic gene expression. Indeed, there are examples of host miRNAs affecting the cell-type-specific gene expression of diverse viruses(Gunasekharan and Laimins, 2013; Trobaugh et al., 2014; Jopling et al., 2005). Despite what is known, many questions remain regarding the extent to which host miRNAs are coopted or antagonized by viruses, and their effect on latency.
An article in this issue of Cell Host & Microbe explores the effect of a host miRNA on the expression of viral gene products, and the consequence of this regulation on the biology and pathology of HSV-1 (Pan et al. this issue; Figure 1). Specifically, Pan and colleagues report that human miR-138, previously identified as a potential tumor suppressor, negatively regulates HSV-1 ICP0 and, in doing so, promotes latency. The authors computationally identify miR-138 binding sites in the 3′ UTR of the ICP0 mRNA and demonstrate that exogenous expression of miR-138 significantly reduces expression of ICP0 from a plasmid containing both its open reading frame and 3′ UTR. The effect of miR-138 on ICP0 levels is sequence-specific, as mutation of the binding site abolishes regulation. To confirm RISC binds the ICP0 transcript, the authors infected cells stably expressing exogenous miR-138, then using crosslinking followed by high throughput sequencing analysis, identified clusters of reads that map to the two predicted binding sites for miR-138 in the 3′ UTR of ICP0. With these techniques, the authors convincingly demonstrate that miR-138 can regulate expression of the ICP0 in a virus-independent context.
Figure 1.
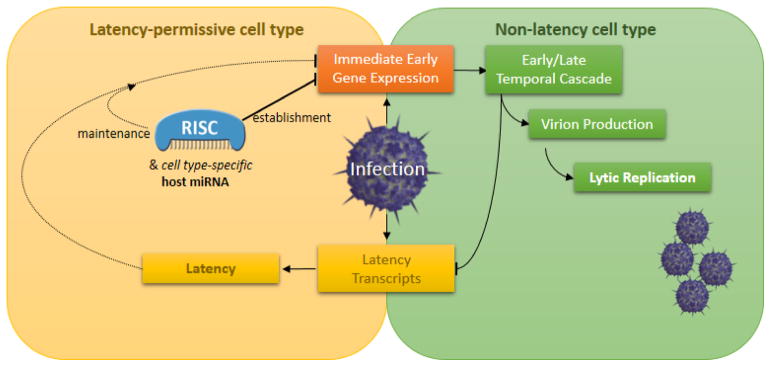
An endogenous host miRNA, with enriched expression in latency-permissive cell types, cooperates with viral and other host factors in both the establishment and maintenance of latency by repressing viral immediate early genes.
Next, the authors focused on miR-138 activity during HSV-1 infection of cultured cells. Treating cells expressing a mimic of miR-138 with an antisense inhibitor of miR-138 resulted in higher ICP0 and IE expression than treating with a control antisense inhibitor. Additionally, infection of neuronal cells (that naturally express miR-138) with a mutant virus containing nucleotide substitutions in both miR-138 docking sites of the ICP0 3′ UTR, results in significantly increased expression of ICP0 and other IE transcripts. However, perhaps unexpectedly, growth of this mutant virus was unaffected. These results demonstrate that the endogenous host miRNA, miR-138, can affect the level of viral gene expression during HSV-1 infection in cultured cells.
To address the biological relevance of miR-138-mediated regulation of ICP0, the authors used a murine model. Following inoculation of the cornea, viral replication at the eye and in the site of latency, the trigeminal ganglion (TG), was reported. Virus yield at both sites was unaffected by mutation of the miR-138 binding sites, and decreased with similar kinetics in the TG, presumably reflecting similar onset of latency. However, at 7 days post-infection, mutant-virus-infected cells had elevated levels of ICP0 and other lytic transcripts. Yet, these mice had wildtype levels of LAT expression and viral genome levels (in the TG), suggesting that the elevated lytic transcript levels were not significantly altering viral replication. These results demonstrate that host miRNA miR-138 regulates lytic gene expression in vivo but, at least under these conditions, with undetectable consequence to virus replication. Remarkably, there was an almost 4-fold increase in mortality of mice by 32 days post-infection with the mutant virus (relative to wild type). One possible explanation for the higher mortality rate associated with the mutant virus could be decreased maintenance of latency accompanying increased lytic virus gene expression. However, analysis of the TG of the mice at 32d post-infection revealed no significant increase in the level of lytic transcripts. Despite the mechanistic basis remaining unclear, these data identify a fascinating role for a host miRNA in preventing pathogenesis of a virus.
Altogether, this work shows that HSV-1 utilizes a host miRNA to regulate lytic gene expression in cell culture and in vivo. Given the enriched expression of miR-138 in neuronal cells, and the restriction of HSV-1 latency to some types of ganglia, the authors’ model of miR-138 as a direct neuronal latency restriction factor seems likely. However, important questions remain regarding this model. For example, how can miR-138 promote latency without altering the amount of virus produced during infection of mice? Why are the kinetics of the onset of latency unaffected for the mutant virus? The use of both host and viral miRNAs to regulate ICP0 clearly underscores its importance, but begs the question of what advantage is conveyed by such multi-miRNA regulation. One possibility is that the host miRNA synergizes with or nucleates viral miRNA regulation. What is clear is that the reduced pathogenicity associated with miR-138-mediated regulation at least indirectly allows for increased latency within the population, since dead hosts are not useful latent reservoirs.
Given the importance of cell-type-specific control of viral gene expression during persistent infection, it is not surprising that other viruses use host miRNAs for similar purposes. Notably, O’Connor and colleagues recently demonstrate that an IE gene from the distantly related HCMV is targeted by miR-200, a miRNA enriched in the CD34+ myeloid lineage that HCMV latently infects (O’Connor et al., 2014). Furthermore, overexpression of miR-200 decreases lytic gene expression and favors latency, while infecting cells with a binding site mutant virus leads to increased viral replication. The prevalence of host miRNA-mediated regulation in diverse viruses suggests intriguing possibilities: RISC activity is diminished by many of the stress signals associated with induction of lytic replication, making it possible for viruses to use RISC activity as a barometer for stress(Seo et al., 2013). In this model, alleviating miRNA-mediated regulation would trigger a feed-forward transcriptional cascade resulting in increased lytic replication. Alternatively, but not mutually exclusive, miRNA regulation may slow lytic replication allowing chromatin modifications to establish latency. Several examples of host miRNAs directly regulating virus gene expression/replication are known (Gunasekharan and Laimins, 2013; Trobaugh et al., 2014; Jopling et al., 2005; O’Connor et al., 2014) and likely many others await discovery. Undoubtedly, the work by Pan et al. makes an important contribution to this growing field, and suggests that other pathogens may also utilize host miRNAs to promote modus vivendi, representing an arrangement that would help host and pathogen coexist.
Acknowledgments
We thank the C.S.S. lab for comments on this manuscript. Support for our work comes from grants: NIHRO1AI077746, NSF Career Award, CPRIT, Burroughs Wellcome Investigators in Pathogenesis Award, and a UT Austin ICMB fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Boutell C, Everett RD. Regulation of alpha herpesvirus infections by the ICP0 family of proteins. J Gen Virol. 2013;94(3):465–81. doi: 10.1099/vir.0.048900-0. [DOI] [PubMed] [Google Scholar]
- Cullen BR. Herpesvirus microRNAs: phenotypes and functions. Curr Opin Virol. 2011 Sep;1(3):211–5. doi: 10.1016/j.coviro.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunasekharan V, Laimins LA. Human papillomaviruses modulate microRNA 145 expression to directly control genome amplification. J Virol. 2013;87(10):6037–43. doi: 10.1128/JVI.00153-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309(5740):1577–81. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- Jurak I, Grittiths A, Coen DM. Mammalian alphaherpesvirus miRNAs. Biochem Biophys Acta. 2011;1809(11–12):641–53. doi: 10.1016/j.bbagrm.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor CM, Vanicek J, Murphy EA. Host miRNA regulation of human cytomegalovirus immediate early protein translation promotes viral latency. J Virol. 2014 doi: 10.1128/JVI.00481-14. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Flores O, Umbach JL, Pesola JM, Bentley P, Rosato PC, Leib DA, Cullen BR, Coen DM. A neuron spefici host microRNA targets herpes simplex virus-1 ICP0 expression and promotes latency. Cell Host Microbe. 2013 doi: 10.1016/j.chom.2014.03.004. This Issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo GJ, Kincaid RP, Phanaksri T, Burke JM, Pare JM, Cox JE, Hsiang TY, Krug RM, Sullivan CS. Reciprocal inhibition between intracellular antiviral signaling and the RNAi machinery in mammalian cells. Cell Host Microbe. 2013;14(4):435–45. doi: 10.1016/j.chom.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trobaugh DW, Gardner CL, Sun C, Haddow AD, Wang E, Chapnik E, Mildner A, Weaver SC, Ryman KD, Klimstra WB. RNA viruses can hijack vertebrate microRNAs to suppress innate immunity. Nature. 2014;506(7487):245–8. doi: 10.1038/nature12869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature. 2008;454(7205):780–83. doi: 10.1038/nature07103. [DOI] [PMC free article] [PubMed] [Google Scholar]