

Abstract
Malignant rhabdoid tumors (MRT) of the kidney are rare in children and even less common in adults, with only six previously reported adult cases. We present the case of a 60-year-old man with an MRT arising in the left kidney with extensive pulmonary micrometastases and thromboembolism resulting in thrombotic pulmonary microangiopathy (pulmonary tumor embolism syndrome). MRT is an extremely aggressive neoplasm with a short survival time.
Malignant rhabdoid tumor (MRT) of the kidney is a highly aggressive, extremely rare neoplasm that is usually seen in children. The term rhabdoid is used because the tumor cells resemble rhabdomyoblasts but lack myogenic markers, and pathologic diagnosis requires familiarity with these microscopic features plus awareness that adult onset is possible. We describe clinical and necropsy findings in an adult with primary renal MRT.
CLINICAL HISTORY
A 60-year-old white man with known hypothyroidism, hypertension, and chronic obstructive pulmonary disease had progressive fatigue and abdominal distension for 6 months and worsening dyspnea for 2 weeks. Imaging showed bilateral pulmonary infiltrates consistent with pneumonia, and he was treated at home with antibiotics and prednisone. Three days later, he presented to an outside emergency department with gross hematuria, left calf pain, hemoptysis, painful testicular swelling, and nontraumatic ecchymoses. He was then transferred to Baylor University Medical Center at Dallas and admitted to the intensive care unit. Examination disclosed bilateral lower-extremity edema and ecchymosis over the left calf and ankle. His body mass index was 30.4 kg/m2, blood pressure, 170/90 mm Hg; heart rate, 96 beats/minute; respiratory rate, 18 breaths/minute; white blood cell count, 31.9 k/μL; hemoglobin, 9.3 g/dL; hematocrit, 25.9%; platelets, 81 k/μL; prothrombin time, 18.0 seconds (reference range 9.0–12.0 seconds); blood urea nitrogen, 46.0 mg/dL; creatinine, 1.6 mg/dL; and arterial blood gas pH, 7.38. He was given heparin and multiple units of red blood cells and cryoprecipitate. Chest radiograph showed bilateral basilar and right upper lobe infiltrates. Computed tomography showed a thrombus within the left renal vein that extended into the inferior vena cava and a mass in the mid to lower pole of the left kidney (Figure 1).
Figure 1.
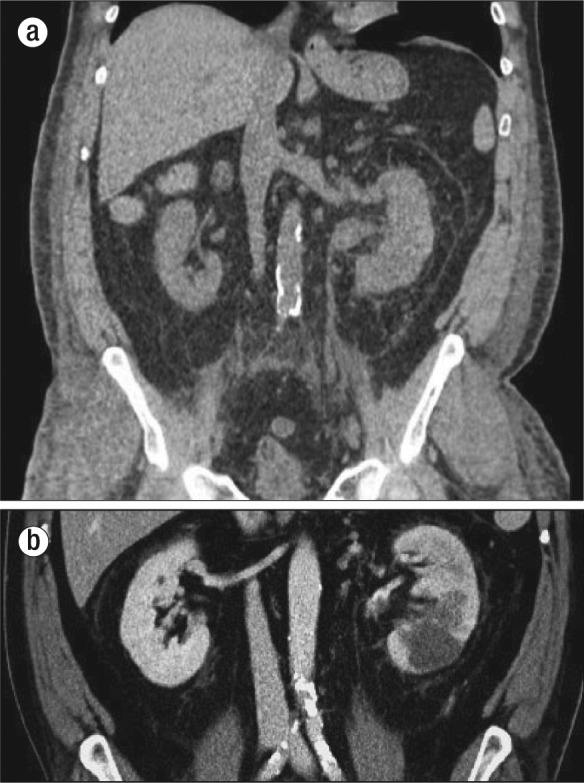
Computed tomography showing (a) a thrombus distending the left renal vein and (b) a mass in the middle to lower pole of the left kidney.
Over the next few days, the patient's oxygen requirements increased, his abdomen became more distended, and his urine output decreased. A left radical nephrectomy and retroperitoneal lymph node dissection was performed. At surgery, there was no residual thrombus in the left main renal vein. Postoperatively, he continued to bleed and developed an abdominal compartment syndrome; the same day at reoperation a splenectomy was performed for apparent “decapsulation,” and he died on the operating table.
The left kidney (surgical specimen) contained a 3.5 × 2.5 × 2.0 cm poorly circumscribed, soft tan lesion in the mid to lower pole that extended from the hilum to the cortex. Numerous tan-gray nodules (0.1–0.4 cm) were also present within perinephric and renal sinus connective tissues. Microscopically, this MRT contained characteristic sheets of anaplastic, noncohesive tumor cells with eccentric nuclei, prominent nucleoli, and eosinophilic, fibrillar cytoplasmic inclusions (Figure 2). There was extensive intravascular tumor within adjacent soft tissues. Immunohistochemically, tumor cells were strongly reactive for vimentin and pancytokeratin (AE 1–3), but negative for desmin, myoglobin, S100, melanoma cocktail, TTF-1, CK7, CK20, CDX2, PAX8, CD31, CD34, Factor VIII, CD30, and CD45. Additionally, tumor cells were negative for integrase interactor 1 (INI-1) nuclear protein expression. Electron microscopy found cytoplasmic whorls of intermediate filaments, some of which displaced the nucleus (Figure 3). Together, these histologic, immunohistochemical, and electron microscopic findings were sufficient for the diagnosis of MRT.
Figure 2.
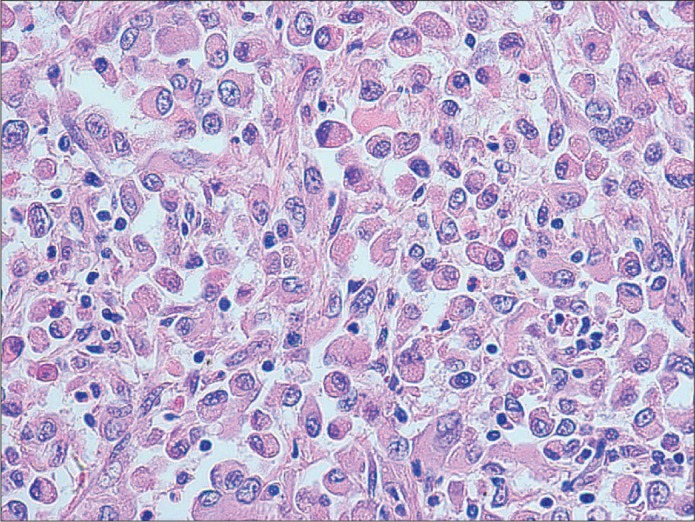
“Rhabdoid” tumor cells with eccentric nuclei and eosinophilic cytoplasm (H&E, 400×).
Figure 3.
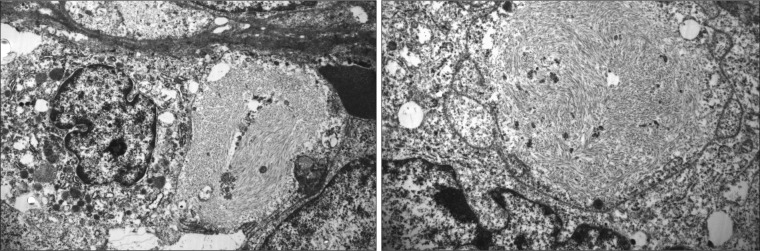
Tumor cells containing cytoplasmic whorls of intermediate filaments (electron micrographs, 3200×/4600×).
At necropsy, the abdomen contained 90 g of clotted blood at the nephrectomy site. Subcentimeter grey-white tumor nodules were present in pulmonary hilar lymph nodes, the left adrenal gland, the left spermatic cord, and the right kidney. The inferior vena cava contained loose fragments of thrombus, and there were large, coiled thromboemboli in bilateral hilar lung vessels. Microscopically, all tumors had the same histologic appearance. The large pulmonary thromboemboli were laminated, and most contained tumor cells. In some sections, the tumor cells extended along endothelial surfaces of the pulmonary arteries, and small, intravascular fibrin thrombi containing tumor cells were widely distributed throughout both lungs (Figure 4). Additional autopsy findings included severe pulmonary emphysema, pulmonary hypertensive vasculopathy, chronic thyroiditis, and chronic gastroesophageal inflammation.
Figure 4.
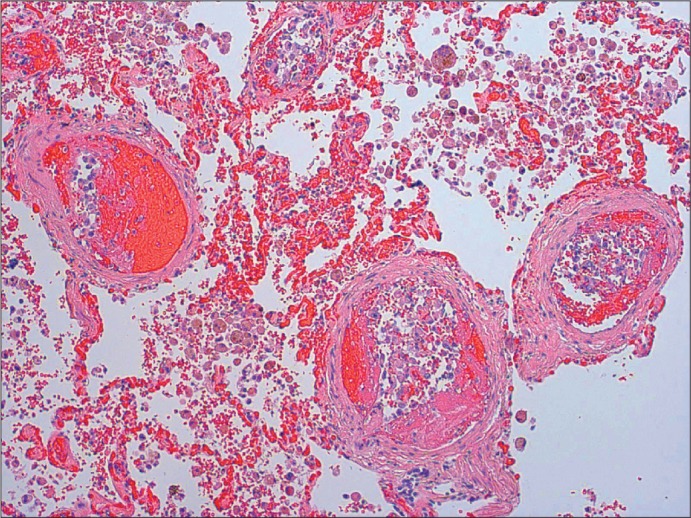
Microthrombi within lung vasculature containing tumor cells (H&E, 100×).
DISCUSSION
MRT of the kidney usually affects children <2 years of age (1). It comprised 1.8% of all pediatric renal tumors in the National Wilms' Tumor Study (2). In 1978, MRT of the kidney was described as a “rhabdomyosarcomatoid variant of Wilms tumor” by Beckwith and Palmer because the cells resembled rhabdomyoblasts (3), but further studies failed to demonstrate myogenic differentiation (1), and eventually MRT was recognized as a distinct clinicopathologic entity (4). These tumors have also been reported to arise in the urinary bladder, gastrointestinal tract, mediastinum, liver, soft tissue, orbit, uterus, and central nervous system (5, 6), and MRT may be “pure” or associated with other malignant patterns (as “composite” tumors) (1).
Adult renal MRT is even less common, and only six cases have been reported (1, 7–11). Age at diagnosis has ranged from 32 to 60 years with no predominant gender. One patient was Asian, one was Caucasian-Hispanic (Spanish), and the remaining four were Caucasian non-Hispanic; all patients presented with symptoms related to the renal mass or to lung metastases. The postdiagnosis survival time was usually only a few months (1). Histologically, all tumors contained noncohesive cells with eccentric vesicular nuclei, prominent nucleoli, and fibrillar, eosinophilic cytoplasmic inclusions.
MRT is immunohistochemically perplexing because of frequent dual reactivity for vimentin, a mesenchymal marker, and cytokeratin, an epithelial marker (12); this dichotomy raises interesting questions regarding the origin of these unusual tumors. Chromosome 22q11.2 deletions or mutations eventually led to the discovery of INI-1 as the tumor suppressor gene mutation involved in renal and extrarenal MRTs (13), and these studies have firmly established MRT as a distinct entity (14). Furthermore, one fascinating hypothesis is that there is no single cell of origin for MRT, and that diverse cell types (even tumor cells) may give rise to an MRT as a consequence of congenital and/or acquired mutations involving the INI-1 gene.
Criteria essential for pathologic diagnosis of MRT include characteristic histology showing rhabdoid features (as described above) and immunohistochemistry indicating loss of INI-1 protein nuclear expression. Although not essential for diagnosis, negative myogenic markers and electron microscopic confirmation of cytoplasmic intermediate filaments in swirls or whorls provide additional support for this diagnosis. Also, loss of INI-1 is helpful to differentiate MRT from other primary renal neoplasms (15) with rhabdoid differentiation including clear cell, transitional cell, papillary, chromophobe, and collecting duct carcinomas (16–18).
Intravascular coagulopathy associated with a pulmonary tumor embolism syndrome also contributed to this patient's death. Pulmonary thrombotic microangiopathy is uncommon, reported in only 3% to 26% of autopsied patients with solid tumors (19–22), and is usually only diagnosed postmortem (23). In this condition, embolic tumor cells obstruct pulmonary vessels, activate the coagulation cascade (24), and produce inflammatory mediators promoting thrombosis and intimal proliferation (23). This disorder results in increased vascular resistance, secondary pulmonary hypertension (24, 25), and right-sided heart failure. Such patients generally have widely disseminated cancer, and their prognosis is dismal.
References
- 1.Peng HQ, Stanek AE, Teichberg S, Shepard B, Kahn E. Malignant rhabdoid tumor of the kidney in an adult: a case report and review of the literature. Arch Pathol Lab Med. 2003;127(9):e371–e373. doi: 10.5858/2003-127-e371-MRTOTK. [DOI] [PubMed] [Google Scholar]
- 2.Weeks DA, Beckwith JB, Mierau GW, Luckey DW. Rhabdoid tumor of kidney. A report of 111 cases from the National Wilms' Tumor Study Pathology Center. Am J Surg Pathol. 1989;13(6):439–458. [PubMed] [Google Scholar]
- 3.Beckwith JB, Palmer NF. Histopathology and prognosis of Wilms tumors: results from the First National Wilms' Tumor Study. Cancer. 1978;41(5):1937–1948. doi: 10.1002/1097-0142(197805)41:5<1937::aid-cncr2820410538>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 4.Palmer NF, Sutow W. Clinical aspects of the rhabdoid tumor of the kidney: a report of the National Wilms' Tumor Study Group. Med Pediatr Oncol. 1983;11(4):242–245. doi: 10.1002/mpo.2950110407. [DOI] [PubMed] [Google Scholar]
- 5.Parham DM, Weeks DA, Beckwith JB. The clinicopathologic spectrum of putative extrarenal rhabdoid tumors. An analysis of 42 cases studied with immunohistochemistry or electron microscopy. Am J Surg Pathol. 1994;18(10):1010–1029. doi: 10.1097/00000478-199410000-00005. [DOI] [PubMed] [Google Scholar]
- 6.Wick MR, Ritter JH, Dehner LP. Malignant rhabdoid tumors: a clinicopathologic review and conceptual discussion. Semin Diagn Pathol. 1995;12(3):233–248. [PubMed] [Google Scholar]
- 7.Lowe W, Weiss RM, Todd MB, True LD. Malignant rhabdoid tumor of the kidney in an adult. J Urol. 1990;143(1):110–112. doi: 10.1016/s0022-5347(17)39882-8. [DOI] [PubMed] [Google Scholar]
- 8.Clausen HV, Horn T, Anagnostaki L, Larsen S. Malignant rhabdoid tumour of the kidney in an adult: a case report with immunohistochemical and ultrastructural investigation. Scand J Urol Nephrol Suppl. 1994;157:123–128. [PubMed] [Google Scholar]
- 9.Ebbinghaus SW, Herrera G, Marshall ME. Rhabdoid tumor of the kidney in an adult: review of the literature and report of a case responding to interleukin-2. Cancer Biother. 1995;10(3):237–241. doi: 10.1089/cbr.1995.10.237. [DOI] [PubMed] [Google Scholar]
- 10.Caballero JM, Collera P, Marti L, Borrat P, Hoyuela C, Rodriguez Santiago J, Ristol J. Malignant rhabdoid tumor of the kidney in the adult. Actas Urol Esp. 1996;20(4):377–379. [PubMed] [Google Scholar]
- 11.Zhao G, Na R, Yang Y, Han R. Pure malignant rhabdoid tumor of the left kidney in an adult: a case report and review of the literature. Oncol Lett. 2013;5(5):1481–1484. doi: 10.3892/ol.2013.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang YN, Kim SP, Jang BK. A primary malignant rhabdoid tumor in adult liver. Korean J Pathol. 2013;47(5):486–488. doi: 10.4132/KoreanJPathol.2013.47.5.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Versteege I, Sévenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A, Delattre O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394(6689):203–206. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
- 14.Ogino S, Ro TY, Redline RW. Malignant rhabdoid tumor: A phenotype? An entity?—A controversy revisited. Adv Anat Pathol. 2000;7(3):181–190. doi: 10.1097/00125480-200007030-00007. [DOI] [PubMed] [Google Scholar]
- 15.Perry A, Fuller CE, Judkins AR, Dehner LP, Biegel JA. INI1 expression is retained in composite rhabdoid tumors, including rhabdoid meningiomas. Mod Pathol. 2005;18(7):951–958. doi: 10.1038/modpathol.3800375. [DOI] [PubMed] [Google Scholar]
- 16.Kumar S, Kumar D, Cowan DF. Transitional cell carcinoma with rhabdoid features. Am J Surg Pathol. 1992;16(5):515–521. doi: 10.1097/00000478-199205000-00011. [DOI] [PubMed] [Google Scholar]
- 17.Weeks DA, Beckwith JB, Mierau GW, Zuppan CW. Renal neoplasms mimicking rhabdoid tumor of kidney. A report from the National Wilms' Tumor Study Pathology Center. Am J Surg Pathol. 1991;15(11):1042–1054. doi: 10.1097/00000478-199111000-00003. [DOI] [PubMed] [Google Scholar]
- 18.Yusuf Y, Belmonte AH, Tchertkoff V. Fine needle aspiration cytology of a recurrent malignant tumor of the kidney with rhabdoid features in an adult. A case report. Acta Cytol. 1996;40(6):1313–1316. doi: 10.1159/000334028. [DOI] [PubMed] [Google Scholar]
- 19.Roberts KE, Hamele-Bena D, Saqi A, Stein CA, Cole RP. Pulmonary tumor embolism: a review of the literature. Am J Med. 2003;115(3):228–232. doi: 10.1016/s0002-9343(03)00305-x. [DOI] [PubMed] [Google Scholar]
- 20.Kane RD, Hawkins HK, Miller JA, Noce PS. Microscopic pulmonary tumor emboli associated with dyspnea. Cancer. 1975;36(4):1473–1482. doi: 10.1002/1097-0142(197510)36:4<1473::aid-cncr2820360440>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 21.Winterbauer RH, Elfenbein IB, Ball WC., Jr. Incidence and clinical significance of tumor embolization to the lungs. Am J Med. 1968;45(2):271–290. doi: 10.1016/0002-9343(68)90044-2. [DOI] [PubMed] [Google Scholar]
- 22.Keenan NG, Nicholson AG, Oldershaw PJ. Fatal acute pulmonary hypertension caused by pulmonary tumour thrombotic microangiopathy. Int J Cardiol. 2008;124(1):e11–e13. doi: 10.1016/j.ijcard.2006.11.162. [DOI] [PubMed] [Google Scholar]
- 23.Sakashita N, Yokose C, Fujii K, Matsumoto M, Ohnishi K, Takeya M. Pulmonary tumor thrombotic microangiopathy resulting from metastatic signet ring cell carcinoma of the stomach. Pathol Int. 2007;57(6):383–387. doi: 10.1111/j.1440-1827.2007.02111.x. [DOI] [PubMed] [Google Scholar]
- 24.Pinckard JK, Wick MR. Tumor-related thrombotic pulmonary microangiopathy: review of pathologic findings and pathophysiologic mechanisms. Ann Diagn Pathol. 2000;4(3):154–157. doi: 10.1016/s1092-9134(00)90038-8. [DOI] [PubMed] [Google Scholar]
- 25.Von Herbay A, Illes A, Waldherr R, Otto HF. Pulmonary tumor thrombotic microangiopathy with hypertension. Cancer. 1990;66(3):587–592. doi: 10.1002/1097-0142(19900801)66:3<587::aid-cncr2820660330>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]