

Abstract
Retinal ischemia–reperfusion (IR) injury remains a common cause of blindness and has a final pathway of retinal ganglion cell (RGC) death by apoptosis and necrosis. RGC apoptosis was intensively studied in IR injury, while RGC necrosis did not receive nearly enough consideration since it was viewed as an accidental and unregulated cellular event. However, there is evidence that necrosis, like apoptosis, can be implemented by a programmed mechanism. In this study, we tested the role of RGC programmed necrosis (necroptosis) in IR-induced retinal injury. We employed the mouse model of retinal IR injury for in vivo experiments. The oxygen and glucose deprivation (OGD) model was used as an IR model in vitro. Primary RGCs were isolated by an immunopanning technique. Necrostatin 1 (Nec1) was used to inhibit necroptosis in in vitro and in vivo experiments. The changes in gene expression were assessed by quantitative RT-PCR. The distribution of proteins in the retina and in RGC cultures was evaluated by immunohistochemistry and immunocytochemistry, respectively. Our data suggest that proteins (Ripk1 and Ripk3), which initiate necroptosis, were present in normal and ischemic RGCs. Treatment with Nec1 significantly reduced retinal damage after IR. Increased RGC survival and reduced RGC necrosis following OGD were observed in Nec1-treated cultures. We found significantly reduced expression of genes coding pro-inflammatory markers Il1b, Ccl5, Cxcl10, Nos2 and Cybb in Nec1-treated ischemic retinas. Thus, our findings suggest that RGC necroptosis contributes to retinal damage after IR through direct loss of cells and induction of associated inflammatory responses.
Keywords: ischemia-reperfusion, retinal damage, necroptosis, retinal ganglion cells, Ripk1, Ripk3, necrostatin 1
1. Introduction
Retinal ischemia-reperfusion (IR) injury is a clinical condition which remains a common cause of visual impairment and blindness, due to relatively ineffective treatments (Osborne et al., 2004). The role of IR was proposed in many retinal disorders including glaucoma, anterior ischemic optic neuropathy, diabetic retinopathy, and traumatic optic neuropathy (Athappilly et al., 2008; Bresnick et al., 1975; Hayreh, 2013; Osborne et al., 1999). Thus, understanding the events involved in IR neuronal injury can provide us with clinically effective treatments for many retinal diseases. Necrosis and apoptosis are noticeable features of retinal damage after IR (Dvoriantchikova et al., 2010a; Fujita et al., 2009; Osborne et al., 2004). Since apoptosis is executed by programmed mechanisms and can be regulated, significant attention was given to this type of cell death (Ko et al., 2011; Lam et al., 1999; Renwick et al., 2006; Rosenbaum et al., 1997). At the same time, necrosis did not receive nearly enough consideration, because it was viewed as an accidental and unregulated cellular event. Accidental necrosis occurs after strong stresses, such as a burn, extremely low temperature, compression, as well as long-term absence of glucose and oxygen in the tissue (ischemia), which are rare events in tissues(Degterev and Yuan, 2008; Galluzzi and Kroemer, 2008). However, necrotic cells can also be observed in tissues when such stresses are absent and, therefore, necrotic cell death in this instance cannot be accidental. It was reported that retinal cells undergo necrosis rather than apoptosis in the first three days after IR in the transient retinal ischemia model (Dvoriantchikova et al., 2010a; Fujita et al., 2009). This result is of great importance because endogenous factors released from the necrotic cells can initiate the prolonged neurotoxic pro-inflammatory response in the retina and, thus, can mediate retinal damage after IR (Challa and Chan, 2010; Dvoriantchikova et al., 2010a; Dvoriantchikova et al., 2011).
Now we know that necrosis, like apoptosis, can be executed by programmed mechanisms (Degterev and Yuan, 2008; Galluzzi and Kroemer, 2008; Takahashi et al., 2012; Vandenabeele et al., 2010). This form of necrotic cell death is called necroptosis (Degterev and Yuan, 2008; Galluzzi and Kroemer, 2008; Takahashi et al., 2012; Vandenabeele et al., 2010). Programmed necrosis (necroptosis) occurs after ligation of death receptors and formation of the necrosome, which is primarily composed of Ripk1 and Ripk3 proteins (Degterev and Yuan, 2008; Galluzzi and Kroemer, 2008; Takahashi et al., 2012; Vandenabeele et al., 2010). Necrostatin 1 (Nec1), an inhibitor of necroptosis, suppresses programmed necrosis by binding to Ripk1 and, thus, preventing the formation of the Ripk1 and Ripk3 complex (Degterev et al., 2008; Degterev et al., 2005). Nec1 was successfully applied previously to prevent IR-induced damage in many tissues, including the retina (Degterev et al., 2005; Rosenbaum et al., 2010; You et al., 2008). Rosenbaum et al. showed that treatment with Nec1 led to significant protection from injury and functional improvement of the inner retina, compared with vehicle-treated controls (Rosenbaum et al., 2010). However, the role of retinal ganglion cell (RGC) necroptosis in retinal IR injury was not investigated in this study. Retinal IR injury has a final pathway of RGC death (Osborne et al., 2004). Although apoptosis was initially implicated to be an important form of RGC death after IR (Ko et al., 2011; Lam et al., 1999; Renwick et al., 2006; Rosenbaum et al., 1997), our data suggest that RGC necrosis should play a role in IR-induced retinal injury (Dvoriantchikova et al., 2010a). We demonstrated in this study that RGC programmed necrosis (necroptosis) contributes to retinal damage after IR.
2. Materials and Methods
2.1. Materials
All chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO), Life Technologies (Grand Island, NY), and Thermo Scientific (Rockford, IL). We use necrostatin 1 derivative 7-Cl-Nec1 (hereinafter referred to as Nec1), which was synthesized as previously described (Degterev et al., 2005).
2.2. Animals
All postsurgical care and experiments were performed in compliance with the NIH Guide for the Care and Use of Laboratory Animals, the Association for Research in Vision and Ophthalmology statement for use of animals in ophthalmic and vision research. The University of Miami Institutional Animal Care and Use Committee (IACUC) specifically approved this study. We used 3-month-old male C57BL/6J mice or 12-day-old pups. Mice were housed under standard conditions of humidity and temperature, with a 12 hours light and dark cycle as well as free access to food and water. Animals were sacrificed by CO2 inhalation under anesthesia.
2.3. Transient retinal ischemia
Anesthesia was induced with isoflurane and maintained for 45 minutes. Body temperature was held constant at 37°C using a temperature-controlled heating pad. Retinal ischemia was induced for 45 min by introducing into the anterior chamber of the left eye a 33-gauge needle attached to a normal saline-filled (0.9% NaCl) reservoir raised above the mouse to increase intraocular pressure (IOP, increased to 120 mmHg). The right eye was cannulated and maintained at normal IOP to serve as a control. Retinal ischemia was considered complete if a whitening of the anterior segment of the eye and blanching of the retinal arteries were observed by microscopic examination. Erythromycin ophthalmic ointment (Fera Pharmaceuticals, Locust Valley, NY) was applied to the conjunctival sac after needle removal.
2.4. Immunohistochemistry of the whole retina flatmounts and counting of ganglion cell layer neurons
Eyes were enucleated upon euthanasia and fixed in a 4% paraformaldehyde (PF). The retinas were removed after 1 hour, and were then cryoprotected overnight in 30% sucrose. Cryoprotected retinas were frozen in liquid nitrogen and were unfrozen at room temperature. After the freeze–thaw cycle was repeated three times, the retinas were rinsed three times with 0.1 M Tris buffer, blocked for 1 hour in buffer (5% donkey serum, 0.1% Triton X-100 in 0.1 M Tris buffer) and incubated overnight with FITC –conjugated Neuronal Nuclei (NeuN) antibody (1:300; Chemicon, Billerica, MA). After rinsing three times with 0.1 M Tris buffer, the retinas were flatmounted, coverslipped, and NeuN positive neurons in the ganglion cell layer (GCL) were imaged using a Leica TSL AOBS SP5 confocal microscope (Leica Microsystems, Exton, PA). Individual retinas were sampled randomly to collect a total of 20 images located at the same eccentricity in the four retinal quadrants. NeuN-positive neurons were counted using ImageJ software. Cell loss in the ischemic retinas was calculated as percentile of the mean cell density in normotensive fellow control eyes.
2.5. Retinal ganglion cell (RGC) primary cultures
Primary retinal ganglion cells (RGCs) were isolated according to the two-step immunopanning protocol as described previously (Dvoriantchikova et al., 2012; Dvoriantchikova et al., 2011). Briefly, the whole retinas were incubated in papain solution (16.5 U/mL) for 30 minutes. In the next step, the macrophage and endothelial cells were removed from the cell suspension by panning with the anti-macrophage antiserum (Accurate Chemical, Westbury, NY). RGCs were bound to the panning plates containing the antibody against Thy1.2 and were then released by incubation with trypsin solution.
2.6. Oxygen and Glucose Deprivation (OGD) Model
Primary RGCs were plated on poly D-lysine and laminin (both from Sigma-Aldrich; St. Louis, MO) treated cover slips in 24-well plate and were cultured in media (Neurobasal/B27; Life Technologies, Grand Island, NY) one day before the experiment. Neurobasal/B27 media was then replaced with OGD media containing: 1.8 mM CaCl2, 0.814 mM MgCl2, 5.33 mM KCl, 26.19 mM NaHCO3, 68.97 mM NaCl, 0.906 mM NaH2PO4–H2O, and 10 mM sucrose (pH 7.4). OGD media was deoxygenated before the experiment by bubbling for at least 1 hour with 95% N2/5% CO2. Primary RGCs were deprived of oxygen using an anaerobic chamber (5% CO2, and 95% N2) for 4 hours at 37°C. After the oxygen and glucose deprivation, OGD media was replaced with “sham media,” which had the same composition, except that sucrose was replaced with 10 mM D-Glucose, and cultures were returned to a normoxic environment. Parallel cultures were exposed to oxygenated “sham media” in a normoxic incubator (37°C; atmosphere 5% CO2) to serve as controls.
2.7. Cell death assay
After treatment, the apoptotic and necrotic neurons were labeled using Vybrant Apoptosis Assay Kit (Life Technologies, Grand Island, NY). Cells were imaged using a Leica TSL AOBS SP5 confocal microscope (Leica Microsystems, Exton, PA). Individual glasses were sampled randomly to collect a total of 6 images using a 20X objective lens. The apoptotic and necrotic RGCs were counted semiautomatically using ImageJ software. The percentage of apoptotic and necrotic RGCs relative to the total number of counted cells on the glass was determined. The experiment was repeated at least three times.
2.8. Immunohistochemistry
Fixed retinas were sectioned to a thickness of 100 μm with a vibratome (Leica Microsystems, Exton, PA) and immunostained as described earlier (Dvoriantchikova et al., 2009a; Dvoriantchikova et al., 2009b; Dvoriantchikova et al., 2010b). Briefly, sections were permeabilized with 0.3% Triton X-100 in PBS for one hour, rinsed three times in PBS, blocked in buffer (5% donkey serum, 2% BSA and 0.15% Tween-20 in PBS) for 1 hour and incubated overnight with anti-Ripk1 (1:250) and anti-Ripk3 (1:250) specific antibodies (both from GeneTex, Inc, Irvine, CA) as well as anti-Tubb3 antibody (β-tubulin III antibody, 1:500; Covance, Princeton, NJ), followed by species-specific secondary fluorescent antibodies (Life Technologies, Grand Island, NY). Control sections were incubated without primary antibodies. Imaging was performed with a Leica TSL AOBS SP5 confocal microscope (Leica Microsystems, Exton, PA).
2.9. Immunocytochemistry
Cultured primary RGCs were fixed in 4% PF and blocked in buffer containing 5% donkey serum, 0.15% Tween-20 and 1xPBS (pH 7.4). Cells were then incubated with anti-Tubb3 antibody (β-tubulin III antibody, 1:500; Covance, Princeton, NJ), anti-Ripk1 and anti-Ripk3 antibodies (1:250; both from GeneTex, Inc, Irvine, CA), followed by species-specific secondary fluorescent antibodies (Life Technologies, Grand Island, NY). Negative controls were incubated with secondary antibody only. Imaging was performed with a confocal microscope (Leica TSL AOBS SP5; Leica Microsystems, Exton, PA).
2.10. Quantitative RT-PCR analysis
Quantitative RT-PCR analysis was accomplished using gene-specific primers (Table 1). Specifically, total RNA was extracted from sham-operated and experimental retinas using Absolutely RNA Microprep kit (Agilent Technologies, Santa Clara, CA), and was reverse transcribed using the Reverse Transcription System (Promega, Madison, WI) to synthesize cDNA. Quantitative RT-PCR was performed in the Rotor-Gene Q Cycler (Qiagen, Valencia, CA) using the SYBR green PCR MasterMix (Qiagen, Valencia, CA). For each gene, relative expression was calculated by comparison with a standard curve, following normalization to the housekeeping gene succinate dehydrogenase, subunit A (Sdha) expression.
Table 1.
List of PCR primers
| Gene | Oligonucleotides | |
|---|---|---|
| Il1b | Forward | GACCTTCCAGGATGAGGACA |
| Reverse | AGGCCACAGGTATTTTGTCG | |
| Tnf | Forward | CAAAATTCGAGTGACAAGCCTG |
| Reverse | GAGATCCATGCCGTTGGC | |
| Ccl2 | Forward | AGGTCCCTGTCATGCTTCTG |
| Reverse | ATTTGGTTCCGATCCAGGTT | |
| Ccl5 | Forward | AGCAGCAAGTGCTCCAATCT |
| Reverse | ATTTCTTGGGTTTGCTGTGC | |
| Cxcl10 | Forward | GCTGCAACTGCATCCATATC |
| Reverse | CACTGGGTAAAGGGGAGTGA | |
| Icam1 | Forward | TGGTGATGCTCAGGTATCCA |
| Reverse | CACACTCTCCGGAAACGAAT | |
| Cybb | Forward | GACTGCGGAGAGTTTGGAAG |
| Reverse | ACTGTCCCACCTCCATCTTG | |
| Nos2 | Forward | CAGAGGACCCAGAGACAAGC |
| Reverse | TGCTGAAACATTTCCTGTGC | |
| Thy1 | Forward | CAAGGATGAGGGCGACTAC |
| Reverse | TCTTGGGGAGGGAGTCAGC | |
| Gfap | Forward | AGAAAGGTTGAATCGCTGGA |
| Reverse | CGGCGATAGTCGTTAGCTTC | |
| Cd11b | Forward | ACAATGTGACCGTATGGGATC |
| Reverse | GCAAACGCAGAGTCATTAAAC | |
| Sdha | Forward | ACACAGACCTGGTGGAGACC |
| Reverse | GCACAGTCAGCCTCATTCAA | |
2.11. Statistical analysis
Statistical analysis was performed using the Student t-test or with the one-way ANOVA test. P-values less than or equal to 0.05 were considered statistically significant.
3. Results
Since the presence of Ripk1 and Ripk3 proteins in the cell is necessary to initiate programmed necrosis, we tested the presence of these proteins in sham-operated and ischemic retinas. Retinal ischemia was induced by unilateral elevation of intraocular pressure for 45 minutes and retinas were collected 6 hours post-reperfusion. The spatial distribution of Ripk1 and Ripk3 proteins was evaluated by immunohistochemistry in fixed retinas. Our data indicate the presence of Ripk1 and Ripk3 proteins throughout the retinal layers (Figure 1A, B). In particular, Ripk1- and Ripk3 specific immunostaining was evident in the ganglion cell layer, which contains retinal ganglion cells (RGCs). To verify RGC-specificity of Ripk1 and Ripk3 labeling, primary RGCs isolated from retinas by an immunopanning technique were deprived of oxygen and glucose (OGD) for 4 hours in an anaerobic chamber. Three hours after reoxygenation, cells were fixed and immunolabeled with Ripk1- and Ripk3-specific antibodies. Tubulin β-III (Tubb3) was used as an RGC-specific marker. Control RGC cultures were maintained in oxygenated and glucose contained “sham media”. As shown in Figure 2(A and B), substantial Ripk1- and Ripk3-specific immunostaining localized to the somata of control and the OGD-treated RGCs. Negative controls incubated with secondary antibody only did not show any specific immunostaining (data not shown).
Figure 1.

The necrosome subunits Ripk1 and Ripk3 are present in RGCs at the protein level: Immunohistochemistry showed accumulation of (A) Ripk1 and (B) Ripk3 proteins in RGCs of sham-operated and ischemic (6 hours after reperfusion) retinas. Anti-Tubb3 (tubulin β-III) antibodies were used to identify RGCs. DAPI was used to label DNA and thus allowed visualization of the nucleus of the cell. The presence of Ripk1 and Ripk3 can be seen in the RGC body as well as in RGC processes (GCL, ganglion cell layer; ONL, outer nuclear layer INL, inner nuclear layer). Scale bar: 100 μm.
Figure 2.

Immunocytochemical results for (A) Ripk1 and (B) Ripk3 proteins show that accumulation of the proteins in control and OGD-treated RGCs was consistent with the immunohistochemistry data. Tubulin β-III (Tubb3) was used as an RGC marker. Scale bar: 50 μm.
Since the inhibitor of necroptosis, Nec1, suppresses programmed necrosis by inhibiting Ripk1 and Ripk3 complex formation, mice subjected to IR were treated with Nec1. The mice were intraperitoneally injected with Nec1 (2 μg/g body of mouse) one hour before IR, and once every 24 hours until sacrifice. Control animals were vehicle-treated. On the seventh day after reperfusion, animals were euthanized and whole retina flatmounts were stained for the neuronal marker, NeuN, to quantify the number of surviving neurons in the ganglion cell layer (GCL). We observed that retinas from experimental eyes of Nec1-treated mice had significantly higher numbers of surviving NeuN-positive neurons (66 ± 6%) in the GCL compared to vehicle-treated mice (48 ± 5%, P < 0.05, Figure 3). Since the ganglion cell layer contains RGCs and displaced amacrine cells, to evaluate the direct role of RGC necroptosis in IR-induced retinal injury, we turned to primary RGC cultures. First, to eliminate the chance of the primary RGC culture being contaminated by other cells, we tested the purity of cells (which were isolated by immunopanning) utilizing specific markers for RGCs (Thy1), astrocytes (Gfap), and microglia/macrophages (Cd11b) in the quantitative RT-PCR reaction. The calculated purity of isolated RGCs was 95% to 99% (Figure 4A). These RGCs were deprived of oxygen and glucose (OGD) for 4 hours in an anaerobic chamber in the presence of Nec1 (20 μM) or vehicle (control). The numbers of live RGCs were determined 24 hours after OGD. We found that only 12% (12±1%, P<0.01) of RGCs survived after OGD compared to RGC cultures maintained in the “sham media” (Figure 4B). At the same time, the level of RGC survival was significantly higher in OGD-treated RGC cultures in the presence of Nec1 (59±7%, P<0.01) compared to RGCs maintained in “sham media” (Figure 4B). We also determined the percentage of live, necrotic and apoptotic RGCs relative to the total number of counted cells on the same cover slip. We found that while OGD promoted significant RGC death predominantly by necrosis (live RGCs: 20±1%; apoptotic RGCs: 27±2%; necrotic RGCs: 53±2%; Figure 4C), treatment with Nec1 increased RGC survival and decreased necrotic cell death following OGD (live RGCs: 45±2%; apoptotic RGCs: 19±3%; necrotic RGCs: 36±2%; Figure 4C). Thus, the level of retinal damage after IR can be significantly reduced by preventing programmed RGC necrosis (necroptosis).
Figure 3.
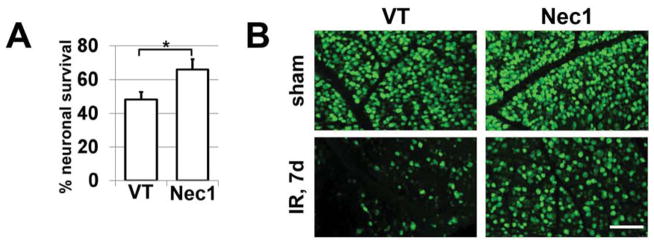
Treatment with necroptosis inhibitor, Nec1, results in neuroprotective effects in the ganglion cell layer (GCL) of retinas after IR. A) Retinal ischemia was induced for 45 min in the left eye of Nec1- and vehicle-treated (VT) animals. Retinas of ischemic and sham-operated (right eye of the same animal) eyes were collected 7 days after reperfusion and immunolabeled with NeuN antibodies. NeuN-positive neurons were counted in flatmounted retinas, and GCL neuron loss in ischemic retinas was calculated as a percentile of the mean cell density in the retinas of normotensive fellow control eyes of the same animals (*P<0.05%). B) Representative confocal images of NeuN-labeled GCLs (green) in flatmounted controls (sham) and ischemic retinas were taken 7 days after reperfusion (VT - vehicle-treated animals). Scale bar: 100 μm.
Figure 4.

Inhibiting necroptosis in RGCs promotes neuronal survival and reduces necrotic cell death after OGD. RGC cultures were deprived of oxygen and glucose for 4 hours followed by 24 hours of re-oxygenation in either the presence of Nec1 (20 μM) or vehicle (control, **P < 0.01, *P < 0.05). A) Primary RGCs isolated by immunopanning were evaluated for the presence of specific markers for RGCs (Thy1), astrocytes (Gfap), and microglia/macrophages (Cd11b) using quantitative RT-PCR. Results are presented as a percentage of Thy1 expression (**P < 0.01, n = 6). B) Total numbers of survived RGCs were counted in OGD-treated cultures in the presence of Nec1 or vehicle. RGC survival in OGD-treated cells was calculated as percentile of the mean number of survived RGCs in the “sham media”. C) The percentage of live, apoptotic and necrotic RGCs relative to the total number of counted cells on the same cover slip was calculated. D) Representative confocal images are presented. Apoptotic RGCs were identified using Annexin V (green) as a marker of apoptotic cells. To recognize necrotic RGCs, we used propidium iodide (PI, red) as a marker for cells, which have lost membrane integrity (necrosis). Scale bar: 100 μm.
Since necrosis triggers a pro-inflammatory response in tissue, we next asked whether RGC programmed necrosis (necroptosis) mediates a neurotoxic pro-inflammatory response in the retina after IR. To this end, mice were intraperitoneally injected with Nec1 (2 μg/g body of mouse) one hour before IR. Vehicle-treated animals were used as controls. Experimental and sham-operated retinas were collected 6 hours post-reperfusion, since most changes in pro-inflammatory activity in the ischemic retina typically occur shortly after reperfusion. The activation of the pro-inflammatory genes coding for Tnf and Il1b cytokines, Ccl2, Ccl5 and Cxcl10 chemokines, the cell adhesion molecule Icam1, inducible nitric oxide synthase Nos2 as well as Nox2 (Cybb) gene encoding catalytic subunit of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase protein complex, was evaluated at the level of RNA expression by quantitative RT-PCR. We detected transcriptional upregulation of the tested genes in ischemic retinas treated with Nec1 and in ischemic retinas of vehicle-treated animals compared to control retinas in sham-operated eyes (Figure 5). However, while reduced expression of all tested pro-inflammatory markers was evident in Nec1-treated ischemic retinas, we found statistically significant reduced expression of Il1b cytokine, Ccl5 and Cxcl10 chemokines, as well as expression of Nos2 and Cybb genes compared to vehicle-treated ischemic retinas (Figure 5). Therefore, we found that RGC necroptosis contributes to retinal inflammation after IR.
Figure 5.

Inhibiting necroptosis with Nec1 suppresses induction of pro-inflammatory response in the retina after IR. Expression of genes coding for markers of inflammation in the retina was measured using quantitative RT-PCR in sham-operated controls and experimental retinas 6 hours after IR. Results are expressed as a percentage of the corresponding value in the sham-operated eye of vehicle-treated (VT) animals (**P < 0.01, *P < 0.05, n = 6).
4. Discussion
Retinal ischemia–reperfusion (IR) injury remains a common cause of blindness, which has a final pathway of RGC death (Osborne et al., 2004). Retinal IR injury results in a prolonged period of RGC death by necrosis and apoptosis (Dvoriantchikova et al., 2010a; Fujita et al., 2009). Previously, necrosis was viewed as an accidental and unregulated cellular event. However, accumulating evidence suggests that necrosis, like apoptosis, can be executed by programmed mechanisms (Degterev and Yuan, 2008; Galluzzi and Kroemer, 2008; Takahashi et al., 2012; Vandenabeele et al., 2010). This form of cell death was called necroptosis (Degterev and Yuan, 2008; Galluzzi and Kroemer, 2008; Takahashi et al., 2012; Vandenabeele et al., 2010). The critical role of necroptosis in retinal IR injury was shown previously (Rosenbaum et al., 2010). However, the role of RGC necroptosis in IR-induced retinal injury was not investigated. Our data suggest that the Ripk1 and Ripk3 proteins, which are required to initiate necroptosis (Degterev and Yuan, 2008; Galluzzi and Kroemer, 2008; Takahashi et al., 2012; Vandenabeele et al., 2010), are present in control and ischemic RGCs. It should be noted that the pattern of Ripk3 protein distribution in sham-operated and IR mouse retinas coincides with Ripk3 protein distribution in sham-operated and ischemic rat retinas (Huang et al., 2013). We found that inhibiting the activation of the Ripk1/Ripk3-pathway in RGCs, using Nec1, significantly reduced IR-induced retinal damage. Treatment with Nec1 also significantly reduced the pro-inflammatory response in the retina after IR. Thus, RGC necroptosis suppression prevents damage and inflammation in retinas challenged by IR.
Apoptotic and necrotic cell death types constitute the damaged region in many retinal neuropathologies for which the role of IR was proposed (Athappilly et al., 2008; Bresnick et al., 1975; Hayreh, 2013; Osborne et al., 1999). Thus, understanding the apoptotic and necrotic pathways involved in IR-induced neuronal injury can provide us with clinically effective treatments for many retinal diseases. While necrotic cell death was considered accidental, rare, and uncontrolled, significant attention was given to apoptosis because it was supposed that retinal damage could be significantly reduced by preventing apoptosis (Ko et al., 2011; Lam et al., 1999; Renwick et al., 2006; Rosenbaum et al., 1997). However, we now know that retinal necrosis considerably surpasses apoptotic cell death over a long period of time after IR (Dvoriantchikova et al., 2010a; Fujita et al., 2009) and, thus, this subtype of necrotic cell death requires considerable attention. The data presented in this study and published literature (Rosenbaum et al., 2010) indicate that this subtype of necrotic cell death occurs in the retina after IR is executed by programmed mechanisms. RGC death is a characteristic feature of many retinal neuropathologies and conditions including IR injury (Osborne et al., 2004; Osborne et al., 1999; You et al., 2013). While many studies have previously focused on RGC death by apoptosis (Ko et al., 2011; Lam et al., 1999; Renwick et al., 2006; Rosenbaum et al., 1997), we demonstrated that RGC programmed necrosis (necroptosis) also significantly contributes to IR-induced retinal damage. Thus, therapy based only on the inhibition of RGC apoptosis may not be sufficient to obtain significant neuroprotection. Importantly, since inhibition of apoptosis in the absence of NF-κB activity does not prevent cell death, but promotes cell death by necroptosis (Degterev and Yuan, 2008; Galluzzi and Kroemer, 2008; Takahashi et al., 2012; Vandenabeele et al., 2010), anti-apoptotic therapy may even have opposite effects: the level of damage in the retina may be higher after anti-apoptotic treatment. Thus, not only apoptosis, but also necroptosis requires significant attention in the IR-challenged retina. It should be noted that the critical role of retinal necroptosis is not limited only to IR injury. The direct role of necroptosis has already been shown in models of retinal detachment (Trichonas et al., 2010) and retinitis pigmentosa (Murakami et al., 2012).
Accumulating evidence suggests that necrotic and apoptotic cells have different influences on the likelihood of death of the cells near them that have survived initial stress. It has been shown that apoptotic cell death turns off the production of pro-inflammatory mediators and stimulates the production of anti-inflammatory and neuroprotective factors from phagocytes (glial cells and infiltrating macrophages in IR retina) that have internalized apoptotic cells (Fadok et al., 2001; Krysko et al., 2006; Minghetti et al., 2005). Therefore, the apoptotic cell dies, but it increases the likelihood of survival for the cells located close to it. We demonstrated previously that a therapeutic strategy based on mimicking apoptotic cell death in the retina significantly reduced IR-induced damage and improved neurologic outcomes (Dvoriantchikova et al., 2009a). At the same time, endogenous factors liberated from necrotic cells are recognized by glial cells (astrocytes and microglia) and infiltrating macrophages as signals for “danger” (so called damage-associated molecular patterns or DAMPs) (Challa and Chan, 2010; Piccinini and Midwood, 2010). After engaging the DAMPs, pattern recognition receptors (PRRs) such as toll like receptors (e.g. TLR4) activate signaling cascades, which trigger inflammation and damage (Challa and Chan, 2010; Piccinini and Midwood, 2010). In other words, if the cell dies by necrosis, this leads to pro-inflammatory toxicity that stimulates death of the surrounding cells. Earlier we showed that DAMPs and PRRs promote retinal injury after IR (Dvoriantchikova et al., 2010b; Dvoriantchikova et al., 2011). In this study, we demonstrated that treatment with the inhibitor of necroptosis, Nec1, reduced expression of all tested genes coding for pro-inflammatory factors. Importantly, expression of genes coding Il1b cytokine, Ccl5 and Cxcl10 chemokines, as well as expression of inducible nitric oxide synthase (Nos2) and Cybb gene coding subunit of reactive oxygen species producing NAD(P)H oxidase, was reduced significantly compared to vehicle-treated controls. The negative role of these pro-inflammatory factors in retinal IR injury was demonstrated previously (Barakat et al., 2012; Tsujikawa et al., 1999; Yoneda et al., 2001). Thus, retinal necroptosis, and particularly RGC necroptosis, can indirectly affect the level of damage after IR.
5. Conclusion
Our findings suggest that suppressing RGC necroptosis in the IR-challenged retina reduces damage and promotes a neuroprotective environment and, thus, effectively protects the retina against IR injury. Therefore, since treatment for retinal IR is limited in part because of a lack of understanding of the molecular events leading to RGC death, a greater understanding of the role of necroptosis in retinal IR injury may aid in the development of specific therapies to treat this condition.
Highlights.
We tested the role of RGC necroptosis in ischemia-reperfusion-induced retinal injury.
The suppressing RGC necroptosis reduces retinal damage after ischemia-reperfusion.
RGC necroptosis mediates pro-inflammatory response in ischemic retina.
Acknowledgments
Supported in part by National Eye Institute/National Institutes of Health (NIH) Grant R01 EY022348 (DI), James and Esther King Biomedical Research Program Bridge Grant 3KB01-50988 (DI), National Institute of General Medical Sciences grants R01GM080356 and R01GM084205 (AD), NIH Center Core Grant P30EY014801, Research to Prevent Blindness Unrestricted Grant, Department of Defense (DOD- Grant#W81XWH-09-1-0675 and Grant# W81XWH-13-1-0048 ONOVA). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Eye Institute or the National Institutes of Health. The authors thank Eleut Hernandez, Andrea Rachelle C. Santos, Dagmara Danek and Gabriel Gaidosh for their expert assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Athappilly G, Pelak VS, Mandava N, Bennett JL. Ischemic optic neuropathy. Neurological research. 2008;30:794–800. doi: 10.1179/174313208X319107. [DOI] [PubMed] [Google Scholar]
- Barakat DJ, Dvoriantchikova G, Ivanov D, Shestopalov VI. Astroglial NF-kappaB mediates oxidative stress by regulation of NADPH oxidase in a model of retinal ischemia reperfusion injury. Journal of neurochemistry. 2012;120:586–597. doi: 10.1111/j.1471-4159.2011.07595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresnick GH, De Venecia G, Myers FL, Harris JA, Davis MD. Retinal ischemia in diabetic retinopathy. Archives of ophthalmology. 1975;93:1300–1310. doi: 10.1001/archopht.1975.01010020934002. [DOI] [PubMed] [Google Scholar]
- Challa S, Chan FK. Going up in flames: necrotic cell injury and inflammatory diseases. Cellular and molecular life sciences : CMLS. 2010;67:3241–3253. doi: 10.1007/s00018-010-0413-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nature chemical biology. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nature chemical biology. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- Degterev A, Yuan J. Expansion and evolution of cell death programmes. Nature reviews Molecular cell biology. 2008;9:378–390. doi: 10.1038/nrm2393. [DOI] [PubMed] [Google Scholar]
- Dvoriantchikova G, Agudelo C, Hernandez E, Shestopalov VI, Ivanov D. Phosphatidylserine-containing liposomes promote maximal survival of retinal neurons after ischemic injury. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2009a;29:1755–1759. doi: 10.1038/jcbfm.2009.95. [DOI] [PubMed] [Google Scholar]
- Dvoriantchikova G, Barakat D, Brambilla R, Agudelo C, Hernandez E, Bethea JR, Shestopalov VI, Ivanov D. Inactivation of astroglial NF-kappa B promotes survival of retinal neurons following ischemic injury. The European journal of neuroscience. 2009b;30:175–185. doi: 10.1111/j.1460-9568.2009.06814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvoriantchikova G, Barakat DJ, Hernandez E, Shestopalov VI, Ivanov D. Liposome-delivered ATP effectively protects the retina against ischemia-reperfusion injury. Molecular vision. 2010a;16:2882–2890. [PMC free article] [PubMed] [Google Scholar]
- Dvoriantchikova G, Barakat DJ, Hernandez E, Shestopalov VI, Ivanov D. Toll-like receptor 4 contributes to retinal ischemia/reperfusion injury. Molecular vision. 2010b;16:1907–1912. [PMC free article] [PubMed] [Google Scholar]
- Dvoriantchikova G, Grant J, Santos AR, Hernandez E, Ivanov D. Neuronal NAD(P)H oxidases contribute to ROS production and mediate RGC death after ischemia. Investigative ophthalmology & visual science. 2012;53:2823–2830. doi: 10.1167/iovs.12-9526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvoriantchikova G, Hernandez E, Grant J, Santos AR, Yang H, Ivanov D. The high-mobility group box-1 nuclear factor mediates retinal injury after ischemia reperfusion. Investigative ophthalmology & visual science. 2011;52:7187–7194. doi: 10.1167/iovs.11-7793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Guthrie L, Henson PM. Differential effects of apoptotic versus lysed cells on macrophage production of cytokines: role of proteases. Journal of immunology. 2001;166:6847–6854. doi: 10.4049/jimmunol.166.11.6847. [DOI] [PubMed] [Google Scholar]
- Fujita R, Ueda M, Fujiwara K, Ueda H. Prothymosin-alpha plays a defensive role in retinal ischemia through necrosis and apoptosis inhibition. Cell death and differentiation. 2009;16:349–358. doi: 10.1038/cdd.2008.159. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Kroemer G. Necroptosis: a specialized pathway of programmed necrosis. Cell. 2008;135:1161–1163. doi: 10.1016/j.cell.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Hayreh SS. Ischemic optic neuropathies - where are we now? Graefe’s archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2013;251:1873–1884. doi: 10.1007/s00417-013-2399-z. [DOI] [PubMed] [Google Scholar]
- Huang JF, Shang L, Zhang MQ, Wang H, Chen D, Tong JB, Huang H, Yan XX, Zeng LP, Xiong K. Differential neuronal expression of receptor interacting protein 3 in rat retina: involvement in ischemic stress response. BMC neuroscience. 2013;14:16. doi: 10.1186/1471-2202-14-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko ML, Chen CF, Peng PH, Peng YH. Simvastatin upregulates Bcl-2 expression and protects retinal neurons from early ischemia/reperfusion injury in the rat retina. Experimental eye research. 2011;93:580–585. doi: 10.1016/j.exer.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Krysko DV, D’Herde K, Vandenabeele P. Clearance of apoptotic and necrotic cells and its immunological consequences. Apoptosis : an international journal on programmed cell death. 2006;11:1709–1726. doi: 10.1007/s10495-006-9527-8. [DOI] [PubMed] [Google Scholar]
- Lam TT, Abler AS, Tso MO. Apoptosis and caspases after ischemia-reperfusion injury in rat retina. Investigative ophthalmology & visual science. 1999;40:967–975. [PubMed] [Google Scholar]
- Minghetti L, Ajmone-Cat MA, De Berardinis MA, De Simone R. Microglial activation in chronic neurodegenerative diseases: roles of apoptotic neurons and chronic stimulation. Brain research Brain research reviews. 2005;48:251–256. doi: 10.1016/j.brainresrev.2004.12.015. [DOI] [PubMed] [Google Scholar]
- Murakami Y, Matsumoto H, Roh M, Suzuki J, Hisatomi T, Ikeda Y, Miller JW, Vavvas DG. Receptor interacting protein kinase mediates necrotic cone but not rod cell death in a mouse model of inherited degeneration. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:14598–14603. doi: 10.1073/pnas.1206937109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne NN, Casson RJ, Wood JP, Chidlow G, Graham M, Melena J. Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Progress in retinal and eye research. 2004;23:91–147. doi: 10.1016/j.preteyeres.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Osborne NN, Ugarte M, Chao M, Chidlow G, Bae JH, Wood JP, Nash MS. Neuroprotection in relation to retinal ischemia and relevance to glaucoma. Survey of ophthalmology. 1999;43(Suppl 1):S102–128. doi: 10.1016/s0039-6257(99)00044-2. [DOI] [PubMed] [Google Scholar]
- Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators of inflammation. 2010 doi: 10.1155/2010/672395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renwick J, Narang MA, Coupland SG, Xuan JY, Baker AN, Brousseau J, Petrin D, Munger R, Leonard BC, Hauswirth WW, Korneluk RG, Tsilfidis C. XIAP-mediated neuroprotection in retinal ischemia. Gene therapy. 2006;13:339–347. doi: 10.1038/sj.gt.3302683. [DOI] [PubMed] [Google Scholar]
- Rosenbaum DM, Degterev A, David J, Rosenbaum PS, Roth S, Grotta JC, Cuny GD, Yuan J, Savitz SI. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. Journal of neuroscience research. 2010;88:1569–1576. doi: 10.1002/jnr.22314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum DM, Rosenbaum PS, Gupta A, Michaelson MD, Hall DH, Kessler JA. Retinal ischemia leads to apoptosis which is ameliorated by aurintricarboxylic acid. Vision research. 1997;37:3445–3451. doi: 10.1016/S0042-6989(96)00328-8. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB, Goossens V, Roelandt R, Van Hauwermeiren F, Libert C, Declercq W, Callewaert N, Prendergast GC, Degterev A, Yuan J, Vandenabeele P. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell death & disease. 2012;3:e437. doi: 10.1038/cddis.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trichonas G, Murakami Y, Thanos A, Morizane Y, Kayama M, Debouck CM, Hisatomi T, Miller JW, Vavvas DG. Receptor interacting protein kinases mediate retinal detachment-induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:21695–21700. doi: 10.1073/pnas.1009179107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujikawa A, Ogura Y, Hiroshiba N, Miyamoto K, Kiryu J, Tojo SJ, Miyasaka M, Honda Y. Retinal ischemia-reperfusion injury attenuated by blocking of adhesion molecules of vascular endothelium. Investigative ophthalmology & visual science. 1999;40:1183–1190. [PubMed] [Google Scholar]
- Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nature reviews Molecular cell biology. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- Yoneda S, Tanihara H, Kido N, Honda Y, Goto W, Hara H, Miyawaki N. Interleukin-1beta mediates ischemic injury in the rat retina. Experimental eye research. 2001;73:661–667. doi: 10.1006/exer.2001.1072. [DOI] [PubMed] [Google Scholar]
- You Y, Gupta VK, Li JC, Klistorner A, Graham SL. Optic neuropathies: characteristic features and mechanisms of retinal ganglion cell loss. Reviews in the neurosciences. 2013;24:301–321. doi: 10.1515/revneuro-2013-0003. [DOI] [PubMed] [Google Scholar]
- You Z, Savitz SI, Yang J, Degterev A, Yuan J, Cuny GD, Moskowitz MA, Whalen MJ. Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2008;28:1564–1573. doi: 10.1038/jcbfm.2008.44. [DOI] [PMC free article] [PubMed] [Google Scholar]