

Abstract
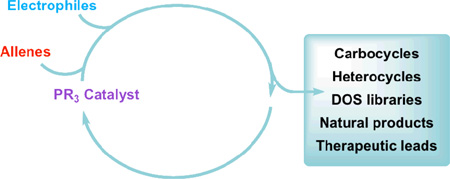
Nucleophilic phosphine catalysis of allenes with electrophiles is one of the most powerful and straightforward synthetic strategies for the generation of highly functionalized carbocycle or heterocycle structural motifs, which are present in a wide range of bioactive natural products and medicinally important substances. The reaction topologies can be controlled through judicious choice of the phosphine catalyst and the structural variations of starting materials. This Tutorial Review presents selected examples of nucleophilic phosphine catalysis using allenes and electrophiles.
1. Introduction
Trivalent phosphines and their derivatives are used widely in organic synthesis. Traditionally, they are applied as stoichiometric reagents in several name reactions, including the Wittig, Staudinger, and Mitsunobu reactions.1 In modern organic chemistry, organophosphorus compounds are often employed as ligands for transition metal–catalyzed processes.2 Although the use of phosphines as catalysts for organic reactions can be traced back to the 1960s, reports of nucleophilic phosphines as organocatalysts are relatively rare in the second half of the last century. In 1963, Rauhut and Currier reported one of the first phosphine-catalyzed reactions: the dimerization of electron-deficient olefins.3 In 1966, Winterfeldt and Dillinger discovered triphenylphosphine-catalyzed annulation for the synthesis of γ-butenolides when using acetylenedicarboxylates and aldehydes as substrates.4 Two years later, Morita et al. described a reaction of an activated olefin and an aldehyde catalyzed by a phosphine.5 This phosphine-catalyzed transformation, together with the similar amine-catalyzed reaction discovered by Baylis and Hillman in 1972, is known as the Morita–Baylis–Hillman (MBH) reaction; it has become one of the most useful and popular methodologies in organic synthesis.
In recent years, recognition of the huge potential of and the determined scientific interest in Lewis base catalysis has led to nucleophilic phosphine catalysis receiving considerable attention.6 Because the catalytic behavior and unique properties of trivalent phosphines differ from those of amines, many novel annulations of electron-deficient alkynes, alkenes, and allenes have been discovered in laboratories worldwide, as described in several recent reviews.7–9 In general, the significant growth of nucleophilic phosphine catalysis can be attributed to several important features: (1) the reactions are highly atom-economical and usually do not produce any by-products; (2) the catalytic system is metal-free, allowing the reactions to performed readily on large scales—a feature that is especially attractive for pharmaceutical applications; and (3) the reaction topologies can be controlled through judicious choice of the phosphine catalyst (i.e., varying its substituents) as well as structural variations of the starting materials.
The main purpose of this Tutorial Review is to present key developments in nucleophilic phosphine catalysis between allenes and electrophiles for application in organic synthesis. Scheme 1 summarizes the diverse range of allenes and electrophiles that can be used in this catalytic reaction. Because of space limitations, this Tutorial Review emphasizes the different kinds of novel reaction types in nucleophilic phosphine catalysis of allenes with electrophiles, rather than providing a comprehensive list of examples. Nevertheless, for certain novel reactions, such as the [3+2] allene–olefin annulation, relatively detailed accounts are presented. Furthermore, some important modifications of the reactions are discussed and very recent progress is mentioned. Some important applications of these methodologies in the construction of combinatorial libraries and the total syntheses of natural products are also highlighted. The literature related to theoretical studies of nucleophilic phosphine catalysis is beyond the scope of this Tutorial Review.
Scheme 1.

Typical allenes and electrophiles used in nucleophilic phosphine catalysis
2. Nucleophilic phosphine catalysis of allenes with electrophiles
2.1. [3+2] Annulation with electron-deficient olefins
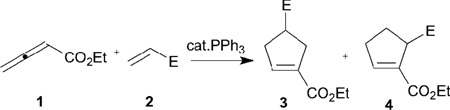
In 1995, Lu discovered a novel phosphine-catalyzed [3+2] annulation of ethyl 2,3-butadienoate (1) with electron-deficient olefins 2 to form the cyclopentene derivatives 3 and 4 (Table 1).10 Although the regioselectivity governing the product distribution was not great, this transformation is the first example of phosphine-catalyzed annulation of an allene. Enlightened by Lu’s pioneering work, the reaction has been investigated by over 20 different research groups, with applications in the syntheses of medicinally important agents and bioactive natural products, including the total syntheses of iridoid β-glucoside-(+)-geniposide and (–)-hinesol.11,12 In the mechanism proposed (Scheme 2), the catalytic cycle is initiated by nucleophilic addition of triphenylphosphine to ethyl 2,3-butadienoate (1), leading to the formation of the resonance-stabilized phosphonium dienolates 5↔6. The nucleophilic addition at the α-carbon atom in 5 to the electron-deficient olefin 2 forms the intermediate 7, which undergoes intramolecular cyclization, proton transfer, and phosphine elimination to yield the α-addition product 3. Alternatively, the γ-addition product 4 can be generated through the γ-addition pathway of 6 to 2. Ethyl 2,3-butadienoate readily serves as a three-carbon synthon in this transformation.
Table 1.
Phosphine-catalyzed [3+2] annulation of ethyl 2,3-butadienoate with electron-deficient olefins
 | |||
|---|---|---|---|
| entry | E | yield (%) | 3 : 4 |
| 1 | CO2Et | 76 | 75 : 25 |
| 2 | CO2Me | 81 | 80 : 20 |
| 3 | COMe | 55 | 63 : 37 |
| 4 | CN | 79 | 83 : 17 |
Scheme 2.

Mechanism of allene–olefin [3+2] annulation
The first intramolecular allene–olefin [3+2] annulation was reported by Kwon in 2007 (Scheme 3).13 The reaction starting materials 14 were synthesized from salicylaldehydes through Wittig reactions to form 13, followed by esterification with 3-butynoic acid under the influence of Mukaiyama’s reagent. The geometric constraint inherent in the substrates can control the reaction pathway so that only the α-addition product is obtained. In this reaction, 2-styrenyl allenoates featuring either electron-donating or -withdrawing substituents on the aromatic ring were readily converted into cyclopentene-fused dihydrocoumarins 15 in excellent to good yields with exclusive diastereoselectivity.
Scheme 3.

Intramolecular allene–olefin [3+2] annulation
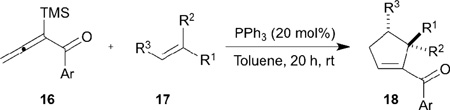
In 2009, Loh reported a highly regioselective synthesis of functionalized cyclopentenes 18 through phosphine-catalyzed [3+2] annulations between α-silyl-substituted allenones 16 and electron-deficient olefins 17 (Table 2).14 The steric bulk of the silyl substituent at the α-position leads to preferential addition at the γ-position, yielding the γ-addition product 18. This method works well with both phenyl and 2-furanyl allenones and a range of electron-deficient olefins, including chalcones, methyl acrylate, diethyl maleate, diethyl fumarate, and ethyl-4,4,4-trifluorocrotonoate. Notably, the trimethylsilyl group in the starting material is not retained in the final product.
Table 2.
Phosphine-catalyzed olefin [3+2] annulation of α-silylallenones
 | |||||
|---|---|---|---|---|---|
| entry | Ar | R1 | R2 | R3 | yield (%) |
| 1 | phenyl | COC6H5 | H | C6H5 | 84 |
| 2 | phenyl | CO2Et | H | CO2Et | 82 |
| 3 | phenyl | CF3 | H | CO2Et | 63 |
| 4 | 2-furanyl | 4-MeCOC6H4 | H | 4-EtOC6H4 | 80 |
| 5 | 2-furanyl | H | CO2Et | CO2Et | 83 |
| 6 | 2-furanyl | CO2Me | H | H | 83 |
Interestingly, a range of polyfunctionalized fused- and spiro-cyclopentenes can besynthesized through Lu’s allene–olefin [3+2] annulation of some designed electron-deficient olefins. A very recent example was reported by the Marinetti group (Scheme 4).15 Several spiro(cyclopentene)oxindoles 21 with trisubstituted cyclopentene units were obtained by using arylideneoxindoles 19 as starting materials. In particular, this synthetic strategy is a straightforward means of synthesizing carbocyclic analogues (22) of an important series of anticancer agents inhibiting MDM2–p53 interactions.
Scheme 4.

Synthesis of spiro(cyclopenten)oxindoles through allene–olefin [3+2] annulation
2.2. [4+2] Annulation with electron-deficient olefins
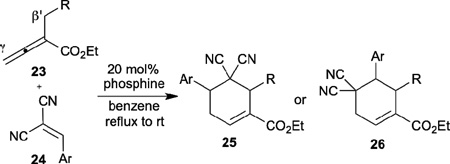
Inspired by the robust Lu’s [3+2] annulation for the synthesis of functionalized cyclopentenes, Kwon disclosed an unprecedented [4+2] annulation of α-substituted allenoates 23 and arylidenemalononitriles 24 (Table 3).16 In this case, the reaction regioselectivity is controlled by the electronic nature of the phosphine catalyst. When hexamethylphosphorous triamide (HMPT) was used as the catalyst, the reaction produced the cyclohexene 25. On the other hand, when a more electron-withdrawing triarylphosphine, such as tris(p-fluorophenyl)phosphine, was used as the catalyst, the reaction provided the alternative regioisomeric cyclohexene 26.
Table 3.
Phosphine-catalyzed [4+2] annulation of α-alkylallenoates with electron-deficient olefins
 | ||||||
|---|---|---|---|---|---|---|
| entry | R | Ar | phosphine | product | yield (%) | cis : trans |
| 1 | H | Ph | HMPT | 25a | 98 | - |
| 2 | H | 4-OMe-Ph | HMPT | 25b | 94 | - |
| 3 | Ph | Ph | HMPT | 25c | 93 | 82 : 18 |
| 4 | Et | Ph | HMPT | 25d | 98 | 92 : 8 |
| 5 | H | 4-Br-Ph | P(4-F-Ph)3 | 26a | 86 | - |
| 6 | H | N-Me-2-indolyl | P(4-F-Ph)3 | 26b | 91 | - |
| 7 | H | Ph | P(4-F-Ph)3 | 26c | 93 | - |
In the mechanism proposed (Scheme 5), the γ-addition reaction pathway begins with conjugate addition of the phosphonium dienolate intermediate 27 to the benzylidenemalononitrile 24a, producing the vinylphosphonium species 28, which undergoes two proton transfer steps to form the allylic phosphonium species 29. Intramolecular cyclization of the intermediate 29 followed by the release of the phosphine catalyst gives the final product 25a. Alternatively, the phosphonium dienolate 27 isomerizes into the vinylogous ylide intermediate 31, leading to the reaction producing the cyclohexene 26c through the β´-addition pathway. The α-substituted allenoate readily serves as a four-carbon synthon in this transformation.
Scheme 5.

Mechanism of allene–alkene [4+2] annulation
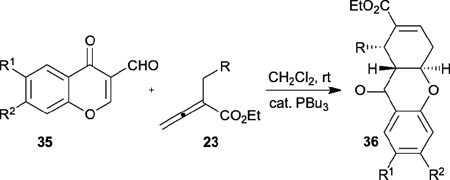
Recently, Kumar expanded the allene–olefin [4+2] annulation to the stereoselective synthesis of common tricyclic benzopyrone compounds 36, which were then efficiently transformed into a diverse range of naturally occurring scaffolds and related compounds (Table 4).17 With n-Bu3P as the phosphine catalyst, the tricyclic benzopyrone derivatives can be prepared, in moderate to good yields and with good diastereoselectivities, from 3-formylchromones 35 and allenoates 23 through a cascade reaction sequence of [4+2] annulation followed by deformylation. Notably, the presence of the formyl group in the chromenone starting materials 35 activates the chromenone double bond and allows the reaction to take place smoothly.
Table 4.
Synthesis of tricyclic benzopyrones through allene–olefin [4+2] annulation
 | ||||
|---|---|---|---|---|
| entry | R1 | R2 | R | yield (%) |
| 1 | H | H | CO2Et | 83 |
| 2 | Cl | Me | CO2Et | 60 |
| 3 | Me | H | Ph | 80 |
| 4 | H | H | Me | 67 |
| 5 | Br | H | Me | 60 |
2.3. [3+2] Annulation with imines
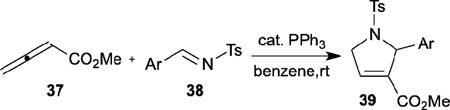
In allene–alkene [3+2] annulations, allenoates react with a variety of electron-deficient olefinic electrophiles to form multiply substituted cyclopentenes. To expand the utility of the [3+2] annulation, Lu developed the phosphine-catalyzed allene–imine [3+2] annulation in 1997 (Table 5).18 They obtained excellent yields of 2-substituted pyrrolines 39 as single annulation products from reactions of methyl 2,3-butadienoate 37 with N-tosyl imines 38 when employing triphenylphosphine as the catalyst. Unlike the allene–alkene [3+2] annulation (Scheme 2), for most aryl imines this reaction proceeds mainly through the α-addition pathway to generate the α-adduct products 39. When 2-furylimine was used as the reaction partner, however, the γ-addition product, methyl 4,5-dihydro-5-furyl-1-tosyl-1H–pyrrole-2-carboxylate, was isolated in 15% yield as a minor product.
Table 5.
Phosphine-catalyzed [3+2] annulation of methyl 2,3-butadienoate with N-tosylimines
 | ||
|---|---|---|
| entry | R | yield (%) |
| 1 | phenyl | 98 |
| 2 | o-methoxyphenyl | 96 |
| 3 | p-chlorophenyl | 97 |
| 4 | 1-naphthyl | 98 |
| 5 | 2-furyl | 83a |
Another adduct, methyl 4,5-dihydro-5-furyl-N-tosyl-1H-pyrrole-2-carboxylate, was isolated in 15% yield.
In 2005, Kwon described the highly diastereoselective syntheses of highly substituted pyrrolines 40 through use of Lu’s allene–imine [3+2] annulation (Scheme 6).19 In this reaction, tributylphosphine was used as the nucleophilic catalyst to improve the reaction efficiency and diastereoselectivity. When γ-substituted allenoates 20a were used as the imine reaction partner, 2,5-cis-substituted pyrrolines 40 were produced exclusively in high yield. The reaction efficiency and selectivity can meet the high standards of diversity-oriented synthesis (DOS) for library construction. A DOS library containing a variety of fused heterocyclic compounds possessing distinctive frameworks was constructed through a sequence of phosphine-catalyzed imine annulation, Tebbe reaction, Diels–Alder reaction, and, in some cases, hydrolysis. From this library, several molecules were identified as antimigratory agents of human breast cancer cells.20
Scheme 6.

Phosphine-catalyzed imine [3+2] annulation of γ-substituted allenoates
Since the first report of Lu’s allene–imine [3+2] annulation, several research groups have applied the reaction in the syntheses of various aza-heterocycles possessing distinctive frameworks. In a very recent example, Wang reported the synthesis of benzo-fused cyclic sulfamidate heterocycles 43 through phosphine-catalyzed [3+2] annulation between the allenoate 42 and the cyclic aldimine/ketimines 41 (Scheme 7).21 This approach allows benzo-fused cyclic sulfamidate heterocycles 43 featuring a variety of substituents at various positions on the aromatic ring to be prepared readily with high isolated yields. Furthermore, these reactions can also be performed conveniently on the gram scale.
Scheme 7.

Synthesis of benzo-fused cyclic sulfamide heterocycles through allene–imine [3+2] annulation
2.4. [4+2] Annulation with imines
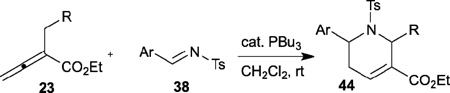
In the imine [3+2] annulation described above, unsubstituted or γ-substituted allenoates react readily with N-sulfonylimines in the presence of a phosphine to generate functionalized pyrrolines. With the vision of exploiting the potential of phosphine catalysis of allenes, in 2003 Kwon disclosed an unprecedented allene–imine [4+2] annulation to produce highly functionalized tetrahydropyridines 44 (Table 6).22 A variety of N-tosylimines 38 and α-substituted allenoates 23 are suitable for this reaction. When 2-benzyl-2,3-butadienoates 23 are used as starting materials, 2,6-diaryltetrahydropyridines 44 are obtained in excellent yields with good diastereoselectivities favoring the cis isomer. This robust allene–imine [4+2] annulation can be employed to generate tetrahydropyridines 44 on large scales.23 The application of this reaction to natural product syntheses, such as the formal syntheses of (±)-alstonerine and (±)-macroline (2005) and the total synthesis of (±)-hirsutine (2012), was reported by the same group (Scheme 8).24,25 Unlike the allene–alkene [4+2] annulation (Scheme 5), this reaction proceeds through the γ-addition pathway to generate γ-addition products 44 as single products. Surprisingly, no β´-addition products were isolated in this case.
Table 6.
Phosphine-catalyzed [4+2] annulation of α-substituted allenoates with imines
 | ||||
|---|---|---|---|---|
| entry | R | Ar | yield (%) | cis : trans |
| 1 | H | Ph | 98 | - |
| 2 | H | 4-MeC6H4 | 95 | - |
| 3 | H | 4-FC6H4 | 95 | - |
| 4 | H | 2-furyl | 97 | - |
| 5 | H | N-Boc-2-pyrroyl | 99 | - |
| 6 | 3-MeOC6H4 | Ph | 99 | 98 : 2 |
| 7 | Ph | 4-NO2C6H4 | 90 | 95 : 5 |
| 8 | 4-CNC6H4 | 2-CF3C6H4 | 80 | 90 : 10 |
Scheme 8.

Synthesis of (±)-alstonerine, (±)-macroline, and (±)-hirsutine through allene–imine [4+2] annulation
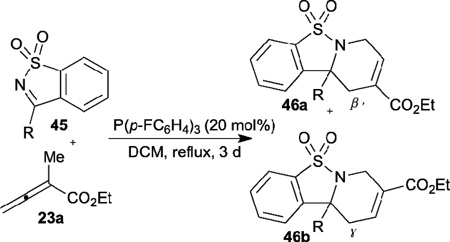
Recently, Ye discovered the phosphine-catalyzed [4+2] annulation of α-substituted allenoate 23a and cyclic ketimines 45 (Table 7).26 This reaction can produce corresponding sultam-fused tetrahydropyridines 46 in good yields and with moderate to excellent regioselectivities. The use of triarylphosphines featuring electron-withdrawing groups, such as tris(4-fluorophenyl)phosphine and tris(4-chlorophenyl)phosphine, can increase the reaction efficiency. Interestingly, in contrast to the annulation with aldimines described above, the ketimine [4+2] annulation proceeds mainly through the β´-addition pathway to generate the β´-adducts 46a as major products.
Table 7.
Synthesis of sultam-fused tetrahydropyridines through allene-imine [4+2] annulation
 | |||
|---|---|---|---|
| entry | R | yield(%) | 46a:46b |
| 1 | Ph | 65 | 5:1 |
| 2 | 4-MeC6H4 | 68 | 6:1 |
| 3 | 4-MeOC6H4 | 73 | 21:1 |
| 4 | 3-BrC6H4 | 69 | 9:1 |
| 5 | 3-CNC6H4 | 72 | 5:1 |
One of the merits of the robust allene–imine [3+2]/[4+2] annulation is that the reaction is extremely compatible with reactions performed in the solid phase, thereby allowing efficient construction of aza-heterocyclic compound libraries for biological screening. In 2007, Kwon described the first solid phase phosphine catalysis of resin-bound allenoates with imines to generate dihydropyrrole and tetrahydropyrodine libraries (Scheme 9).27 The resin-bound allenoates 50 were prepared from SynPhase lanterns of Wang resin 48 and allenoic acid in the presence of Mukaiyama’s reagent. A library of 4288 carboxylic acids 54 and 55 was obtained, with good to excellent yields and high diastereoselectivities, through phosphine-catalyzed [3+2]/[4+2] annulations of the resin-bound allenoates with the N-sulfonylimines and subsequent cleavage of the resins 52 and 53 using 2.5% trifluoroacetic acid in CH2Cl2. After biological screening of the 4288 analogues, two potent inhibitors of protein geranylgeranyltransferase type I with submicromolar IC50 values were identified. An octahydro-1,6-naphthyridin-4-one library was reported by the same group in 2011. The library was built through a sequence of phosphine- catalyzed imine [4+2] annulation, Tebbe reaction, Diels–Alder reaction, and hydrolysis, using the solid phase split-and-pool technique; this approach led to the identification of octahydro-1,6-naphthyridin-4-ones as activators of endothelium-driven immunity.28
Scheme 9.

Construction of nitrogen heterocycle libraries through phosphine-catalyzed allene–imine [3+2] and [4+2] annulations
2.5. [3+2]/[4+2] Annulation with ketones
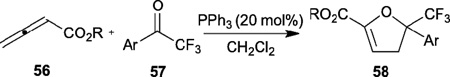
Inspired by the robust allene–olefin/imine [3+2] and [4+2] annulations pioneered by Lu and Kwon, Ye reported the [3+2] and [4+2] annulations between allenoates and ketones in 2010 and 2011 (Tables 8 and 9).29,30 The use of trifluoromethyl ketones was key for the success of these transformations. Various dihydrofurans 58 were generated through phosphine-catalyzed [3+2] annulation of 2,3-butadienoates 56 and trifluoromethyl ketones 57 in good yields with excellent γ-regioselectivities (Table 8).29 Furthermore, hydrogenation of the dihydrofurans produced the corresponding 2,5,5-trisubstituted tetrahydrofurans in good yields and with exclusive cis-selectivities.
Table 8.
Phosphine-catalyzed [3+2] annulaion of allenoates with trifluoromethyl ketonos
 | |||
|---|---|---|---|
| entry | R | Ar | yield(%) |
| 1 | Et | Ph | 72 |
| 2 | Et | 4-MeC6H4 | 79 |
| 3 | Et | 4-ClC6H4 | 99 |
| 4 | Cy | 4-MeOC6H4 | 61 |
| 5 | Cy | 2-thienyl | 54 |
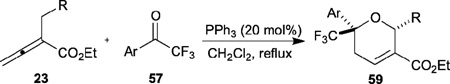
Table 9.
Phosphine-catalyzed [4+2] annulation of allenoates with trifluoromethyl ketones
 | ||||
|---|---|---|---|---|
| entry | R | Ar | yield (%) | dr |
| 1 | Ph | Ph | 76 | 14:1 |
| 2 | 4-MeOC6H4 | Ph | 67 | 14:1 |
| 3 | 2-BrC6H4 | Ph | 85 | 14:1 |
| 4 | 2-MeC6H4 | Ph | 68 | 12:1 |
| 5 | Ph | 4-ClC6H4 | 85 | >25:1 |
| 6 | Ph | 2-thienyl | 58 | 13:1 |
Moreover, when ethyl α-benzylallenoates 23 were used as the allene reaction partners, [4+2] annulation occurred through a γ-addition pathway, very similar to the catalytic cycle proposed by Kwon. This transformation provided a straightforward means for the highly diastereoselective synthesis of 6-trifluoromethyl-5,6-dihydropyrans 59, reduction of which with H2 gave the corresponding tetrahydropyrans in high yields and with exclusive diastereoselectivity (Table 9).30
2.6. Annulations with aldehydes
In contrast to the successful transformations observed when using electron-deficient olefins, imines, and trifluoromethyl ketones as electrophiles, subjecting aldehydes to the reaction did not result in any corresponding [3+2] and [4+2] annulations, but rather in interesting alternative transformations. Indeed, the reaction topologies are controlled by the nature of the phosphine catalyst, the reaction conditions, as well as structural variations of the starting materials (Scheme 10).31–34 When a small tertiary phosphine (e.g., trimethylphosphine) was employed in the reaction, the dioxanes 65 were produced through the intermediate 60 having s-trans geometry, due to electrostatic association of the dienolate oxygen anion with the phosphonium cation. When a bulky tertiary phosphine or a hydrogen-bond donor was present, the pyrones (in presence of tricyclopentylphosphine) or dihydropyrones (in presence of MeOH) were generated through the s-cis-phosphonium dienolate intermediate 61. When γ-methyl allenoate was used as the reaction partner instead of an unsubstituted allenoate, the tetrahydrofurans 74 were obtained through the phosphorus ylide 70, which was generated from the phosphonium dienoate 62 by an overall 1,4-hydrogen shift.
Scheme 10.

Nucleophilic phosphine catalysis of allenes with aldehydes
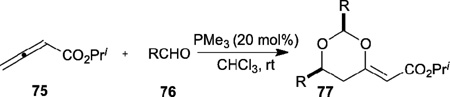
In 2005, Kwon reported the first phosphine-catalyzed reaction of aldehydes 76 with allenoates 75 to generate 2,6-disubstituted-1,3-dioxan-4-ylidene-acetates 77 (Table 10).31 Pyridine aldehydes and benzaldehydes with electron-withdrawing groups both afforded the corresponding 1,3-dioxan-4-ylidenes 77 in excellent yields and with good stereoselectivities favoring the E isomers. Less-reactive benzaldehydes bearing electron-donating substituents, however, afforded their products in moderate yields. Furthermore, some ubiquitous δ-hydroxyl-β-ketoesters were synthesized from the 1,3-dioxan-4-ylidenes through acid-mediated hydrolysis of the acetal functionality.
Table 10.
Synthesis of 1,3-dioxan-4-ylidenes through nucleophilic phosphine catalysis
 | |||
|---|---|---|---|
| entry | R | yield(%) | E/Z ratio |
| 1 | 4-pyridyl | 99 | 8:1 |
| 2 | 4-CF3C6H4 | 99 | 7:1 |
| 3 | 2-CF3C6H4 | 65 | 6:1 |
| 4 | 3-NO2C6H4 | 97 | 7:1 |
| 5 | Ph | 54 | only E |
| 6 | 3-MeOC6H4 | 47 | only E |
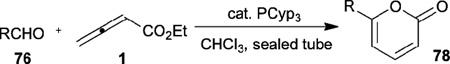
On the other hand, when bulky tricyclopentylphosphine was used as the catalyst, the 2-pyrones 78 were isolated from the same starting materials (Table 11).32 Various aromatic aldehydes 76, including benzaldehyde, 2-naphthaldehyde, and 2-furaldehyde, gave the 6-aryl-2-pyrones 78 in good yields. Although the reaction yields were not satisfactory when using aliphatic aldehydes as reaction partners, the reaction afforded a valuable compound, 6-propyl-2-pyrone, which possesses a sweet, creamy, coumarin-like herbal flavor, in one step from a commercially available aldehyde.
Table 11.
Synthesis of 6-aryl-2-pyrones through nucleophilic phosphine catalysis
 | |||
|---|---|---|---|
| entry | R | PCyp3 (mol%) | yield (%) |
| 1 | Ph | 10 | 60 |
| 2 | 3-ClC6H4 | 10 | 91 |
| 3 | 2-ClC6H4 | 10 | 73 |
| 4 | 2-CF3C6H4 | 20 | 71 |
| 5 | 2-naphthyl | 30 | 66 |
| 6 | 2-furyl | 30 | 61 |
| 7 | n-propyl | 30 | 34 |
In 2008, Kwon also described the alcohol-assisted phosphine-catalyzed reaction of aldehydes 76 and allenoates 37 to provide disubstituted dihydropyrones 79 in moderate to good yields (Table 12).33 They found that the addition of a base, such as n-BuLi, allowed generation of methoxide in situ and led to the promising formation of the dihydropyrones 79 in good yields, without any undesired products. Benzaldehydes possessing a variety of electron-withdrawing or -donating groups provided the desired dihydropyrones 79 in moderate to good yields.
Table 12.
Synthesis of disubstituted dihydropyrones through nucleophilic phosphine catalysis
 | ||
|---|---|---|
| entry | R | yield (%) |
| 1 | 4-NCC6H4 | 58 |
| 2 | 3-NCC6H4 | 83 |
| 3 | 3-NO2C6H4 | 74 |
| 4 | 2-CF3C6H4 | 58 |
| 5 | Ph | 37 |
| 6 | 2-MeC6H4 | 36 |
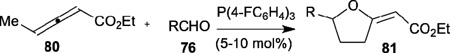
Recently, He disclosed the phosphine-catalyzed [3+2] annulation of allenoates 80 with aldehydes 76 as a simple and efficient pathway for the synthesis of 2-arylidenetetrahydrofurans 81 (Table 13).34 The presence of the γ-methyl group on the allenoates 80 changed the regioselectivity of the addition of the aldehyde to the non-substituted allenoates and played a critical role in the occurrence of the [3+2] annulation with aldehydes. The reaction conditions (i.e., catalyst, solvent, and temperature) significantly influence the chemoselectivity and efficiency of the [3+2] annulations.
Table 13.
Synthesis of 2-arylidenetetrahydrofurans through nucleophilic phosphine catalysis
 | |||||
|---|---|---|---|---|---|
| entry | R | 80 (equiv) | t(h) | yield (%) | E/Z ratio |
| 1 | 2-ClC6H4 | 2.4 | 18 | 94 | 4:1 |
| 2 | 2-FC6H4 | 1.8 | 24 | 81 | 4:1 |
| 3 | 2,4-Cl2C6H4 | 1.8 | 22 | 98 | 6:1 |
| 4 | 4-NO2C6H4 | 1.8 | 5 | 84 | 5:1 |
| 5 | 4-CF3C6H4 | 2.0 | 17 | 81 | 4:1 |
| 6 | 3-pyridyl | 1.8 | 5 | 81 | 20:1 |
| 7 | 2-furyl | 2.0 | 24 | 72 | 2:1 |
2.7. Miscellaneous
Since Lu’s initial discovery of phosphine-catalyzed allene–alkene [3+2] annulation, many research groups have expanded the substrate scope of the electrophiles, increasing the molecular complexity and structural variety of the products. Currently, more than eight allenes (shown in Scheme 1) with different substituents are known to be suitable substrates for nucleophilic phosphine catalysis with various electrophiles containing carbon, oxygen, nitrogen, sulfur, phosphorus, and fluorine atoms (Scheme 1). Several unusual reaction types of allenes with electrophiles, such as [8+2], [4+3], [3+3], and [3+2+3] annulations, have also been discovered in this research area. Some of the most interesting examples are outlined below (for more details, see ref. 35 and references therein).
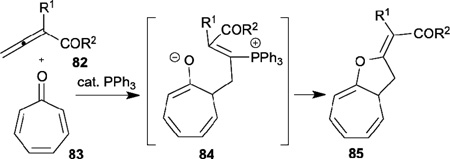
In 2000, Ishar reported phosphine-catalyzed [8+2] annulation of allenic esters/ketones 82 with tropone 83, leading to the 8-oxa-9-(ethoxycarbonyl/acylalkylidene)-bicylo[5.3.0]deca-1,3,5-trienes 85 (Table 14).36 Notably, the [8+2] cycloadducts 85 were obtained as the only products in excellent yields; the normal electron-deficient allene–olefin [3+2]/[4+2] annulation products were not observed. Mechanistically, γ-addition of the phosphonium dienolate at the C2 atom of tropone formed the intermediate 84; subsequent intramolecular cyclization and elimination of phosphine provided the desired product 85.
Table 14.
Phosphine-catalyzed [8+2] annulation of allenes with tropone
 | ||||
|---|---|---|---|---|
| entry | R1 | R2 | t(h) | yield (%) |
| 1 | H | OEt | 3.5 | 82 |
| 2 | H | Me | 3.0 | 95 |
| 3 | H | Ph | 5.0 | 90 |
| 4 | H | CH2Ph | 8.0 | 90 |
| 5 | Me | OEt | 14 | 70 |
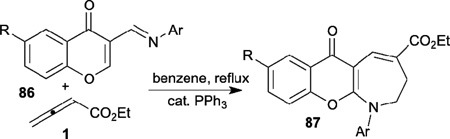
In the same year, Ishar also described the synthesis of novel N-aryl-2,3-dihydro-4-ethoxycarbonylchromano[2,3-b]azepine-6-ones 87 from 3-(N-arylimino-methyl)chromones 86 through phosphine-catalyzed [4+3] annulation followed by tandem rearrangement (Table 15).37 In the mechanism proposed (Scheme 11), α-addition of the phosphonium dienolate 5 to the chromon 86 forms the intermediate 88, which undergoes intramolecular cyclization and phosphine elimination to afford the [4+3] annulation product 90. Thermal opening of the chromone ring in compound 90 generates the intermediate 91; subsequent rotation around a C–C single bond, recyclization of the chromone ring, and a 1,5-H shift in 93 affords the final product 87.
Table 15.
Phosphine-catalyzed [4+3] annulation of allenes with 3-(N-aryliminomethyl)chromones
 | ||||
|---|---|---|---|---|
| entry | R | Ar | t(h) | yield (%) |
| 1 | H | p-anisyl | 80 | 55 |
| 2 | H | p-chlorophenyl | 90 | 57 |
| 3 | Me | p-anisyl | 85 | 64 |
| 4 | Cl | p-chlorophenyl | 80 | 64 |
Scheme 11.

Mechanism of chromone–allene [4+3] annulation
In 2009, Kwon disclosed a novel [3+3] annulation of allenoates 95 with aziridines 94 to generate highly functionalized tetrahydropyridines 96 (Table 16).38 Aryl aziridines 94 featuring both electron-rich and -deficient substituents are suitable for this reaction, giving the desired products 96 in good to excellent yields. In the mechanism proposed (Scheme 12), β´-addition of the vinylogous ylide 98 to the phenyl aziridine 94 occurs to form the intermediate 99, which is converted to the phosphonium dienolate 100 through proton transfer. Intramolecular nucleophilic aromatic substitution and desulfonylation of 100 affords 101; subsequent intramolecular conjugate addition and phosphine elimination afford the final product 96. Although the reaction is catalytic, one equivalent of triphenylphosphine was used to expedite the reaction.
Scheme 12.

Mechanism of allene–aziridine [3+3] annulation
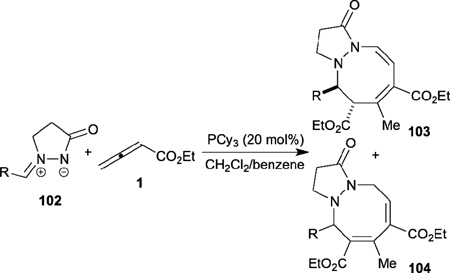
Recently, Guo and Kwon reported a phosphine-catalyzed [3+2+3] annulation of two molecules of an allenoate 1 and an azomethine imine 102, leading to the tetrahydropyrazolo-diazocinone products 103 and 104 (Table 17).35 Both electron-rich and -deficient aryl azomethine imines, as well as a polyaromatic azomethine imine, are compatible substrates. Mechanistically, the key event is the formation of the trimeric zwitterionic intermediate 106, which gives the intermediate 107 after addition to the azomethine imine 102 (Scheme 13). Intramolecular cyclization of 107 generates 108, which gives the final products 103 and 104 as a mixture of tautomers after proton transfer, phosphine elimination, and olefin isomerization.
Table 17.
Phosphine-catalyzed [3+2+3] annulation of allenes with azomethine imines
 | |||
|---|---|---|---|
| entry | R1 | yield (%) | radio of 103:104 |
| 1 | Ph | 81 | 34:66 |
| 2 | 4-l-PrC6H4 | 67 | 42:58 |
| 3 | 4-MeOC6H4 | 76 | 44:56 |
| 4 | 4-BrC6H4 | 77 | 29:71 |
| 5 | 4-ClC6H4 | 90 | 24:76 |
| 6 | 4-FC6H4 | 88 | 25:75 |
| 7 | 2-naphthyl | 76 | 32:68 |
Scheme 13.

Mechanism of allene–allene–azomethine imine [3+2+3] annulation
3. Enantioselective nucleophilic phosphine catalysis of allenes with electrophiles
Since Lu reported the first phosphine-catalyzed allene–alkene [3+2] annulation in 1995, the nucleophilic phosphine catalysis of allenes with electrophiles has become one of the most powerful and straightforward synthetic strategies for generation of the highly functionalized carbocyclic and heterocyclic structural motifs found widely in bioactive natural products and medicinally important substances. Despite the many very promising results that have been achieved in phosphine catalysis of allenes with electrophiles, enantioselective variants of these transformations has, however, remained relatively rare. A handful of chiral phosphine organocatalysts, which can be divided into two categories (chiral phosphines without additional functionalities and chiral phosphines with hydrogen bond donors), have been found to be effective for these reactions (Scheme 14). In this Section, we provide a brief overview of these enantioselective nucleophilic phosphine-catalyzed reactions of allenes with electrophiles and also point the reader to several comprehensive reviews for more details.9,39–41
Scheme 14.

Typical chiral phophines used in nucleophilic phosphine catalysis
3.1. Chiral phosphines without additional functionality
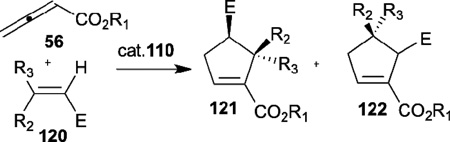
In 1997, Zhang reported the first enantioselective phosphine-catalyzed [3+2] annulation of 2,3-butadienoates 56 with electron-deficient olefins 120, affording the cyclopentene products 121 and 122 with excellent regioselectivity and good enantioselectivity (Table 18).42 After initial screening of known and new chiral phosphines, they found that the newly designed bicyclic monodentate chiral phosphine 110 was most suitable for this reaction. Furthermore, using acrylates with bulky ester substituents (e.g., tert-butyl) improved the regio- and enantioselectivity.
Table 18.
Enantioselection allene-olefin [3+2] annulation reported by Zhang
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | E | R1 | R2 | R3 | yield (%) | 121:122 | % ee of 121 |
| 1 | CO2Me | Et | H | H | 87 | 96 : 4 | 79 |
| 2 | CO2iBu | Et | H | H | 88 | 100 : 0 | 93 |
| 3 | CO2Et | tBu | H | H | 84 | 94 : 6 | 69 |
| 4 | CO2Et | Et | CO2Et | H | 49 | 79 | |
| 5 | CO2Me | Et | H | CO2Me | 84 | 36 | |
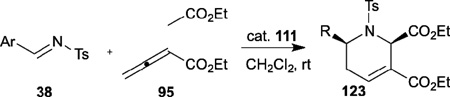
Despite the great synthetic potential of the results described above, no further progress was made in enantioselective nucleophilic phosphine catalysis of allenes with electrophiles until 2005, when Fu demonstrated the asymmetric allene–imine [4+2] annulation to give the piperidine derivatives 123 when employing Gladiali’s phosphine 111 as the catalyst (Table 19).43 Under the optimized conditions, they obtained a range of useful functionalized tetrahydropyridines 123 in high yields and enantioselectivities. This asymmetric [4+2] reaction is a novel and efficient means of access to a common framework found in several important natural products, including 6-oxoalstophyllal and 6-oxoalstophylline.
Table 19.
Enantioselective allene–imine [4+2] annulation reported by Fu
 | ||||
|---|---|---|---|---|
| entry | Ar | ee (%) | cis:trans | yield (%) |
| 1 | Pr | 98 | 91:9 | 93 |
| 2 | 3-MeC6H4 | 98 | 93:7 | 98 |
| 3 | 2-NO2C6H4 | 68 | 96:4 | 98 |
| 4 | 2-naphthyl | 99 | 93:7 | 96 |
| 5 | 2-furyl | 97 | 87:13 | 98 |
| 6 | 3-pyridyl | 97 | 91:9 | 76 |
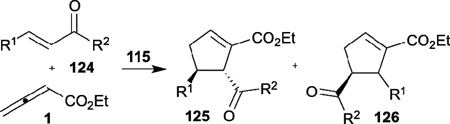
In 2008, Marinetti reported the enantioselective allene–alkene [3+2] annulation by using the phosphine 115, exhibiting planar chirality, as the catalyst (Table 20).44 When using the chalcones 124 as reaction partners, the γ-addition products 125 were afforded in moderate to good yields, with excellent enantioselectivities favoring the trans isomers. Very recently, the same group employed this chiral phosphine as the catalyst for the enantioselective syntheses of spirocyclic compounds.45
Table 20.
Enantioselective allene–alkene [3+2] annulation reported by Marinetti
 | |||||
|---|---|---|---|---|---|
| entry | R1 | R2 | 125 : 126 ratio | yield (%) | ee (%) |
| 1 | H | OEt | 1 : 1.5 | 53 | 88/84 |
| 2 | Ph | Ph | 20 : 1 | 65 | 92 |
| 3 | 1-naphthyl | Ph | >20 : 1 | 87 | 96 |
| 4 | 4-MeOC6H4 | Ph | >20 : 1 | 71 | 93 |
| 5 | 4-NO2C6H4 | Ph | 10 : 1 | 63 | 92 |
| 6 | 2-furyl | Ph | 8 : 1 | 71 | 93 |
In 2012, Kwon demonstrated the first enantioselective total synthesis of the monoterpene indole alkaloid (+)-ibophyllidine 132, featuring an asymmetric phosphine-catalyzed allene–imine [3+2] annulation as the key reaction step (Scheme 15).46 Using their P-chiral [2.2.1]bicyclic phosphine 112 as the catalyst, they obtained the syn-pyrrolidine 129 as a synthetic intermediate from 4-ethyl-2,3-butadienoate and N-tosylbenzaldimine in 93% yield and with 99% ee. Hydrogenation of 129 afforded the stereochemically dense pyrrolidine ring compound 130 in excellent yield with exceptionally high degrees of both diastereo- and enantioselectivity. A subsequent 12-step sequence of chemical manipulations provided the target natural product (+)-ibophyllidine 132.
Scheme 15.

Enantioselective total synthesis of (+)-ibophyllidine through asymmetric phosphine-catalyzed allene-imine [3+2] annulation
3.2. Chiral phosphines with hydrogen-bond donors
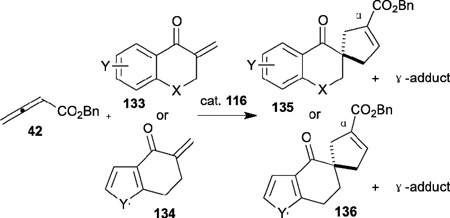
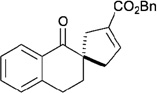
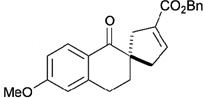
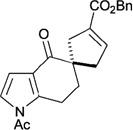
In contrast to chiral phosphines lacking additional functionality, which induce asymmetry through steric interactions, chiral phosphines with hydrogen-bonding donors instigate high enantioselectivity through hydrogen bonding. The first example of asymmetric allene–olefin [3+2] annulation employing the multifunctional chiral phosphine 116 as the catalyst was reported by Miller in 2007 (Table 21).47 When using benzyl 2,3-budienoate 42 as the reaction partner, they obtained various spirocyclic compounds 135 and 136 from corresponding electron-deficient olefins in good yields with good to moderate enantioselectivities. The presence of a heteroatom-substituted ring system in the starting material resulted in significantly lower yields and enantioselectivities.
Table 21.
Enantioselective allene-olefin [3+2] annulation reported by Miller
 | |||||
|---|---|---|---|---|---|
| entry | substrate | product() | yield +(%) | α : | ee(%) |
| 1 | 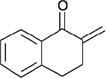 |
 |
95 | > 99 : 1 | 80 |
| 2 | 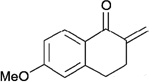 |
 |
95 | > 99:1 | 84 |
| 3 | 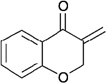 |
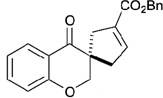 |
68 | 94 : 6 | 65 |
| 4 | 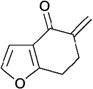 |
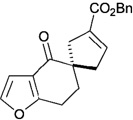 |
75 | > 99 : 1 | 76 |
| 5 | 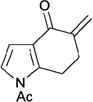 |
 |
53 | > 99 : 1 | 71 |
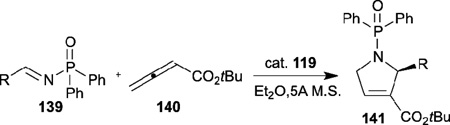
Encouraged by Miller’s pioneering work, Zhao and Lu independently developed their amino acid–derived bifunctional chiral phosphines in 2010 and 2011, respectively.48,49 Zhao reported the highly enantioselective [4+2] annulation of the allenoate 95 with the alkylidene cyanoacetate 137 catalyzed by his N-acyl aminophosphine 118 (Table 22);48 a range of highly functionalized cyclohexene derivatives 138, bearing three contiguous chiral centers, were synthesized with good to excellent diastereoselectivities and enantioselectivities. On the other hand, Lu described versatile enantioselective [3+2] annulations of imines and allenoates catalyzed by his dipeptide-based chiral phosphine 119 (Table 23).49 Notably, this reaction is the first example of aliphatic imines 139 being applied successfully in phosphine-catalyzed [3+2] annulation. Moreover, this transformation was applied as a key step in the concise formal synthesis of (+)-trachelanthamidine.
Table 22.
Enantioselective allene-olefin [4+2] annulation reported by Zhao
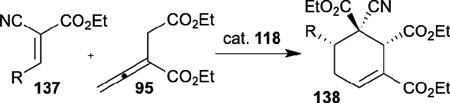 | ||||
|---|---|---|---|---|
| entry | R | t(h) | yield (%) | ee (%) |
| 1 | Ph | 4 | 94 | 96 |
| 2 | Isopropyl | 3 | 92 | 97 |
| 3 | 2-nathphyl | 3 | 95 | 95 |
| 4 | 4-ClC6H4 | 6 | 99 | 97 |
| 5 | 3-MeOC6H4 | 6 | 88 | 96 |
| 6 | 2-thienyl | 4 | 87 | 93 |
Table 23.
Enantioselective allence-imine [3+2] annulation reported by Lu
 | |||
|---|---|---|---|
| entry | R | yield (%) | ee (%) |
| 1 | methyl | 75 | 95 |
| 2 | ethyl | 78 | 96 |
| 3 | n-hexyl | 84 | 97 |
| 4 | iso-propyl | 81 | 99 |
| 5 | PhCH2CH2 | 81 | 95 |
| 6 | cyclohexyl | 83 | 99 |
Another significant example of asymmetric allene–imine [3+2] annulation catalyzed by a chiral phosphine featuring a hydrogen-bonding donor was discovered by Jacobsen et al. in 2008 (Table 24).50 They developed a new family of chiral phosphinothiourea derivatives 117 for highly enantioselective syntheses of disubstituted dihydropyrroles 143. Notably, the use of diphenylphosphinoyl (DPP) imines 142 and the addition of 0.2 equiv of H2O and 0.05 equiv of Et3N improved the reaction efficiency.
Table 24.
Enantioselective allene-imine [3+2] annulation reported by Jacobsen
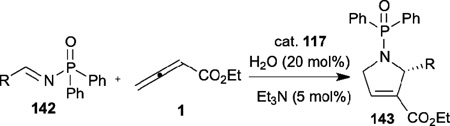 | ||||
|---|---|---|---|---|
| entry | R | 7a (mol%) | yield (%) | ee (%) |
| 1 | Ph | 10 | 84 | 98 |
| 2 | p-FC6H4 | 10 | 72 | 95 |
| 3 | p-MeOC6H4 | 20 | 80 | 97 |
| 4 | m-NO2C6H4 | 10 | 70 | 95 |
| 5 | o-BrC6H4 | 10 | 90 | 95 |
| 6 | 4-pyridyl | 10 | 70 | 94 |
| 7 | 3-furyl | 20 | 68 | 94 |
| 8 | 2-thienyl | 20 | 77 | 97 |
4. Conclusions
The use of nucleophilic phosphines as Lewis base catalysts for organic reactions has grown over the last decade and is now an active area of organocatalysis. Ten modes of nucleophilic phosphine catalysis with allenes and electrophiles have been demonstrated in this Tutorial Review; these transformations take advantage of the unique and highly tunable properties of trivalent phosphine. A variety of unique carbo/heterocyclic frameworks, some of which are present in a wide range of bioactive natural products and medicinally important substances, can be obtained through these transformations. Some examples of asymmetric nucleophilic phosphine catalysis with high enantioselectivities have also been developed, with promising applications in the total synthesis of natural products. Future advances in phosphine catalysis are likely to occur along a number of lines, including the development of novel reaction modes and the synthesis of novel chiral phosphines that perform their catalytic functions with high efficiency. In addition, much work remains to develop asymmetric variants of several recently discovered transformations.
Table 16.
Phosphine-catalyzed [3+3] annulation of allenes with aziridines
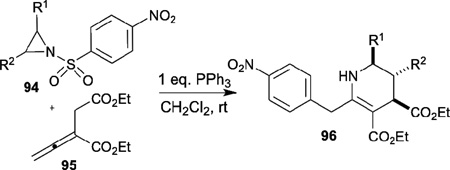 | ||||
|---|---|---|---|---|
| entry | R1 | R2 | yield (%) | dr (trans:cis) |
| 1 | H | 2-MeC6H4 | 88 | 90:10 |
| 2 | H | 4-FC6H4 | 76 | 88:12 |
| 3 | H | 3-ClC6H4 | 86 | 83:17 |
| 4 | H | 2-naphthyl | 58 | 85:15 |
| 5 | Me | H | 66 | 41:59 |
| 6 | H | Ph | 73 | 9:1 |
Key learning points.
An overview of the main history of nucleophilic phosphine catalysis.
Electron-deficient allenes can be used as three- or four-carbon synthons in phosphine-catalyzed annulations.
A diverse range of electrophiles—including olefins, imines, aldehydes, ketones, aziridines, and azomethine imines—can be coupled with allenes in phosphine-catalyzed annulations.
Reaction topologies of nucleophilic phosphine catalysis can be controlled through judicious choice of the catalyst.
Reaction outcomes of nucleophilic phosphine catalysis can be controlled through structural variations of the starting materials.
Acknowledgements
Financial support from the NIH (GM071779, to O.K.), the Natural Science Foundation of China (nos. 21102009 and 21372033, to Z.W.), the Priority Academic Program Development of Jiangsu Higher Education Institutions, and the Natural Science Foundation of Jiangsu Province (no. BK2011230, to Z.W.) is gratefully acknowledged.
References
- 1.Valentine DH, Hillhouse JH. Synthesis. 2003;317 [Google Scholar]
- 2.Quin LD, editor. A Guide to Organophosphorus Chemistry. New York: Wiley; 2000. [Google Scholar]
- 3.Rauhut MM, H Currier. U. S. Patent 3 074 999, 1963; Chem. Abstr. 1963;58:11224a. [Google Scholar]
- 4.Winterfeldt E, Dillinger HJ. Chem. Ber. 1966;99:1558. [Google Scholar]
- 5.Morita K, Suzuki Z, Hirose H. Bull. Chem. Soc. Jpn. 1968;41:2815. [Google Scholar]
- 6.Denmark SE, Beutner GL. Angew. Chem. Int. Ed. 2008;47:1560. doi: 10.1002/anie.200604943. [DOI] [PubMed] [Google Scholar]
- 7.Ye L-W, Zhou J, Tang Y. Chem. Soc. Rev. 2008;37:1140. doi: 10.1039/b717758e. [DOI] [PubMed] [Google Scholar]
- 8.Cowen BJ, Miller SJ. Chem. Soc. Rev. 2009;38:3102. doi: 10.1039/b816700c. [DOI] [PubMed] [Google Scholar]
- 9.Fan YC, Kwon O. Chem. Commun. 2013;49:11588. doi: 10.1039/c3cc47368f. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang C, Lu X. J. Org. Chem. 1995;60:2906. [Google Scholar]
- 11.Jones RA, Krische MJ. Org. Lett. 2009;11:1849. doi: 10.1021/ol900360h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Du Y, Lu X. J. Org. Chem. 2003;68:6463. doi: 10.1021/jo034281f. [DOI] [PubMed] [Google Scholar]
- 13.Henry CE, Kwon O. Org. Lett. 2007;9:3069. doi: 10.1021/ol071181d. [DOI] [PubMed] [Google Scholar]
- 14.Sampath M, Loh T. Chem. Commun. 2009:1568. doi: 10.1039/b819959k. [DOI] [PubMed] [Google Scholar]
- 15.Gomez C, Gicquel M, Carry J, Schio L, Retailleau P, Voituriez A, Marinetti A. J. Org. Chem. 2013;78:1488. doi: 10.1021/jo302460d. [DOI] [PubMed] [Google Scholar]
- 16.Tran YS, Kwon O. J. Am. Chem. Soc. 2007;129:12632. doi: 10.1021/ja0752181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baskar B, Dakas P, Kumar K. Org. Lett. 2011;13:1988. doi: 10.1021/ol200389p. [DOI] [PubMed] [Google Scholar]
- 18.Xu Z, Lu X. Tetrahedron Lett. 1997;38:3461. [Google Scholar]
- 19.Zhu X, Henry CE, Kwon O. Tetrahedron. 2005;61:6276. [Google Scholar]
- 20.Wang Z, Castellano S, Kinderman SS, Argueta CE, Beshir AB, Fenteany G, Kwon O. Chem. Eur. J. 2011;17:649. doi: 10.1002/chem.201002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Zhang Y, Dong H, Zhang J, Zhao J. Eur. J. Org. Chem. 2013:3764. [Google Scholar]
- 22.Zhu X, Lan J, Kwon O. J. Am. Chem. Soc. 2003;125:4716. doi: 10.1021/ja0344009. [DOI] [PubMed] [Google Scholar]
- 23.Lu K, Kwon O, Brummond KM, Davis MM. Org. Synth. 2009;86:212. doi: 10.1002/0471264229.os086.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tran YS, Kwon O. Org. Lett. 2005;7:4289. doi: 10.1021/ol051799s. [DOI] [PubMed] [Google Scholar]
- 25.Villa RA, Xu Q, Kwon O. Org. Lett. 2012;14:4634. doi: 10.1021/ol302077n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen X, Ye S. Eur. J. Org. Chem. 2012:5723. [Google Scholar]
- 27.Castellano S, Fiji HDG, Kinderman SS, Watanabe M, Leon P, Tamanoi F, Kwon O. J. Am. Chem. Soc. 2007;129:5843. doi: 10.1021/ja070274n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Daniel C, Wang Z, Kibbie J, Modlin R, Kwon O. Proc. Nat. Acad. Sci. U.S.A. 2011;108:6769. doi: 10.1073/pnas.1015254108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang T, Ye S. Org. Biomol. Chem. 2011;9:5260. doi: 10.1039/c1ob05444a. [DOI] [PubMed] [Google Scholar]
- 30.Wang T, Ye S. Org. Lett. 2010;12:4168. doi: 10.1021/ol101762z. [DOI] [PubMed] [Google Scholar]
- 31.Zhu X, Henry CE, Wang J, Dudding T, Kwon O. Org. Lett. 2005;7:1387. doi: 10.1021/ol050203y. [DOI] [PubMed] [Google Scholar]
- 32.Zhu X, Schaffner A, Li RC, Kwon O. Org. Lett. 2005;7:2977. doi: 10.1021/ol050946j. [DOI] [PubMed] [Google Scholar]
- 33.Creech GS, Kwon O. Org. Lett. 2008;10:429. doi: 10.1021/ol702462w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu S, Zhou L, Ma R, Song H, He Z. Chem. Eur. J. 2009;15:8698. doi: 10.1002/chem.200901276. [DOI] [PubMed] [Google Scholar]
- 35.Na R, Jing C, Xu Q, Jiang H, Wu X, Shi J, Zhong J, Wang M, Benitez D, Tkatchouk E, Goddard WA, III, Guo H, Kwon O. J. Am. Chem. Soc. 2011;133:13337. doi: 10.1021/ja200231v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar K, Kapur A, Ishar MS. Org. Lett. 2000;2:787. doi: 10.1021/ol000007l. [DOI] [PubMed] [Google Scholar]
- 37.Kumar K, Kapoor R, Kapur A, Ishar MS. Org. Lett. 2000;2:2023. doi: 10.1021/ol0000713. [DOI] [PubMed] [Google Scholar]
- 38.Guo H, Xu Q, Kwon O. J. Am. Chem. Soc. 2009;131:6318. doi: 10.1021/ja8097349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou Z, Wang Y, Tang C. Curr. Org. Chem. 2011;15:4083. [Google Scholar]
- 40.Wang S-X, Han X, Zhong F, Wang Y, Lu Y. Synlett. 2011:2766. [Google Scholar]
- 41.Zhao Q-Y, Lian Z, Wei Y, Shi M. Chem. Commun. 2012;48:1724. doi: 10.1039/c1cc15793k. [DOI] [PubMed] [Google Scholar]
- 42.Zhu G, Chen Z, Jiang Q, Xiao D, Cao P, Zhang X. J. Am. Chem. Soc. 1997;119:3836. [Google Scholar]
- 43.Wurz RP, Fu GC. J. Am. Chem. Soc. 2005;127:12234. doi: 10.1021/ja053277d. [DOI] [PubMed] [Google Scholar]
- 44.Voituriez A, Panossian A, Fleury-Brégeot N, Retailleau P, Marinetti A. J. Am. Chem. Soc. 2008;130:14030. doi: 10.1021/ja806060a. [DOI] [PubMed] [Google Scholar]
- 45.Duvvuru D, Pinto N, Gomez C, Betzer JF, Retailleau P, Voituriez A, Marinetti A. Adv. Synth. Catal. 2012;354:408. [Google Scholar]
- 46.Andrews IP, Kwon O. Chem. Sci. 2012;3:2510. doi: 10.1039/C2SC20468A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cowen BJ, Miller SJ. J. Am. Chem. Soc. 2007;129:10988. doi: 10.1021/ja0734243. [DOI] [PubMed] [Google Scholar]
- 48.Xiao H, Chai Z, Cao D, Wang H, Chen J, Zhao G. Org. Biomol. Chem. 2012;10:3195. doi: 10.1039/c2ob25295c. [DOI] [PubMed] [Google Scholar]
- 49.Han X, Zhong F, Wang Y, Lu Y. Angew. Chem. Int. Ed. 2012;51:767. doi: 10.1002/anie.201106672. [DOI] [PubMed] [Google Scholar]
- 50.Fang Y, Jacobsen EN. J. Am. Chem. Soc. 2008;130:5660. doi: 10.1021/ja801344w. [DOI] [PubMed] [Google Scholar]