

Abstract
Abnormal excitatory glutamate neurotransmission and plasticity have been implicated in schizophrenia and affective disorders. Gria1−/− mice lacking GluA1 subunit (encoded by Gria1 gene) of AMPA-type glutamate receptor show robust novelty-induced hyperactivity, social deficits and heightened approach features, suggesting that they could be used to test for anti-manic activity of drugs. Here, we tested the efficacy of chronic treatment with established anti-manic drugs on behavioural properties of the Gria1−/− mice. The mice received standard mood stabilizers (lithium and valproate) and novel ones (topiramate and lamotrigine, used more as anticonvulsants) as supplements in rodent chow for at least 4 weeks. All drugs attenuated novelty-induced locomotor hyperactivity of the Gria1−/− mice, especially by promoting the habituation, while none of them attenuated 2-mg/kg amphetamine-induced hyperactivity as compared to control diet. Treatment with lithium and valproate reversed the elevated exploratory activity of Gria1−/− mice. Valproate treatment also reduced struggling behaviour in tail suspension test and restored reciprocally-initiated social contacts of Gria1−/− mice to the level shown by the wild-type Gria1+/+ mice. Gria1−/− mice consumed slightly more sucrose during intermittent sucrose exposure than the wild-types, but ran similar distances on running wheels. These behaviours were not consistently affected by lithium and valproate in the Gria1−/− mice. The efficacy of various anti-manic drug treatments on novelty-induced hyperactivity suggests that the Gria1−/− mouse line can be utilized in screening for new therapeutics.
Introduction
Abnormal major excitatory neurotransmission and neuroplasticity, driven by glutamatergic neurotransmitter system, have been implicated in schizophrenia and mood and anxiety disorders [1], [2]. Psychotic, cognitive and emotional disturbances are linked to hyperactive glutamatergic neurotransmission in the brain [3]. These disturbances can be reproduced in animals and human subjects by blockade of N-methyl-D-aspartate (NMDA) receptors, with a mechanism thought to involve enhanced non–NMDA receptor-mediated glutamate transmission [4], [5] and to be attenuated by agents inhibiting presynaptic glutamate release [6], [7]. Hyperglutamatergic state in the frontal cortical areas [8] and upregulated markers of excitotoxicity and neuroinflammation in the post-mortem frontal cortex [9] have been also observed in bipolar patients. Several susceptibility genes encoding for glutamate receptor subunits, including the Gria1 gene encoding for GluA1 subunit of AMPA-type glutamate receptor (previously named GLUA1, GluR1, GluRA, GluR-A [10]), have been identified for bipolar disease [11], [12]. Most of them are overlapping with schizophrenia [13], [14], as these two illnesses share many behavioural characteristics.
An interesting finding has been the decreased GluA1 subunit expression in the post-mortem hippocampus, thalamus and frontal cortex of schizophrenic patients [15]–[19] and in the striatum of bipolar patients [20]. Mice lacking the GluA1 subunit [21] have been suggested to mimic some features of schizoaffective disorder and schizophrenia [22], [23]. These animals are abnormally active in the open field [21], [22], [24]–[26], but have a similar locomotor profile as wild-type animals in familiar home-cage environment [24]. Exaggerated exploration response provoked by a new object in the cage further point to abnormal reactivity to novel situations, although the Gria1−/− animals are known to habituate and recognize a familiar object [25]. Furthermore, abnormalities in working memory [21], [27], [28] and increased impulsive behaviour have been observed in the Gria1−/− mice [23]. These mice also exhibit a deficiency in pre-pulse inhibition and aberrant social interaction [25], [29], which resemble characteristic features seen in schizophrenic patients.
Here, we have focused on pharmacological features of the Gria1−/− mouse line, relevant for some positive symptoms of schizophrenia and/or mania, using a battery of behavioural tests to assess the predictive validity of the mouse model.
Materials and Methods
Ethics
All animal testing procedures were approved by the State Provincial Government of Southern Finland (ESAVI-0010026/041003/2010). All efforts were made to minimize the number and suffering of animals.
Animals
Gria1−/− mice and their Gria1+/+ wild-type (WT) controls were from heterozygous breeding, generated previously by inactivation of the Gria1 gene [21] and genotyped as reported elsewhere [26]. The Gria1−/− mouse line is available at the Jackson Laboratory (B6N.129-Gria1 tm2Rsp/J, stock number: 019012). During experiments, mice were individually-housed or grouped-housed in same-sex cages under standard laboratory conditions (12-h light-dark cycle; lights on at 6∶00 A.M.; temperature 20–23°C; relative humidity 50–60%; aspen chip beddings).
Drugs
The powdered laboratory chow (R36, Lantmännen Lantbruk, Stockholm, Sweden and RM1 (E) SQC FG, 811004, Special Diet Services, Essex, UK) was available ad libitum and it was mixed homogenously with drugs as follows: lithium carbonate (Sigma-Aldrich Corp St. Louis, MO USA) was added 1.2 g/kg chow for the first week and 2.4 g/kg until the end of treatment; sodium valproate (Deprakine, Sanofi Aventis Oy) 10 g/kg for the total treatment time; topiramate (Topiramat Ratiopharm) 27 mg/kg for the total treatment time; lamotrigine (Lamotrigin Ratiopharm) 75 mg/kg for the total treatment time. Doses aimed at human therapeutic levels and were based on the literature [30], [31] or pilot studies. Lithium and valproate group had free access to additional saline bottles to prevent possible ion imbalances during chronic treatments [32]. Control chow was made in the same way but without drugs. Animals were observed daily for any significant body weight changes or toxicity signs during the treatments.
Drug Concentrations
Lithium and lamotrigine concentrations were analysed in hospital laboratories from blood samples of mice taken after the test of locomotor activity in novel environment. Trunk blood was collected by decapitation and serum separated by centrifugation. Concentration of lithium was determined by a colorimetric method [33] in NordLab Oulu (Oulu, Finland) and that of lamotrigine by a liquid chromatography after solid-phase extraction [34] in Rinnekoti-Foundation Laboratory (Espoo, Finland).
Experimental Design
Animals received drug treatments in their diet for 28 days, followed by behavioural testing while they still continued on the diets. Experimental design is presented in Fig. 1. We used test batteries to measure sets of specific behavioural features. The test order as well as a recovery break between the tests was designed to minimize the effect of previous test on subsequent ones. The first battery consisted of elevated-plus maze, forced swimming and locomotor activity tests in this order for two cohorts. In the second test battery, the animals were tested for sucrose preference using the two-bottle choice test to evaluate hedonistic behaviour towards sucrose, followed by open field test combined with new object installation, tail-suspension test, social interaction test and locomotor activity test after acute psychostimulant injection as the last test (the drug treatments lasted up to 2 months). Locomotor activity test was performed in several independent cohorts of WT and Gria1−/− mice and at the end of the first test battery and the results were pooled. All tests were performed between 08∶00 and 14∶00 h. In order to study sucrose drinking and wheel running, mice were separated to individual cages when the treatments began (4 weeks before the beginning of test) to minimize the effect of acute social isolation on hedonistic behavior [35] and this housing was kept throughout this battery of tests. The nesting material was provided in cages [36], [37]. Between the tests, the animals were handled at least two times weekly for body weight measurement or for changing to clean cages. Also, for certain time period the environment was enriched with running wheels to assess hedonistic behavior.
Figure 1. Experimental schedule for different cohorts of mice.
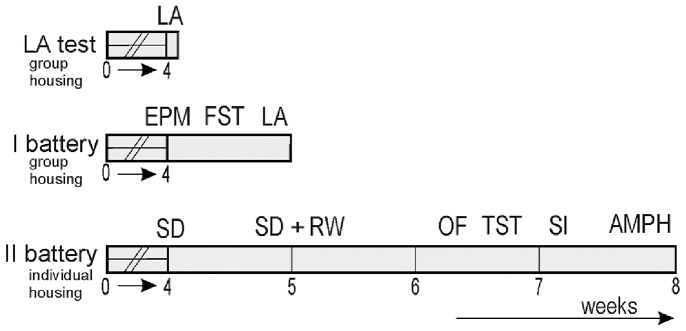
LA, locomotor activity test; EPM, elevated plus maze test; FST, forced swimming test; SD, sucrose drinking; RW, running wheel access; OF, open field test; TST, tail suspension test; SI, social interaction test; AMPH, D-amphetamine-induced LA test.
Locomotor activity (LA) in a novel environment was observed in plastic cages (40×30×20 cm) as described in detail [24]. Animals were habituated to the experimental room at least for 1 h before the test. Horizontal movements of eight to ten mice, placed in visually isolated cages in a sound-attenuated room at light intensity of 175 lx, were simultaneously recorded for 2 h using EthoVision Color-Pro 3.0 video tracking software (EthoVision System, Noldus Information Technology, Wageningen, Netherlands). All treatment groups are listed in Table 1.
Table 1. Characteristics of the treatment groups and drug concentrations.
| Treatment | Gender | Genotype (numberof animals) | Age(weeks) | Initial bodyweight (g) | Final bodyweight (g) | Drug concentrationin blood |
| Control | Male | WT (16) | 29.4±3.1 | 31.6±0.9 | 33.9±0.9*** | / |
| Gria1−/− (10) | 30.1±2.2 | 29.1±0.9 | 31.6±0.9*** | / | ||
| Female | WT (11) | 15.7±0.7 | 20.2±0.2 | 22.0±0.2*** | / | |
| Gria1−/− (10) | 22.3±4.0 | 21.1±1.0 | 23.7±0.9*** | / | ||
| Lithium | Male | WT (8) | 21.0±1.8 | 29.6±0.6 | 29.7±0.5 | 0.7±0.1 |
| Gria1−/− (7) | 20.6±2.4 | 28.0±0.9 | 28.2±0.6 | 0.8±0.1 | ||
| Female | WT (13) | 23.7±2.3 | 22.5±0.6 | 23.4±0.7 | 0.7±0.1 | |
| Gria1−/− (12) | 23.2±2.0 | 23.9±1.0 | 24.0±0.7 | 0.9±0.1 | ||
| Valproate | Male | WT (13) | 14.7±0.3 | 24.7±0.6 | 25.7±0.4 | / |
| Gria1−/− (10) | 25.7±5.6 | 24.9±1.4 | 26.7±1.0 | / | ||
| Female | WT (4) | 14.6±1.3 | 17.8±1.5 | 21.1±1.0 | / | |
| Gria1−/− (4) | 17.3±2.8 | 18.5±1.1 | 20.1±0.7 | / | ||
| Topiramate | Male | WT (8) | 16.7±0.7 | 28.0±1.0 | 30.1±1.2*** | / |
| Gria1−/− (8) | 21.4±2.8 | 27.6±0.7 | 29.0±0.7** | / | ||
| Female | WT (9) | 17.9±0.7 | 22.4±0.8 | 23.9±0.7** | / | |
| Gria1−/− (10) | 16.4±1.0 | 20.2±0.6 | 21.9±0.8** | / | ||
| Lamotrigine | Female | WT (8) | 33.4±7.4 | 23.4±1.0 | 23.1±0.7 | 3.6±0.4# |
| Gria1−/− (9) | 33.8±4.1 | 25.1±1.0 | 24.3±0.7 | 5.0±0.4 |
Treatment groups, ages at the time of locomotor activity tests in novel cages, and body weights at the beginning and end of treatments are showed. Drug concentrations were analysed from serum samples collected after the locomotor activity tests and presented in mM (lithium) or µM (lamotrigine). Data are means ± SEM. *p<0.05, **p<0.01, ***p<0.001 compared to body weight at the beginning of treatments (paired t-test), # p<0.05 for the genotype difference (unpaired t-test).
Elevated plus maze (EPM) test was used to assess mouse anxiety [38]. The maze was made of grey plastic, elevated 50 cm from the floor level. It consisted of a central platform (5×5 cm), two open arms (5×40 cm with a 0.2 cm edge) and two enclosed arms (5×40×20 cm). The mice were placed individually on the central platform facing the open arm and allowed free exploration of the maze for 5 min. Central square was defined until 2 cm out of the central platform, allowing detection of the centre of animal. Movements were recorded and analysed automatically with the EthoVision software.
Forced swimming test (FST) and tail suspension test (TST) were done to assess animal’s coping with despair-like condition [39]. FST [24], [40], [41] was conducted so that each mouse was placed for 6 min individually in transparent cylindrical beakers (height 25 cm, diameter 15 cm) containing 3 l of water (23°C). In TST [24], [42], the mice were tape-attached individually by their tails on elevated metal bar for 6 min. Behavior of each mouse was video-recorded and analysed later by data acquisition program Ethograph (Ethograph software 2.06, RITEC, St. Petersburg, Russia) for the last 4 min of the tests. Behavioural analysis was focused on the mouse immobility which was indicated when the animal floated passively, making only small movements with the hind paws and/or tail to prevent sinking in the water during FST or when no struggling signs were obvious during TST.
Sucrose preference alone (SD) or in combination with running wheel (RW) was performed to assess hedonic propensity of the mice [43]. Voluntary sucrose drinking [8% weight/volume (w/v)] was evaluated using short-term intermittent protocol [44], [45]: on alternate days the mice obtained access to choose between water and sucrose. The alternation of sucrose-free and sucrose days (S1, S2 and S3) was repeated 3 times, and thereafter the alternation was repeated 3 times (S4, S5 and S6) with running wheels (RW; ENV-044 model; Med Associates, Inc., St. Albans, Vermont, USA). The position of water and sucrose bottle was switched pseudo-randomly to prevent the development of place preference towards the sides. Fluid intake and body weights were monitored daily. Sucrose solution intake (in ml) was calculated by dividing loss of sucrose bottle weight with 1.08 [weight in grams of 1 ml of 8% (w/v) sucrose solution]. Sucrose preference (%) was calculated as a percentage of sucrose intake out of the total fluid intake (sucrose plus water).
Open field (OF) test combined with new object exploration was performed to evaluate anxiety and mouse explorative activity [46]. The animals were placed individually on the centre of empty cage (33×55×19 cm) divided into fifteen squares (11×11 cm) initially for 3 min. Testing was performed at the light intensity of 175 lx. Then, a round, textured object (diameter = 4 cm) with three 1-cm holes was placed on the centre for the next 3 min [41]. Mouse behaviour was video-recorded and analysed later by Ethograph for the last 3 min of the test. Arena of the OF was divided into peripheral and central zones in the video tracking software (EthoVision), which was used to track the mouse automatically for the whole 6 min duration of the test. Object-related behaviours (sniffing, manipulation and nose-pokes of the object) were counted as total object interactions. Other behaviours including the rearing, locomotion and individual behaviours (any other behavior which does not include locomotion) were divided to central and peripheral behaviours according to the virtual zones of the OF.
Social interaction (SI) was evaluated for 10 min on a new territory among animals receiving the same treatment [41]. Two or three animals were simultaneously introduced to a novel cage with fresh bedding. The behaviour of animals was video-recorded. Behavior of every mouse in the temporarily-formed group was analysed by the Ethograph software for (1) individual behavior without contacts with other members, such as locomotor activity, (2) reciprocal (simultaneously-initiated) contacts, and (3) passive contacts initiated by other group members. All observed contacts were non-aggressive, although aggressive behaviour was expected.
Response to psychostimulants was tested 30 min after a single i.p injection of 2 mg/kg amphetamine diluted in saline (D-amphetamine sulphate, Dexedrine, GlaxoSmithKline, Brentford, UK) in a volume of 10 ml/kg. Mouse locomotion was monitored for 1 h in a novel arena by using the EthoVision system and software.
Statistics
Statistics were carried out using PASW Statistic 18 software (SPSS Inc., Chicago, IL, USA). Multivariate ANOVA (two-way) followed by a Bonferroni (p<0.05) post hoc test was used to analyse the data obtained from EPM, FST, TST, new object exploration and social activity tests. For the repeated measurements such as locomotor activity and sucrose consumption, analysis for repeated measurements followed by a Bonferroni post hoc test (p<0.05) was applied. Kaplan-Meier survival analysis with a Mantel-Cox non-parametric test (p<0.05) was used to analyse the latencies to immobility, to the contact with the new object and to the social contact. Spearman’s correlation coefficient was used to test correlation between sucrose drinking and activity on running wheels. Two-tailed t-test was used to compare differences in results for body weights (paired), drug concentrations (unpaired) and LA (unpaired) obtained in different cohorts. All behavioural elements or group of the elements obtained by the Ethograph software were statistically analysed in four measurements: total duration (sum of the duration of the element during the test), medial duration (ratio of the element duration to its total frequency), total frequency, and relative frequency (ratio of the element frequency to the sum of all frequencies of the observed elements).
Results
Chronic Treatment with Mood Stabilizers Lithium and Valproate and Effects on Hyperactivity of Gria1−/− Mice
As locomotor activity tests were performed in several cohorts with different experimental schedules (as an independent test and at the end of the first battery, Fig. 1), we analysed statistically whether the previous tests affected the later ones, by using t-tests within each 6 experimental groups. No difference was observed in the total 2-h locomotor activity within the control-treated Gria1−/− mice (t18 = 0.02, p>0.05) between the separate test [571±45 m (10), mean ± SEM, (n)] and the last test of the first battery [572±42 m (10)], nor within the corresponding groups of control-treated WT mice [t25 = 0.25, p>0.05, 308±25 m (12) and 300±22 m (15)]. Nor were there any differences in LA between independently performed and after EPM and FST performed tests within the lithium-treated Gria1−/− mice [t17 = 1.47, p>0.05, 417±51 m (7) and 509±38 m (12)], lithium-treated WT mice [t19 = 0.47, p>0.05, 281±23 m (8) and 302±31 m (13)], valproate-treated Gria1−/− mice [t12 = 1.17, p>0.05, 426±24 m (7) and 375±36 m (7)] nor valproate-treated WT mice [t15 = 0.33, p>0.05, 240±18 m (7) and 229±25 m (10)]. We conclude that previous testing experience did not influence the novelty-induced LA.
In the first 60-min of LA test, there was a significant genotype × treatment × gender interaction (F2,106 = 3.20, p<0.05) after three-way ANOVA. Bonferroni post-hoc comparisons showed that female and male Gria1−/− mice travelled longer distances than WT mice, in all treatment groups (p<0.05). There were no significant gender differences in LA scores within Gria1−/− mice on control-, lithium- or valproate-diets (p>0.05), nor within the WT mice on these diets (p>0.05). Both lithium (p<0.05) and valproate (p<0.05) treatments were efficient in reducing locomotor hyperactivity of female Gria1−/− mice compared to control treatment. The drugs did not affect LA of WT mice in comparison to control diet. Valproate-treated Gria1−/− (p = 0.075) and WT (p = 0.064) male mice tended to show reduced LA compared to respective control-treated mice. Thus, only a slight difference in response of female and male Gria1−/− mice to the lithium treatment appeared during the first 60-min of the test. Statistical analysis showed that gender did not interact with other factors in the second 60-min of exposure to novel cages, when the effect of the drugs on the locomotor activity was predominant (Fig. 2). Because of that, we analyzed below pooled female and male data also for the first 60-min of the test using two-way ANOVA and these results are presented on Fig. 2C.
Figure 2. Effects of chronic lithium and valproate on hyperactivity of the Gria1−/− mice.

Locomotor activities of WT and Gria1−/− mice treated chronically with control diet and lithium- and valproate-supplemented diets in 5-min time intervals for the whole 2-h period (A, B) and cumulative activities during the first and last 60 min of exposures to novel arena (C, D). Data are means ± SEM (n = 14–27). ***p<0.001 for the differences between genotypes after the same treatment; ## p<0.01, ### p<0.001 for the differences from controls of the same genotype (two-way ANOVA followed by Bonferroni post-hoc test). In panel B, the earliest significant reduction from the control activity has been marked by #(p<0.05).
Locomotor activity of Gria1−/− mice during the first 60-min exposures to novel environment after the chronic drug treatments was markedly increased as compared to that of WT mice (F1,112 = 101.17, p<0.001) (Fig. 2A–C). Only valproate-treated Gria1−/− had reduced LA as compared to control Gria1−/− mice (F2,112 = 10.18, p<0.001). The LA of the Gria1−/− mice continued to be higher than that of WT mice till the end of the 2-h monitoring period, but cumulative LA during the last 60 min showed that both lithium and valproate were efficient in reducing LA in Gria1−/− mice (F2,112 = 4.82, p<0.05 for genotype × treatment interaction), but not in WT animals (Fig. 2D). Thus, chronic treatments led to increased habituation of the Gria1−/− mice. Indeed, the effects were observed only after 15 min by valproate and after 40 min by lithium (Fig. 2B), and the reductions were more pronounced for 60–120 min than 0–60 min periods (Fig. 2C and D). The differences between treatments were indistinguishable after 70 min (Fig. 2B).
Effects on Elevated Plus-maze Test and Open Field Exploration
Gria1−/− mice visited the centre of the elevated plus-maze more frequently than the WT mice (F1,54 = 5.86, p<0.01) and spent less time in the closed arms (F1,54 = 16.10, p<0.001) (data not shown), which measures were not affected by drug treatments. Control Gria1−/− mice spent more time in the open arms than the control WT mice (F1,54 = 5.20, p<0.05) independent of the treatments (Fig. 3A). No genotype or treatment effects were observed on number of entries to open arms or on distance travelled in the open arms (Fig. 3B–C).
Figure 3. Effects of chronic drug treatments on behaviour of Gria1−/− mice in elevated-maze test of anxiety.

Control Gria1−/− spent more time in the open arms than control WT mice. Chronic lithium and valproate did not affect the time spent in (A) or entries to (B) or distances travelled in open arms (C). Data are means ± SEM (n = 7–12). **p<0.01 for the difference between genotypes after the same treatment (two-way ANOVA followed by Bonferroni post-hoc test).
Gria1−/− mice moved more than WT mice in the whole open field arena during 3 min before the object was introduced (Fig. 4A; F1,61 = 19.94, p<0.001), and chronic treatments with lithium and valproate did not affect total movements (F2,61 = 1.10, p>0.05). Gria1−/− mice visited the OF centre more frequently (Fig. 4C; F1,61 = 4.85, p<0.05) than WT mice, but travelled similar distances in the centre as them (Fig. 4B; F1,61 = 1.51, p>0.05). Valproate increased the time spent in the central zone in WT mice, but not in Gria1−/− mice (Fig. 4D; genotype × treatment interaction F2,61 = 3.78, p<0.05).
Figure 4. Effects of chronic drug treatments on open field activity and interaction with a novel object.

Distance travelled in the whole arena (A, E) and in the centre (B, F), entries to the centre (C, G) and time spent in the centre (D, H) during the first 3 min before the object was introduced (A–D) and during the next 3 min when a novel object was located in the centre of arena (E–H). (I) Representative tracking paths for Gria1−/− and WT mice during the 2nd 3-min period with an object, with a square in the centre delineating visual centre of arena. (J) Frequencies of object interactions, scored using Ethograph software. Data are means ± SEM (n = 6–15). *p<0.05, **p<0.01, ***p<0.001 for the differences between genotypes after the same treatment; # p<0.05, ## p<0.01 for the differences from the control within the same genotype (two-way ANOVA followed by Bonferroni post-hoc test).
After the object was introduced in the OF for the next 3 min, Gria1−/− mice kept moving more in the whole arena (Fig. 4E; F1,61 = 92.90, p<0.001) as well as in the arena centre (Fig. 4F; F1,61 = 55.23, p<0.001), independently of chronic treatments. Gria1−/− mice visited the central zone with the object more often (Fig. 4G; F1,61 = 81.15, p<0.001) and stayed there longer (Fig. 4H; F1,61 = 22.32, p<0.001). Control Gria1−/− mice were in contact with the object more frequently than WT mice, while treatments of the Gria1−/− mice with valproate and lithium decreased the frequency to the level of the corresponding WT mice (Fig. 4J; F2,61 = 3.97, p<0.05).
Effects on Tests for Goal-directed Behaviours
Forced swimming test (FST) has been validated to examine increased vigour and goal-directed behavioural pattern of mania [39]. The increased goal-directed behaviour in both FST and tail suspension test (TST) has been already reported in Gria1−/− mice [24]. In the FST, Gria1−/− mice were less immobile as compared to the WT mice (Fig. 5; F1,60 = 23.87, p<0.001), especially the control and valproate-treated groups. Lithium-treated WT mice showed a trend towards being less immobile than other WT groups (F2,60 = 3.13, p = 0.051). Kaplan-Meier analysis showed that immobility was observed later in the control and lithium-treated Gria1−/− mice than in the corresponding WT mice (14.15, p<0.0001 and 5.73, p = 0.017, respectively, data not shown).
Figure 5. Effects of chronic drug treatments in behavioural despair paradigms.

Gria1−/− mice were less immobile than the WT mice in both the forced swimming (FST) and tail suspension (TST) tests. Data are means ± SEM (n = 7–13). **p<0.01, ***p<0.001 for the differences between genotypes after the same treatment; # p<0.05 for the differences from the control within the same genotype (two-way ANOVA followed by Bonferroni post-hoc test).
In the TST, again the Gria1−/− mice were less immobile than the WT mice (Fig. 5; F1,61 = 22.79, p<0.001), especially the control and lithium-treated Gria1−/− mice. Valproate prolonged the immobility in the Gria1−/− mice (F2,61 = 5.34, p<0.01). Kaplan-Meier analysis showed that lithium-treated Gria1−/− mice demonstrated immobility later than control Gria1−/− mice (log-rank Mantel-Cox 14.32, p<0.0001) and lithium-treated WT mice (13.50, p<0.0001) (data not shown).
We compared the immobility times in FST and TST tests that were carried out in group- or individually-housed mice, respectively. Within the Gria1−/− mice, no difference (t18 = 0.39, p>0.05) was observed in the total immobility time of between FST and TST [86.4±15.1 s (12), mean ± SEM, (n) and 96.1±20.3 s (9), respectively], and similarly, the immobility times of WT mice in FST [176.5±8.8 s (12)] and TST [152.9±12.2 s (14)] were identical (t24 = 1.52, p>0.05). Also, lithium- (t20 = 0.46, p>0.05) and valproate-treated (t11 = 1.03, p>0.05) Gria1−/− mice spent similar times immobile in these tests, as did lithium- (t26 = 1.75, p>0.05) and valproate-treated (t21 = 0.63, p>0.05) WT mice. It seems that the housing conditions (individual or group-housing) hardly affected the behavior of Gria1−/− and WT mice.
Effects on Social Interaction
Reciprocally initiated contacts were shorter-lasting but more frequent in Gria1−/− mice than in WT mice (F2,60 = 4.64, p<0.05), while valproate treatment levelled the Gria1−/− mice behaviour to that of the WT mice (Fig. 6AB; treatment effect F2,60 = 3.46, p<0.05 and F2,60 = 3.76, p<0.05 for the mean time and frequency, respectively). Unlike WT, Gria1−/− mice spent more time in passive interaction receiving contacts from other mice (F1,60 = 4.24, p<0.05; data not shown), independently of the treatments with lithium and valproate. They were engaged more frequently in individual behaviour than WT mice (F1,60 = 31.22, p<0.001) and lithium increased it over the control and valproate-induced levels (F2,60 = 9.91, p<0.001; data not shown). Lithium delayed the appearance of reciprocal contacts between the group members in WT mice (log-rank Mantel-Cox 5.11, p<0.05), and reciprocal contacts were observed later in control Gria1−/− than WT mice (log-rank Mantel-Cox 4.70, p<0.05; data not shown).
Figure 6. Effects of chronic drug treatments on social activity on a new territory.

Two or three animals on the same treatment were observed for 10(simultaneously-initiated) contacts. The contacts were shorter (A) but more frequent (B) among the control Gria1−/− mice than among the WT mice. Chronic valproate, but not lithium, moderated the activity of the Gria1−/− mice to the level found in the WT mice. Data are means ± SEM (n = 6–15). *p<0.05, ***p<0.001 for the differences between genotypes after the same treatment; # p<0.05 for the differences from the control within the same genotype (two-way ANOVA followed by Bonferroni post-hoc test).
Effects on Amphetamine-induced Hyperactivity
Gria1−/− and WT mice are similarly activated by acute amphetamine challenge [26]. Here, we studied whether this dopaminergic challenge would be affected by chronic drug treatments. Gria1−/− mice preserved their higher locomotor activity after 2 mg/kg amphetamine challenge throughout the 60-min monitoring period as compared to WT mice, independently of the chronic treatments with lithium and valproate (F11,671 = 12.37, p<0.001 and F1,61 = 50.29, p<0.001 for time intervals and genotype, respectively). Locomotor activity distances (in meters as estimated by using Ethovision video-tracking) were for the control, lithium and valproate groups of the WT mice: 311±18 (mean ± SEM, n = 14), 342±22 (15) and 295±11 (13), respectively, and for the corresponding treatment groups of the Gria1−/− mice: 507±59 (9), 489±32 (10) and 462±24 (6), respectively.
Effects on Sucrose Preference and Activity on Running Wheels
To compare the mouse lines and to assess the effects of lithium and valproate on hedonic behaviour, we tested the Gria1−/− and WT mice for preference of sucrose-containing solution and for running wheel activity. The Gria1−/− mice had higher preference for sweet taste than WT mice (F1,57 = 8.87, p<0.01), although all animals preferred sucrose solution over plain water. Animals receiving lithium (Fig. 7A) and valproate (Fig. 7C) increased sucrose consumption as compared with respective control animals (F2,57 = 8.86, p<0.001). Interestingly, the control WT mice reduced their consumption of sucrose during the first (S4) and third (S6) session in the presence of running wheels (F5,285 = 5.12, p<0.01). Otherwise the access to running wheels little affected the preference for sucrose, although particularly the valproate-treated WT mice clearly increased their activity on running wheel on the last session (S6, Fig. 7D; F2,250 = 5.83, p<0.01). Also lithium treatment increased running activity in the WT mice on session S6 (Fig. 7B). Thus, no correlation (p>0.05) was observed between running wheel activity and sucrose preference in WT (0.01) or Gria1−/− mice (0.091).
Figure 7. Effects of chronic lithium and valproate on sucrose preference and running wheel activity Gria1−/− mice.

Preference for sucrose-containing solution over plain water, with and without an access to running wheels (RW) on sucrose-choice days (S1, S2 and S3) in animals treated with lithium (A) and valproate (C). Running wheel activity during the sucrose-choice days (S1, S2, S3) in animals treated with lithium (B) and valproate (D). Data are means ± SEM (n = 6–14). *p<0.05, **p<0.01 for the differences between genotypes after the same treatment; # p<0.05, ### p<0.001 for the differences from the control treatment within the same genotype (two-way ANOVA followed by Bonferroni post-hoc test).
Effects of Anticonvulsants Topiramate and Lamotrigine on Behavior of Gria1−/− Mice
In WT and Gria1−/− females, lamotrigine reduced cumulative 2-h novelty-induced LA in Gria1−/− mice (Fig. 8A–B; treatment effect F1,35 = 5.93, p<0.05, genotype effect F1,35 = 46.38, p<0.001), with the earliest significant reduction taking place at 20 min after starting the test (Fig. 8A).
Figure 8. Effects of chronic treatments with anticonvulsants lamotrigine and topiramate on behavior of Gria1−/− mice.

Cumulative locomotor activities of Gria1−/− and WT mice treated chronically with lamotrigine- or topiramate-supplemented chow for 5-min time intervals (A, C) and for the whole 2-h experiment (B, D). Data are means ± SEM (n = 8–27). In A and C, the earliest significant point from the control activity has been marked by #(p<0.05). Topiramate treatment reduced the time spent in open arms of elevated-plus maze (E; means ± SEM, n = 9–12) and increased immobility time in forced swimming test (F; means ± SEM, n = 11–12) in Gria1−/− mice. **p<0.01, ***p<0.001 for the difference between genotypes after the same treatment; # p<0.05, ### p<0.001 for the difference from the control within the same genotype (two-way ANOVA followed by Bonferroni post-hoc test).
There was no difference (t16 = 1.58, p>0.05) in LA of topiramate-treated Gria1−/− mice between the two LA tests [performed independently (351±31 m (6), mean ± SEM (n)) or after other tests in the battery (457±44 m (12)], nor within the WT mice [t15 = 0.15, p>0.05; 271±12 m (6) vs. 265±30 m (11)]. Therefore, the data were pooled, analysed and presented in Fig. 7C–D. The gender effect was not significant for either two 1-h time intervals (F1,74>0.01, p>0.05, three-way ANOVA) and, therefore, female and male data were pooled. Chronic treatment with topiramate reduced 2-h novelty-induced hyperactivity specifically in Gria1−/− mice in a time-dependent manner, without affecting WT animals (Fig. 8C–D; F23,1794 = 3.09, p<0.01 for time interval × genotype × treatment interaction). The first effect of topiramate on locomotor activity was observed after 20 min (Fig. 8C). In the EPM, treatment with topiramate reduced the time that Gria1−/− mice stayed on the open arms (Fig. 8E; F1,39 = 7.37, p<0.05 for genotype × treatment interaction), suggesting anxiogenic or reduced risk-taking effects. Topiramate increased total immobility time in Gria1−/− mice, but not in WT mice (Fig. 8F, F 1,43 = 5.16, p<0.05 for genotype × treatment interaction) in the FST. Kaplan-Meier analysis showed that topiramate-treated Gria1−/− mice demonstrated immobility earlier than the control Gria1−/− mice (log-rank Mantel-Cox 4.26, p = 0.039).
Discussion
In this study, we confirmed the elevated activity and highly exploratory phenotype of Gria1−/− animals and conducted a predictive assessment of efficacy of chronic treatments of standard and novel, structurally and mechanistically different mood stabilizers on a battery of behavioural tests for hyperactive aspects of disorders, such as bipolar mania-like illness, schizophrenia and schizoaffective disorder. We found that the treatments with lithium, valproate, topiramate and lamotrigine all reduced hyperactivity by increasing habituation of the Gria1−/− animals in a novel environment and variably affected other behaviours.
Locomotor over-activity and increased exploration in open space as well as over-reactivity to new objects were characteristic for the Gria1−/− mice, indicative of high novelty seeking and risk-taking behaviour. This behaviour was particularly sensitive to both lithium and valproate. Dysfunctional exploration pattern has been observed in bipolar manic and schizophrenic patients [47], [48]. Interestingly, bipolar manic patients are more mobile and habituate faster than schizophrenic patients. Moreover, bipolar patients are more interested in new objects and spend more time near to the object [47], [49]. Thus, specific behavioural features differentiate bipolar patients from schizophrenic ones and could be used as discriminating criteria between these disorders. Moreover, behavioural disinhibition pattern was suggested as endophenotype of bipolar disorder [50]. In the present work, chronic treatment with the mood stabilizers studied here attenuated the 2nd-h hyperlocomotion in a novel environment in the Gria1−/− mice, but were not effective in reducing the initial hyperactivity or the acutely exacerbated locomotion by amphetamine challenge. Gria1−/− mice show normal home-cage activity pattern and diurnal rhythm [24] and amphetamine elevates the activity similarly as is the WT mice [26], which suggest that their abnormal novelty-induced hyperactivity resembles more mania-type behaviour than attention deficit hyperactivity disorder-type of behaviour.
Novelty-induced hyperactivity of the Gria1−/− mice is greatly reduced by acute blockade of AMPA receptors with NBQX [24] and by mGlu2/3 receptor agonist LY354740 [51], which results are consistent with hyperactive glutamate system in the Gria1−/− mice. Our recent c-Fos mapping data suggest overactivity of the dorsal hippocampus of the Gria1−/− animals in a novel situation [24], [51]. Of the mood stabilizers studied here, topiramate and lamotrigine reduce glutamate functions by inhibiting glutamate receptors and glutamate release, respectively [52]–[54], which could explain their efficacy. Also treatment with lithium is known to indirectly affect NMDA-type glutamate receptor function, subunit expression and phosphorylation and activation of the related intracellular signalling cascades, such as phospholipase PLA2 and nitric oxide (NO) pathways [55]–[58], while the NMDA receptor antagonists potentiate the actions of lithium [59]. However, other mechanisms than glutamate antagonism are likely to be involved also, since valproate is not known to antagonize the glutamate system, and indeed valproate has failed to protect from NMDA-induced seizures [60]. Protein kinase C and extracellular signal-regulated kinase cascades constitute as shared targets for lithium and valproate and are involved in mediating their anti-manic actions on various facets of the disease [61]. These pathways may also be among the mediators of the anti-hyperactive effect of lithium and valproate in the Gria1−/− model of abnormal hyperactivity.
Chronic treatment with lithium could also suppress hyperlocomotion via presynaptic mechanisms that decrease release of catecholamines or inhibit their synthesis [62]. Indeed, dopamine D2 receptor antagonist haloperidol somewhat reduces hyperactivity in the Gria1−/− and WT mice [25]. However, depletion of dopamine levels by inhibition of tyrosine hydroxylase did not affect the locomotor phenotype of these mice [22], neither was there any differential activation of the ventral tegmental area dopaminergic (VTA DA) neurons or striatal neurons between Gria1−/− and WT mice after 2 h in novel environment [24], and therefore, dopaminergic mechanisms are unlikely to decisively contribute to the hyperlocomotor phenotype. On the other hand, glutamate receptor neuroplasticity in VTA DA neurons is associated with the effects of rewarding drugs of abuse [63], [64], and this neuroplasticity process might be deficient in Gria1−/− mice as the opioid morphine failed to induce an increase of AMPA/NMDA ratio of VTA DA neurons in Gria1−/− mice like it did in WT animals [65]. State-dependent place conditioning with morphine is abnormal in Gria1−/− mice [65]. In the present study, of the two naturally rewarding stimuli, sucrose drinking and running wheel activity, only sucrose drinking test was useful to discriminate between the Gria1−/− and WT mice, while running activity did not differ between the genotypes. Dopaminergic projection from the VTA is important for appetitive behaviour and hedonic responses to palatable food [66], with a lesion of this projection decreasing sucrose intake [67]. Activation of VTA DA neurons by disinhibition via cannabinoid CB1 receptor-dependent mechanism has been linked to rewarding properties of voluntary running wheel activity [68]. However, we found no interaction between accesses to sucrose drinking and running wheels in Gria1−/− and WT mice on control diet, and unexpectedly, treatment with lithium and valproate rather increased than suppressed sucrose preference. Unlike sucrose consumption, which increased in both Gria1−/− and WT mice by valproate and lithium, running activity increased significantly only in WT animals. Rewarding responses were not consistently affected by the drugs in the present study.
The main effects of the drugs studied here were on hyperactivity of the Gria1−/− animals, but they produced also some effects on other behaviours. Chronic lithium reduced the frequency of contacts to new objects in the open field, as did valproate. Valproate induced an anxiolytic-like effect in the WT mice that was not observed in the Gria1−/− mice. In social interaction test, valproate prolonged reciprocal social contacts only in the Gria1−/− mice. The effects of lithium and valproate in different tests might be related to increased habituation. Topiramate had an anxiogenic-like effect in the Gria1−/− animals. Topiramate and lamotrigine were not studied here as widely as lithium and valproate for other behaviours than hyperactivity. Even though we did not find any significant difference in the behavior of mice differently housed (see the immobility times in FST vs. TST in the Results section), deprivation of social contacts is known to impair behavior [69]. Isolation of rats increases motivational value of sucrose [35] and number of social contacts [70], [71]. Multiple testing can also have a substantial influence on behavior [72]. In the present study, locomotor activity of mice with previous testing history was similar compared to those naïve to experimentation. However, this does not straightforwardly imply that the behavior observed in other tests in a multiple testing battery would not be affected by the prior testing and associated stress. Still, behavioural test batteries in rodents are widely used and favoured for wide behavioural screening and designed to cover many distinct behavioural domains relevant for human neuropsychiatric disorders [73]. We conclude that in the present experiments, the main effects that regulated the behaviour, especially the hyperactivity, were the mouse genotype and chronic drug treatments.
Rodent models in neuropsychiatry usually phenocopy only some aspects of a disease, such as hyperactivity. Hyperactivity is a shared feature among several psychiatric conditions, such as mania, anxiety, attention deficit hyperactivity disorder and autism spectrum disorders, in addition to some forms of schizophrenia. Animal models are valuable tools for studying predictive validity of treatments, but even with partial face validity, they usually have weak construct validity [74]. In Gria1−/− mouse line, hyperactivity is pronounced and concomitant with other behavioral abnormalities indicative of highly disinhibited behavior. Moreover, as we show here, their behavioral abnormalities and deficits are controlled by mood stabilizing medications. Importantly, AMPA receptors, as determinants of synaptic plasticity might have a critical role during late adolescence for the onset of cognitive and behavioral abnormalities for neuropsychiatric disorders. Ablation of GluA1 subunit from the hippocampus in late adolescence reproduced all behavioural abnormalities of mice with global deletion of GluA1 subunit, except for social deficits [75]. However, aggressiveness as a correlate of irritability and easily provoked behavior is lacking in Gria1−/− mice [29]. There are two other interesting glutamate-linked models for mania-like hyperactivity. They include the mice deficient in kainate receptor GluK2 subunit (Grik2−/−) [76], in which chronic treatment with lithium reduces hyperactivity, risk-taking behaviour and aggressive manic displays, and the Shank3 overexpressing mice [77], in which acute valproate, but not chronic lithium, reduces hyperactivity. While SHANK proteins are postsynaptic density scaffolding proteins in glutamate synapses and implicated in a number of neuropsychiatric disorders [78], heteromeric kainate receptors, assembling also GluK2 subunits, regulate presynaptically and postsynaptically neurotransmission of both interneurons and principal neurons [79], [80]. Together with the present results on Gria1−/− mouse line, these models suggest an important role for excitatory glutamate transmission in disorders with hyperactivity.
In conclusion, this is the first report to describe behavioural effects of chronic treatments with different clinically used mood stabilizers in the AMPA receptor GluA1 subunit-deficient mice using test batteries specific to some hyperactive facets of bipolar illness, schizophrenia and schizoaffective disorder. Gria1−/− mice showed disinhibited risk-taking behaviour, hyperlocomotion and social deficits, which were at least partially reversed by mood stabilizers, and therefore, we suggest that this mouse line can be used as a model for screening for novel drugs to treat hyperactive neuropsychiatric disorders.
Supporting Information
The ARRIVE Guidelines Checklist.
(PDF)
Funding Statement
The study was supported by the Academy of Finland, the Sigrid Juselius Foundation, the Orion research foundation and the Jane and Aatos Erkko Foundation (ERK). The support of the FinPharma Doctoral Program Drug Discovery section (MM) is gratefully acknowledged. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Inta D, Monyer H, Sprengel R, Meyer-Lindenberg A, Gass P (2010) Mice with genetically altered glutamate receptors as models of schizophrenia: a comprehensive review. Neurosci Biobehav Rev 34: 285–294. [DOI] [PubMed] [Google Scholar]
- 2. Coyle JT, Duman RS (2003) Finding the intracellular signaling pathways affected by mood disorder treatments. Neuron 38: 157–160. [DOI] [PubMed] [Google Scholar]
- 3. Krystal JH, Belger A, D’Souza DC, Anand A, Charney DS, et al. (1999) Therapeutic implications of the hyperglutamatergic effects of NMDA antagonists. Neuropsychopharmacology 21: S143–S157. [Google Scholar]
- 4. Newcomer JW, Farber NB, Jevtovic-Todorovic V, Selke G, Melson AK, et al. (1999) Ketamine-induced NMDA receptor hypofunction as a model of memory impairment and psychosis. Neuropsychopharmacology 20: 106–118. [DOI] [PubMed] [Google Scholar]
- 5. Maeng S, Zarate CA Jr (2007) The role of glutamate in mood disorders: results from the ketamine in major depression study and the presumed cellular mechanism underlying its antidepressant effects. Curr Psychiatry Rep 9: 467–474. [DOI] [PubMed] [Google Scholar]
- 6. Moghaddam B, Adams BW (1998) Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science 281: 1349–1352. [DOI] [PubMed] [Google Scholar]
- 7. Anand A, Charney DS, Oren DA, Berman RM, Hu XS, et al. (2000) Attenuation of the neuropsychiatric effects of ketamine with lamotrigine - Support for hyperglutamatergic effects of N-methyl-D-aspartate receptor antagonists. Arch Gen Psychiatry 57: 270–276. [DOI] [PubMed] [Google Scholar]
- 8. Gigante AD, Bond DJ, Lafer B, Lam RW, Young LT, et al. (2012) Brain glutamate levels measured by magnetic resonance spectroscopy in patients with bipolar disorder: a meta-analysis. Bipolar Disord 14: 478–487. [DOI] [PubMed] [Google Scholar]
- 9. Rao JS, Harry GJ, Rapoport SI, Kim HW (2010) Increased excitotoxicity and neuroinflammatory markers in postmortem frontal cortex from bipolar disorder patients. Mol Psychiatry 15: 384–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Collingridge GL, Olsen RW, Peters J, Spedding M (2009) A nomenclature for ligand-gated ion channels. Neuropharmacology 56: 2–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ginsberg SD, Hemby SE, Smiley JF (2012) Expression profiling in neuropsychiatric disorders: emphasis on glutamate receptors in bipolar disorder. Pharmacol Biochem Behav 100: 705–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kerner B, Jasinska AJ, DeYoung J, Almonte M, Choi OW, et al. (2009) Polymorphisms in the GRIA1 gene region in psychotic bipolar disorder. Am J Med Genet B Neuropsychiatr Genet 150B: 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bramon E, Sham PC (2001) The common genetic liability between schizophrenia and bipolar disorder: a review. Curr Psychiatry Rep 3: 332–337. [DOI] [PubMed] [Google Scholar]
- 14. Lin CY, Sawa A, Jaaro-Peled H (2012) Better understanding of mechanisms of schizophrenia and bipolar disorder: from human gene expression profiles to mouse models. Neurobiol Dis 45: 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eastwood SL, McDonald B, Burnet PW, Beckwith JP, Kerwin RW, et al. (1995) Decreased expression of mRNAs encoding non-NMDA glutamate receptors GluR1 and GluR2 in medial temporal lobe neurons in schizophrenia. Brain Res Mol Brain Res 29: 211–223. [DOI] [PubMed] [Google Scholar]
- 16. Sokolov BP (1998) Expression of NMDAR1, GluR1, GluR7, and KA1 glutamate receptor mRNAs is decreased in frontal cortex of “neuroleptic-free” schizophrenics: evidence on reversible up-regulation by typical neuroleptics. J Neurochem 71: 2454–2464. [DOI] [PubMed] [Google Scholar]
- 17. Eastwood SL, Kerwin RW, Harrison PJ (1997) Immunoautoradiographic evidence for a loss of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate-preferring non-N-methyl-D-aspartate glutamate receptors within the medial temporal lobe in schizophrenia. Biol Psychiatry 41: 636–643. [DOI] [PubMed] [Google Scholar]
- 18. Harrison PJ, Mclaughlin D, Kerwin RW (1991) Decreased Hippocampal Expression of a Glutamate Receptor Gene in Schizophrenia. Lancet 337: 450–452. [DOI] [PubMed] [Google Scholar]
- 19. Ibrahim HM, Hogg AJ, Healy DJ, Haroutunian V, Davis KL, et al. (2000) Ionotropic glutamate receptor binding and subunit mRNA expression in thalamic nuclei in schizophrenia. Am J Psychiat 157: 1811–1823. [DOI] [PubMed] [Google Scholar]
- 20. Meador-Woodruff JH, Hogg AJ, Smith RE (2001) Striatal ionotropic glutamate receptor expression in schizophrenia, bipolar disorder, and major depressive disorder. Brain Res Bull 55: 631–640. [DOI] [PubMed] [Google Scholar]
- 21. Zamanillo D, Sprengel R, Hvalby O, Jensen V, Burnashev N, et al. (1999) Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science 284: 1805–1811. [DOI] [PubMed] [Google Scholar]
- 22. Fitzgerald PJ, Barkus C, Feyder M, Wiedholz LM, Chen YC, et al. (2010) Does gene deletion of AMPA GluA1 phenocopy features of schizoaffective disorder? Neurobiol Dis 40: 608–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barkus C, Feyder M, Graybeal C, Wright T, Wiedholz L, et al. (2012) Do GluA1 knockout mice exhibit behavioral abnormalities relevant to the negative or cognitive symptoms of schizophrenia and schizoaffective disorder? Neuropharmacology 62: 1263–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Procaccini C, Aitta-aho T, Jaako-Movits K, Zharkovsky A, Panhelainen A, et al. (2011) Excessive novelty-induced c-Fos expression and altered neurogenesis in the hippocampus of GluA1 knockout mice. Eur J Neurosci 33: 161–174. [DOI] [PubMed] [Google Scholar]
- 25. Wiedholz LM, Owens WA, Horton RE, Feyder M, Karlsson RM, et al. (2008) Mice lacking the AMPA GluR1 receptor exhibit striatal hyperdopaminergia and ‘schizophrenia-related’ behaviors. Mol Psychiatry 13: 631–640. [DOI] [PubMed] [Google Scholar]
- 26. Vekovischeva OY, Zamanillo D, Echenko O, Seppala T, Uusi-Oukari M, et al. (2001) Morphine-induced dependence and sensitization are altered in mice deficient in AMPA-type glutamate receptor-A subunits. J Neurosci 21: 4451–4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bannerman DM, Deacon RM, Brady S, Bruce A, Sprengel R, et al. (2004) A comparison of GluR-A-deficient and wild-type mice on a test battery assessing sensorimotor, affective, and cognitive behaviors. Behav Neurosci 118: 643–647. [DOI] [PubMed] [Google Scholar]
- 28. Sanderson DJ, Good MA, Skelton K, Sprengel R, Seeburg PH, et al. (2009) Enhanced long-term and impaired short-term spatial memory in GluA1 AMPA receptor subunit knockout mice: evidence for a dual-process memory model. Learn Mem 16: 379–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vekovischeva OY, Aitta-aho T, Echenko O, Kankaanpaa A, Seppala T, et al. (2004) Reduced aggression in AMPA-type glutamate receptor GluR-A subunit-deficient mice. Genes Brain Behav 3: 253–265. [DOI] [PubMed] [Google Scholar]
- 30. Shaldubina A, Einat H, Szechtman H, Shimon H, Belmaker RH (2002) Preliminary evaluation of oral anticonvulsant treatment in the quinpirole model of bipolar disorder. J Neural Transm 109: 433–440. [DOI] [PubMed] [Google Scholar]
- 31. O’Donnell KC, Gould TD (2007) The behavioral actions of lithium in rodent models: Leads to develop novel therapeutics. Neurosci Biobehav Rev 31: 932–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lan MJ, Yuan P, Chen G, Manji HK (2008) Neuronal peroxisome proliferator-activated receptor gamma signaling: regulation by mood-stabilizer valproate. J Mol Neurosci 35: 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chapoteau E, Czech BP, Zazulak W, Kumar A (1992) First practical colorimetric assay of lithium in serum. Clin Chem 38: 1654–1657. [PubMed] [Google Scholar]
- 34. Yamashita S, Furuno K, Kawasaki H, Gomita Y, Yoshinaga H, et al. (1995) Simple and Rapid Analysis of Lamotrigine, a Novel Antiepileptic, in Human Serum by High-Performance Liquid-Chromatography Using a Solid-Phase Extraction Technique. J Chromatogr B 670: 354–357. [DOI] [PubMed] [Google Scholar]
- 35. Hall FS, Humby T, Wilkinson LS, Robbins TW (1997) The effects of isolation-rearing on sucrose consumption in rats. Physiol Behav 62: 291–297. [DOI] [PubMed] [Google Scholar]
- 36. VandeWeerd HA, VanLoo PLP, VanZutphen LFM, Koolhaas JM, Baumans V (1997) Preferences for nesting material as environmental enrichment for laboratory mice. Lab Anim 31: 133–143. [DOI] [PubMed] [Google Scholar]
- 37. Van Loo PLP, de Weerd HAV, Van Zutphen LFM, Baumans V (2004) Preference for social contact versus environmental enrichment in male laboratory mice. Lab Anim 38: 178–188. [DOI] [PubMed] [Google Scholar]
- 38. Linden AM, Aller MI, Leppa E, Vekovischeva O, Aitta-Aho T, et al. (2006) The in vivo contributions of TASK-1-containing channels to the actions of inhalation anesthetics, the alpha(2) adrenergic sedative dexmedetomidine, and cannabinoid agonists. J Pharmacol Exp Ther 317: 615–626. [DOI] [PubMed] [Google Scholar]
- 39. Flaisher-Grinberg S, Einat H (2009) A possible utilization of the mice forced swim test for modeling manic-like increase in vigor and goal-directed behavior. J Pharmacol Toxicol Methods 59: 141–145. [DOI] [PubMed] [Google Scholar]
- 40. Porsolt RD, Bertin A, Jalfre M (1977) Behavioral despair in mice: a primary screening test for antidepressants. Arch Int Pharmacodyn Ther 229: 327–336. [PubMed] [Google Scholar]
- 41. Vekovischeva OY, Peuhkuri K, Backstrom P, Sihvola N, Pilvi T, et al. (2013) The effects of native whey and alpha-lactalbumin on the social and individual behaviour of C57BL/6J mice. Br J Nutr 110: 1336–1346. [DOI] [PubMed] [Google Scholar]
- 42. Steru L, Chermat R, Thierry B, Simon P (1985) The tail suspension test: a new method for screening antidepressants in mice. Psychopharmacology 85: 367–370. [DOI] [PubMed] [Google Scholar]
- 43. Flaisher-Grinberg S, Overgaard S, Einat H (2009) Attenuation of high sweet solution preference by mood stabilizers: A possible mouse model for the increased reward-seeking domain of mania. J Neurosci Meth 177: 44–50. [DOI] [PubMed] [Google Scholar]
- 44. Rada P, Avena NM, Hoebel BG (2005) Daily bingeing on sugar repeatedly releases dopamine in the accumbens shell. Neuroscience 134: 737–744. [DOI] [PubMed] [Google Scholar]
- 45. Simms JA, Steensland P, Medina B, Abernathy KE, Chandler LJ, et al. (2008) Intermittent access to 20% ethanol induces high ethanol consumption in Long-Evans and Wistar rats. Alcohol Clin Exp Res 32: 1816–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ramos A (2008) Animal models of anxiety: do I need multiple tests? Trends Pharmacol Sci 29: 493–498. [DOI] [PubMed] [Google Scholar]
- 47. Perry W, Minassian A, Paulus MP, Young JW, Kincaid MJ, et al. (2009) A reverse-translational study of dysfunctional exploration in psychiatric disorders: from mice to men. Arch Gen Psychiatry 66: 1072–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Minassian A, Henry BL, Geyer MA, Paulus MP, Young JW, et al. (2010) The quantitative assessment of motor activity in mania and schizophrenia. J Affect Disorders 120: 200–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Perry W, Minassian A, Henry B, Kincaid M, Young JW, et al. (2010) Quantifying over-activity in bipolar and schizophrenia patients in a human open field paradigm. Psychiatry Res 178: 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Henry BL, Minassian A, Patt VM, Hua J, Young JW, et al. (2013) Inhibitory deficits in euthymic bipolar disorder patients assessed in the human behavioral pattern monitor. J Affect Disorders 150: 948–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Procaccini C, Maksimovic M, Aitta-Aho T, Korpi ER, Linden AM (2013) Reversal of novelty-induced hyperlocomotion and hippocampal c-Fos expression in GluA1 knockout male mice by the mGluR2/3 agonist LY354740. Neuroscience 250: 189–200. [DOI] [PubMed] [Google Scholar]
- 52. Li XH, Frye MA, Shelton RC (2012) Review of Pharmacological Treatment in Mood Disorders and Future Directions for Drug Development. Neuropsychopharmacology 37: 77–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gibbs JW, Sombati S, DeLorenzo RJ, Coulter DA (2000) Cellular actions of topiramate: Blockade of kainate-evoked inward currents in cultured hippocampal neurons. Epilepsia 41: S10–S16. [DOI] [PubMed] [Google Scholar]
- 54. Leach MJ, Baxter MG, Critchley MAE (1991) Neurochemical and Behavioral-Aspects of Lamotrigine. Epilepsia 32: S4–S8. [DOI] [PubMed] [Google Scholar]
- 55. Basselin M, Chang L, Bell JM, Rapoport SI (2006) Chronic lithium chloride administration attenuates brain NMDA receptor-initiated signaling via arachidonic acid in unanesthetized rats. Neuropsychopharmacology 31: 1659–1674. [DOI] [PubMed] [Google Scholar]
- 56. Valdes JJ, Weeks OI (2009) Estradiol and lithium chloride specifically alter NMDA receptor subunit NR1 mRNA and excitotoxicity in primary cultures. Brain Res 1268: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hashimoto R, Hough C, Nakazawa T, Yamamoto T, Chuang DM (2002) Lithium protection against glutamate excitotoxicity in rat cerebral cortical neurons: involvement of NMDA receptor inhibition possibly by decreasing NR2B tyrosine phosphorylation. J Neurochem 80: 589–597. [DOI] [PubMed] [Google Scholar]
- 58. Ghasemi M, Dehpour AR (2011) The NMDA receptor/nitric oxide pathway: a target for the therapeutic and toxic effects of lithium. Trends Pharmacol Sci 32: 420–434. [DOI] [PubMed] [Google Scholar]
- 59. Ghasemi M, Shafaroodi H, Nazarbeiki S, Meskar H, Heydarpour P, et al. (2010) Voltage-dependent calcium channel and NMDA receptor antagonists augment anticonvulsant effects of lithium chloride on pentylenetetrazole-induced clonic seizures in mice. Epilepsy Behav 18: 171–178. [DOI] [PubMed] [Google Scholar]
- 60. Kubová H, Mares P (2010) Vigabatrin but not valproate prevents development of age-specific flexion seizures induced by N-methyl-D-aspartate (NMDA) in immature rats. Epilepsia 51: 469–472. [DOI] [PubMed] [Google Scholar]
- 61. Zarate CA, Singh J, Manji HK (2006) Cellular plasticity cascades: Targets for the development of novel therapeutics for bipolar disorder. Biol Psychiatry 59: 1006–1020. [DOI] [PubMed] [Google Scholar]
- 62. Berggren U (1985) Effects of Chronic Lithium Treatment on Brain Monoamine Metabolism and Amphetamine-Induced Locomotor Stimulation in Rats. J Neural Transm 64: 239–250. [DOI] [PubMed] [Google Scholar]
- 63. Saal D, Dong Y, Bonci A, Malenka RC (2003) Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 38: 359–359. [DOI] [PubMed] [Google Scholar]
- 64. Vashchinkina E, Panhelainen A, Vekovischeva OY, Aitta-aho T, Ebert B, et al. (2012) GABA site agonist gaboxadol induces addiction-predicting persistent changes in ventral tegmental area dopamine neurons but is not rewarding in mice or baboons. J Neurosci 32: 5310–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Aitta-Aho T, Moykkynen TP, Panhelainen AE, Vekovischeva OY, Backstrom P, et al. (2012) Importance of GluA1 subunit-Containing AMPA glutamate receptors for morphine state-dependency. PLoS One 7(5): e38325 doi:10.1371/journal.pone.0038325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kelley AE, Berridge KC (2002) The neuroscience of natural rewards: Relevance to addictive drugs. J Neurosci 22: 3306–3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shibata R, Kameishi M, Kondoh T, Torii K (2009) Bilateral dopaminergic lesions in the ventral tegmental area of rats influence sucrose intake, but not umami and amino acid intake. Physiol Behav 96: 667–674. [DOI] [PubMed] [Google Scholar]
- 68. Dubreucq S, Durand A, Matias I, Benard G, Richard E, et al. (2013) Ventral Tegmental Area Cannabinoid Type-1 Receptors Control Voluntary Exercise Performance. Biol Psychiatry 73: 895–903. [DOI] [PubMed] [Google Scholar]
- 69. Voikar V, Polus A, Vasar E, Rauvala H (2005) Long-term individual housing in C57BL/6J and DBA/2 mice: assessment of behavioral consequences. Genes Brain Behav 4: 240–252. [DOI] [PubMed] [Google Scholar]
- 70. Varlinskaya EI, Spear LP, Spear NE (1999) Social behavior and social motivation in adolescent rats: Role of housing conditions and partner’s activity. Physiol Behav 67: 475–482. [DOI] [PubMed] [Google Scholar]
- 71. Niesink RJ, van Ree JM (1982) Short-term isolation increases social interactions of male rats: a parametric analysis. Physiol Behav 29: 819–825. [DOI] [PubMed] [Google Scholar]
- 72. Voikar V, Vasar E, Rauvala H (2004) Behavioral alterations induced by repeated testing in C57BL/6J and 129S2/Sv mice: implications for phenotyping screens. Genes Brain Behav 3: 27–38. [DOI] [PubMed] [Google Scholar]
- 73. Takao K, Yamasaki N, Miyakawa T (2007) Impact of brain-behavior phenotypying of genetically-engineered mice on research of neuropsychiatric disorders. Neurosci Res 58: 124–132. [DOI] [PubMed] [Google Scholar]
- 74. Kato T, Kubota M, Kasahara T (2007) Animal models of bipolar disorder. Neurosci Biobehav Rev 31: 832–842. [DOI] [PubMed] [Google Scholar]
- 75.Inta D, Vogt MA, Elkin H, Weber T, Lima-Ojeda JM, et al.. (2013) Phenotype of mice with inducible ablation of GluA1 AMPA receptors during late adolescence: Relevance for mental disorders. Hippocampus. [DOI] [PubMed]
- 76. Shaltiel G, Maeng S, Malkesman O, Pearson B, Schloesser R, et al. (2008) Evidence for the involvement of the kainate receptor subunit GluR6 (GRIK2) in mediating behavioral displays related to behavioral symptoms of mania. Mol Psychiatry 13: 858–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Han K, Holder JL Jr, Schaaf CP, Lu H, Chen H, et al. (2013) SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature 503: 72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Guilmatre A, Huguet G, Delorme R, Bourgeron T (2014) The emerging role of SHANK genes in neuropsychiatric disorders. Dev Neurobiol 74: 113–122. [DOI] [PubMed] [Google Scholar]
- 79. Bureau I, Bischoff S, Heinemann SF, Mulle C (1999) Kainate receptor-mediated responses in the CA1 field of wild-type and GluR6-deficient mice. J Neurosci 19: 653–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Fisahn A, Contractor A, Traub RD, Buhl EH, Heinemann SF, et al. (2004) Distinct roles for the kainate receptor subunits GluR5 and GluR6 in kainate-induced hippocampal gamma oscillations. J Neurosci 24: 9658–9668. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The ARRIVE Guidelines Checklist.
(PDF)