

Abstract
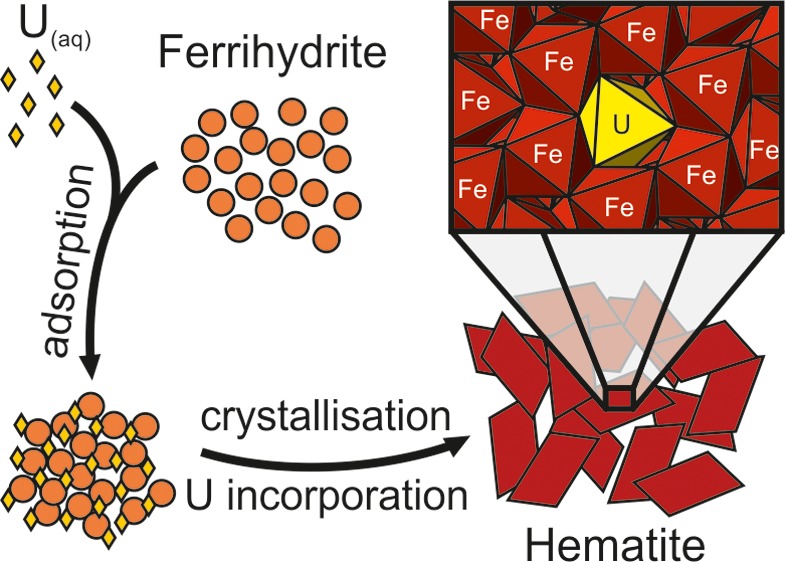
Ferrihydrite was exposed to U(VI)-containing cement leachate (pH 10.5) and aged to induce crystallization of hematite. A combination of chemical extractions, TEM, and XAS techniques provided the first evidence that adsorbed U(VI) (≈3000 ppm) was incorporated into hematite during ferrihydrite aggregation and the early stages of crystallization, with continued uptake occurring during hematite ripening. Analysis of EXAFS and XANES data indicated that the U(VI) was incorporated into a distorted, octahedrally coordinated site replacing Fe(III). Fitting of the EXAFS showed the uranyl bonds lengthened from 1.81 to 1.87 Å, in contrast to previous studies that have suggested that the uranyl bond is lost altogether upon incorporation into hematite. The results of this study both provide a new mechanistic understanding of uranium incorporation into hematite and define the nature of the bonding environment of uranium within the mineral structure. Immobilization of U(VI) by incorporation into hematite has clear and important implications for limiting uranium migration in natural and engineered environments.
Introduction
Uranium is an environmental contaminant that arises as a result of authorized and accidental releases at various stages in the nuclear fuel cycle, including from uranium ore mining activities and post-reactor operations. Additionally, in many countries, uranium-containing radioactive wastes, including spent nuclear fuel and intermediate-level waste, are likely to be disposed in deep geological disposal facilities (GDF). Here, uranium will typically be the most significant radionuclide by mass in the waste inventory. After deep disposal has been implemented, it is inevitable that, on geological time scales, uranium (and other radionuclides) will be released from within the waste containers and, importantly, due to its long half-life (4.5 Ga), the behavior of uranium and of its resultant decay chain will be important to any safety case for geological disposal over extended time frames. It is, therefore, crucial that we understand the fate of uranium in these natural and engineered environments to be able to both predict and constrain its environmental impact.
Iron (oxyhydr)oxides (e.g., hematite α-Fe2O3) are ubiquitous and are known to be effective at reducing the mobility of U(VI) through either their high sorption capacity (e.g., surface adsorption) or, where Fe(II) is present, via reductive precipitation to poorly soluble U(IV) phases. Studies of uranium retardation mechanisms in the environment have tended to focus on adsorption of U(VI) to various mineral phases1,2 or reduction of U(VI) to U(IV) either directly or indirectly as a result of microbial3−6 or abiotic pathways.7,8 However, a change in the geochemical conditions may reverse these processes (e.g., reduction in pH leading to desorption or reoxidation of U(IV)) and cause remobilization of the contaminant.9−11 Incorporation of uranium into stable mineral phases, such as iron (oxyhydr)oxides, offers a pathway for sequestration with the potential for long-term immobilization. It has been shown that goethite and hematite are able to accommodate various impurities (e.g., Si, Ti, Mn, Ni) into their structure.12,13 Specifically, U(VI) and reportedly even U(V) may be incorporated into goethite (α-FeOOH) during Fe(II)-catalyzed crystallization of ferrihydrite,14−16 and evidence for U(VI) incorporation into hematite during coprecipitation has been reported.17−19 Notably, Duff et al.18 precipitated ferrihydrite from a solution containing U(VI) and Fe(III) and induced hematite formation by aging at pH 11 and 60 °C. Here, they reported incorporation of U(VI) into hematite in a uranate-like coordination environment with the resultant loss of the short uranyl bonds. Ilton et al.19 followed the method of Duff et al.18 and reported a similar structure for incorporated U(VI). Atomistic simulations of U(IV), U(V), and U(VI) incorporation into hematite using various different charge compensation mechanisms, based on the Duff et al.18 incorporation model, indicated that U(VI) maintained octahedral coordination in most cases but that the predicted interatomic distances differed from the experimental data.20 Furthermore, in a similar study, chemical extractions on U(VI) associated with ferrihydrite showed a decrease in leachable uranium as the solid phase aged and the formation of U(VI)-labeled crystalline goethite and hematite occurred, suggesting a change in speciation during crystallization.21 The lack of agreement between the spectroscopic and atomistic modeling approaches in the literature to date indicates that the mechanism of uranium incorporation, and the details of the molecular-level bonding environment within the hematite structure warrant further investigation.
In addition to forming in soil and sediments, predominantly as a weathering product of iron-bearing minerals, iron (oxyhydr)oxides form as corrosion products of steel22 and are present in intermediate level radioactive wastes.23,24 They are also reported to form in deep geological systems on tunnel walls due to biological oxidation of Fe(II).25 Many geological disposal concepts utilize cementitious materials (often within the wasteform itself or in the engineered barrier system) and many contaminated soils at nuclear facilities will be in contact with cements and concrete construction materials. Leaching of the cementitious materials will buffer the pH to hyperalkaline conditions, creating a chemically disturbed zone (CDZ) in the host rock or local environment.26,27 Thus, understanding the changes in speciation (i.e., adsorbed versus incorporated) of actinides during crystallization of iron (oxyhydr)oxides under these geochemical conditions is key to predicting their long-term stability and mobility in natural and engineered environments. Ferrihydrite crystallizes to hematite or goethite depending upon solution conditions, with pH, ionic strength, and temperature all having an influence.28 Hematite formation is favored under near-neutral conditions and higher temperature and ionic strength, whereas goethite forms under extremes of pH (less than 4, greater than 10) and at lower temperature and ionic strength.28,29 The hematite formation process begins with ferrihydrite particle aggregation,30 followed by recrystallization within the aggregate via dissolution and reprecipitation processes that occur at the nanoscale.31 This crystallization involves a variety of processes including dehydration of the ferrihydrite particles, deprotonation of hydroxyl groups, creation of oxy-linkages, and redistribution of cation vacancies.32 During this process, adsorbed uranium has the potential to become incorporated into the structure of the hematite. However, the mechanism of this reaction is poorly constrained, and how much of the adsorbed uranium is incorporated, at which stage in the crystallization process uranium is incorporated, and what the final site of uranium is within the hematite structure are all worthy of attention.
In this contribution, we provide a detailed insight into the mechanism(s) of uranium incorporation during hematite formation under conditions relevant to both geological disposal and contaminated land to determine whether significant amounts of uranium could be sequestered into this phase in the long term. We have combined aqueous chemical data with X-ray diffraction (XRD), transmission electron microscopy (TEM), and X-ray absorption spectroscopy (XAS) to characterize the solid phase crystallization at elevated pH (10.5). Throughout, we have focused on the fate of uranium during ferrihydrite transformation to hematite to determine the mechanism(s) of uranium incorporation, and our aim was to define the atomic scale bonding environment of uranium within this environmentally important phase.
Experiments and Analyses
Batch experiments were used to follow the crystallization of U(VI)-adsorbed ferrihydrite in a synthetic cement leachate (0.015 g L–1 Ca(OH)2; pH 10.5) system. Full experimental setup and sampling and analysis details are given in Supporting Information. Briefly, batch experiments were set up at a solid solution concentration of 0.4 g L–1 and spiked with U(VI) to give an initial U(aq) concentration of 1 ppm (4.2 × 10–6 mol L–1), which was thermodynamically modeled (PHREEQC) to be below the solubility of any U(VI) phase in the synthetic leachate. The experiments were placed in an oven at 60 °C for up to 70 days. Some experiments were also placed into an oven at 105 °C for up to 45 days to suppress the formation of goethite and favor hematite formation. All experiments were maintained between pH 10.3–10.7 and were purged with CO2-free air throughout. Partitioning of uranium between the solid and the solution was determined by analysis of uranium in solution (U(aq)). Chemical extractions were performed to assess the partitioning of uranium to the solid phase.33 The surface-bound uranium (U(ads)) was determined by titration of the iron (oxyhydr)oxide suspension to pH 2.5, below the U adsorption edge,34 using HCl. The resulting supernatant was analyzed for uranium, and U(ads) was calculated by subtracting U(aq). The nonleachable uranium (U(s)) was then calculated from the mass balance according to
Aqueous samples were analyzed for 238U by ICP-MS, and solids were characterized by powder XRD and surface area using the BET method. Particle morphologies were characterized via TEM. Uranium LIII-edge XAS spectra were collected on beamline B18, Diamond Light Source, at room temperature in fluorescence mode using a nine-element Ge detector.35 Reference spectra from U(VI) and U(IV) standards (schoepite ((UO2)8O2(OH)12·12(H2O)) and uraninite (UO2), respectively) were collected in transmission mode. In-line yttrium foil reference spectra were also collected for each sample for energy calibration. Background subtraction, data normalization, and fitting to the EXAFS spectra were performed using the software packages Athena and Artemis.36
Results and Discussion
Characterization of Experimental Products
XRD patterns for the products from the 60 °C crystallization experiment show hematite formed rapidly from 2-line ferrihydrite over the first 24–48 h of aging (Figure 1). Quantitative analysis of the XRD patterns (QXRD) (Supporting Information Table SI-1) reveals that greater than 80% of the 24 h sample was hematite, decreasing to 55% hematite with the remaining phase 45% goethite after 30 days. Hematite formation is favored over goethite with increasing temperature,28 therefore, to obtain a high purity end-member hematite sample, a 105 °C crystallization at pH 10.5 was performed. Quantitative analysis of the XRD patterns from the high temperature experiment confirmed that the sample was greater than 90% hematite after 45 days aging.
Figure 1.

XRD pattern time series for the 60 °C crystallization and the end-point of the 105 °C crystallization. Observable Peaks are indexed with F, H, or G signifying ferrihydrite, hematite, and goethite, respectively.
TEM photomicrographs of the solid samples (Supporting Information Figure SI-1) illustrate the crystallization pathway of hematite and goethite from ferrihydrite at 60 °C.30 At 0 h, the ferrihydrite nanoparticles were 3–5 nm and had no visible structure within the particles. After 24 h at 60 °C, the ferrihydrite had aggregated and clumps of nanocrystalline hematite and acicular goethite became evident (Supporting Information Figure SI-1). Thereafter, the amount of ferrihydrite rapidly decreased and the size of the hematite/goethite crystals gradually increased. Over the first 48 h of crystallization, there was a rapid decrease in surface area from 164 ± 3 to 21 ± 1 m2 g–1, followed by a slow continued decrease to 17 ± 1 m2 g–1 after 30 days (Figure 2). The XRD and QXRD, TEM, and BET data indicated that during the first 24–48 h, there was rapid aggregation of ferrihydrite and crystallization of hematite/goethite, causing a large and rapid decrease in surface area. This was followed by a stage of crystal ripening, with some transformation of hematite to goethite (Supporting Information Table SI-1). No evidence for discrete uranium phases was detected using XRD or TEM, as expected from PHREEQC modeling of the system.
Figure 2.
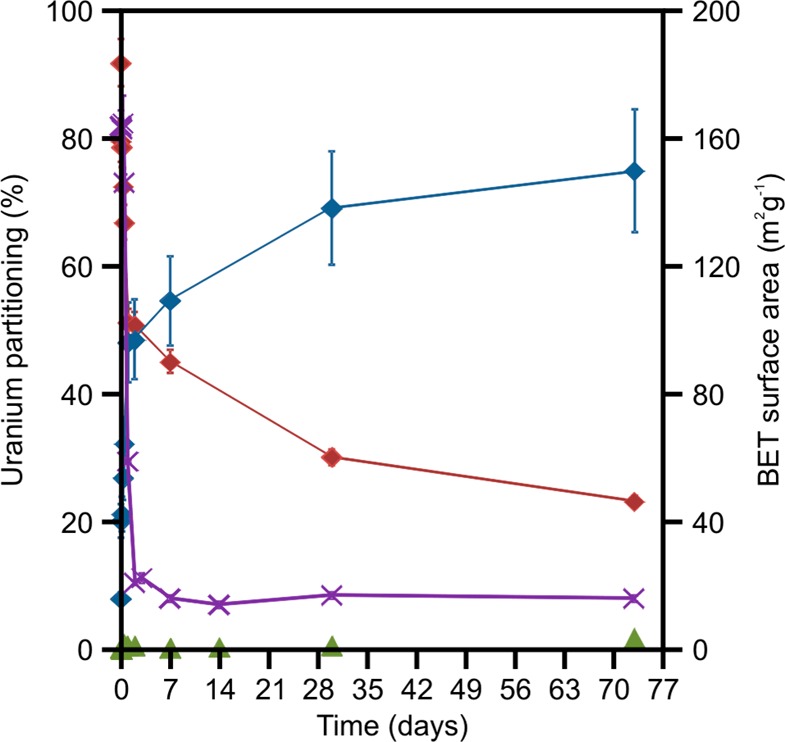
Partitioning of uranium (%) and BET surface area (m2 g–1) during the crystallization of ferrihydrite at 60 °C. Green triangles = uranium in solution (U(aq)); red diamonds = surface bound uranium (U(ads)); blue diamonds = nonleachable uranium (U(s)); purple crosses = BET surface area.
The majority of the uranium (91.9 ± 0.2%) was instantaneously adsorbed to the solid phase on its addition to the ferrihydrite slurry (Figure 2). During the aggregation and initial crystallization phase, U(ads) rapidly decreased to 79.6 ± 3.2% at 1 h and 51.3 ± 2.1% at 48 h, with a continued decrease to 23.2 ± 0.9% after 70 days of aging. On the basis of the uranium mass balance, this shows an increase in U(s) from 20.2 ± 2.6% at 1 h to 75.0 ± 9.6% after 70 days (Figure 2). Thus, the chemical extraction data strongly suggest that a significant proportion of the uranium is becoming increasingly strongly associated with, and possibly structurally incorporated into, hematite/goethite during crystallization. Reflecting this, we suggest that uranium adsorbed to the surface of the ferrihydrite particles is trapped within the solid phase during the aggregation process at the early stages of the crystallization process, consistent with the incorporation mechanism of Pb into hematite during hydrothermal crystallization of ferrihydrite.33,37 The gradual increase in U(s) during crystallization and ripening indicates that U(VI) then continues to be incorporated as the iron (oxyhdr)oxide crystals form and grow. This is in contrast to the behavior observed for Pb, where the contaminant was slowly released from the hematite structure during ripening because of its incompatibility (i.e., located within defect sites) with the mineral structure.33,37 This does not occur with uranium, suggesting that it may become located within a stable crystallographic site within the newly formed mineral, in agreement with modeling simulations.20
X-ray Absorption Spectroscopy
XANES spectra from a time series of samples spanning the crystallization of ferrihydrite at 60 and 105 °C, along with U(VI) and U(IV) reference spectra are shown in Figure 3.
Figure 3.
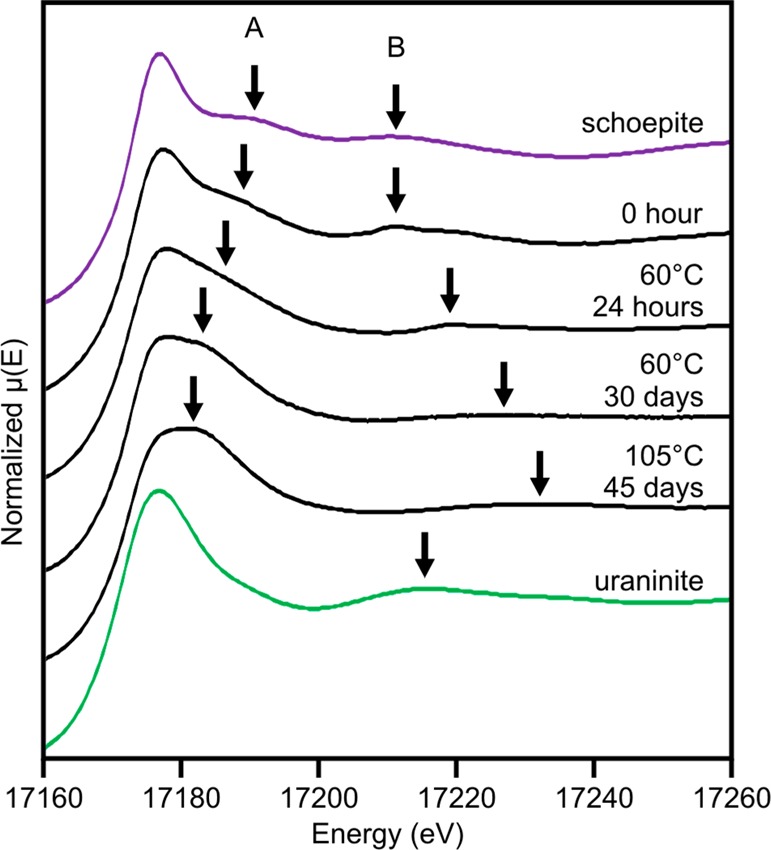
Uranium LIII-edge XANES spectra during the crystallization of ferrihydrite. Reference spectra for U(VI) (schoepite ((UO2)8O2(OH)12·12(H2O)); purple) and U(IV) (uraninite (UO2); green) are shown for comparison. Arrows A and B show features related to axial and equatorial U—O bonds, respectively.
The edge positions of all XANES spectra from the four iron (oxyhydr)oxide samples aligned to the U(VI) schoepite standard (∼17 172 eV) indicating that uranium remained as U(VI) during crystallization. Two prominent resonance features are visible in the XANES spectra of uranyl-containing compounds (A and B, Figure 3) and are due to resonance from the different U—O bonds. Feature A (∼17 190 eV) is attributable to the short axial U=O bonds in the dioxygenyl species, and feature B (∼17 210–17 215 eV) is attributable to the longer U—O equatorial bonds.38 Feature A is absent from the XANES spectra of compounds that do not have the axial U=O bonds, such as uraninite, but is clearly present in all our U-bearing Fe sample spectra (Figure 3). The XANES spectrum of the 0 h solid associated sample is very similar to that of schoepite, indicating uranyl coordination. However, during the experiment the resonance features migrate in energy with time indicating a change in the local bonding environment of uranium throughout crystallization. Here, feature A migrates to a lower energy (17 188 to 17 182 eV), whereas feature B migrates to a higher energy (17 211 to 17 228 eV) over time (Figure 3). Changes in the energy of these resonance features in the XANES region are reportedly inversely proportional to the changes in the corresponding bond length.38 The time series XANES data, therefore, suggest that during reaction, the axial uranyl oxygen bond elongates while the average bond length in the equatorial plane shortens. The XANES spectra of the 60 °C, 30 day sample and the 105 °C, 45 day sample are very similar to reported XANES for alkali metal uranate compounds.39 This indicates that the U(VI) associated with the crystalline iron (oxyhydr)oxide is likely in a uranate-like coordination and presumably relates to the uranium becoming structurally incorporated. It is important to note that feature A remains during incorporation, indicating retention of the uranyl bonds, albeit with an increase in the bond distance inferred from its migration to lower energy. The XANES spectra from previous studies of U(VI) incorporation into hematite18,19 are also very similar to our data, suggesting the same U(VI) local environment is favored in several experimental systems.
Further information on the bonding environment of the uranium can be determined from analysis of the EXAFS spectra and their Fourier transform (Figure 4a and b respectively). The model of Waite et al.34 for U(VI) adsorption to ferrihydrite in a mononuclear bidentate complex was applied to the 0 h data and provided a good fit (Figure 4, Table 1). To test our hypothesis of uranium incorporation into hematite during crystallization, fitting was performed using the hematite structure40 with U(VI) substituted for Fe(III) in the mineral structure. The model was then applied to the 105 °C, 45 day data first because this was 90.8 ± 0.8% hematite and had only limited potential contributions from goethite (Figure 1). The refined fit model from the 105 °C system was then applied to the 60 °C, 30 day data (55% hematite) to assess the goodness of fit to hematite-incorporated uranium in a more environmentally relevant system.
Figure 4.

(a) Uranium LIII-edge EXAFS spectra and (b) corresponding Fourier transforms of the EXAFS data from U(VI) during hematite crystallization from ferrihydrite. Black lines are k3-weighted data, and red lines are model fits to the data. The Fourier transform is plotted with a phase correction calculated from Oax. Fit parameters are given in Table 1.
Table 1. Details of EXAFS Fit Parameters from Uranium Adsorbed to Ferrihydrite (t = 0 h) and Uranium Associated with Crystalline Hematite (t = 30 days, 60 °C; t = 45 days, 105 °C)a.
| sample | path | CN | R (Å) | σ2 (Å2) | ΔE0 (eV) | S02 | Xv2 | R |
|---|---|---|---|---|---|---|---|---|
| 0 h | Oax | 2 | 1.81 (1) | 0.003 (1) | 6.9 ± 2.0 | 1.05 (1) | 75.1 | 0.027 |
| Oeq 1 | 3 | 2.28 (4) | 0.009 (6) | |||||
| Oeq 2 | 2 | 2.40 (4) | 0.005 (6) | |||||
| FeF | 1 | 3.40 (4) | 0.008 (5) | |||||
| Oax MSb | 2 | 3.63 (2) | 0.005 (1) | |||||
| 105 °C 45 day | Oax | 2 | 1.87 (2) | 0.007 (2) | –4.4 ± 6.0 | 0.85 (6) | 27.5 | 0.018 |
| Oeq 1 | 2 | 2.07 (2) | 0.003 (1) | |||||
| Oeq 2 | 2 | 2.23 (3) | 0.005 (2) | |||||
| FeF | 1 | 2.87 (3) | 0.007 (2) | |||||
| FeE | 3 | 3.11 (2) | 0.010 (2) | |||||
| FeC1 | 3 | 3.45 (6) | 0.016 (7) | |||||
| FeC2 | 6 | 4.01 (6) | 0.024 (7) | |||||
| Oax MSb | 2 | 3.74 (4) | 0.014 (3) | |||||
| 60 °C 30 day | Oax | 2 | 1.84 (1) | 0.009 (1) | 6.5 ± 2.9 | 0.85 (3) | 30.7 | 0.013 |
| Oeq 1 | 2 | 2.17 (4) | 0.008 (4) | |||||
| Oeq 2 | 2 | 2.31 (6) | 0.013 (9) | |||||
| FeF | 1 | 2.91 (2) | 0.006 (2) | |||||
| FeE | 3 | 3.16 (2) | 0.011 (1) | |||||
| Oax MSb | 2 | 3.68 (3) | 0.017 (3) | |||||
| hematite40 | O1 | 3 | 1.95 | |||||
| O2 | 3 | 2.12 | ||||||
| FeF | 1 | 2.90 | ||||||
| FeE | 3 | 2.97 | ||||||
| FeC1 | 3 | 3.36 | ||||||
| FeC2 | 6 | 3.71 |
CN denotes coordination number; R denotes atomic distance; σ2 denotes Debye–Waller factor; ΔE0 denotes the shift in energy from the calculated Fermi level; S02 denotes the amplitude factor which was constrained to between 0.85 and 1.05; Xv2 denotes the reduced χ square value; R denotes the “goodness of fit” factor; the subscript MS denotes multiple scattering paths in the axial O—U—O unit.
The multiple scattering paths considered were linear paths and their ΔR and σ2 parameters were evaluated as multiples of the corresponding single scattering path parameter. Numbers in parentheses are the standard deviation on the last decimal place.
In the hematite structure (Table 1), each Fe is octahedrally coordinated by oxygen and is surrounded by Fe—O octahedra which are face, edge, or corner sharing (Table 1, Figure 5). We assumed an octahedral U—O coordination for our EXAFS fits based on our XANES, previous modeling,20 and the working hypothesis that the Fe(III) that was replaced by U(VI) was octahedrally coordinated. The best fit to the EXAFS data showed that the optimal coordination was a fit with three separate U—O shells, each with a coordination of 2 at U—O distances of 1.87 Å, 2.07 Å, and 2.23 Å. These U—O distances are similar to those in barium uranate (BaUO4), in which uranium is also octahedrally coordinated with oxygen, with 2 at U—O distances of 1.89 Å and 4 at 2.20 Å.41 We were then able to fit all of the four closest neighboring Fe shells expected from the hematite structure with a good level of statistical significance (Supporting Information Table SI-7). The U—Fe bond distances for face sharing Fe (FeF) and the nearer corner sharing Fe (FeC1) are at approximately the same distance as the Fe—Fe distance in hematite, whereas an increased atomic distance to the other two Fe shells (FeE and FeC2) is observed suggesting some strain in the structure (Table 1). U(VI) has a larger crystal radius than Fe(III) (0.870 Å versus 0.785 Å),42 and thus, upon incorporation, it would be expected to cause expansion and distortion to the host octahedral site.
Figure 5.
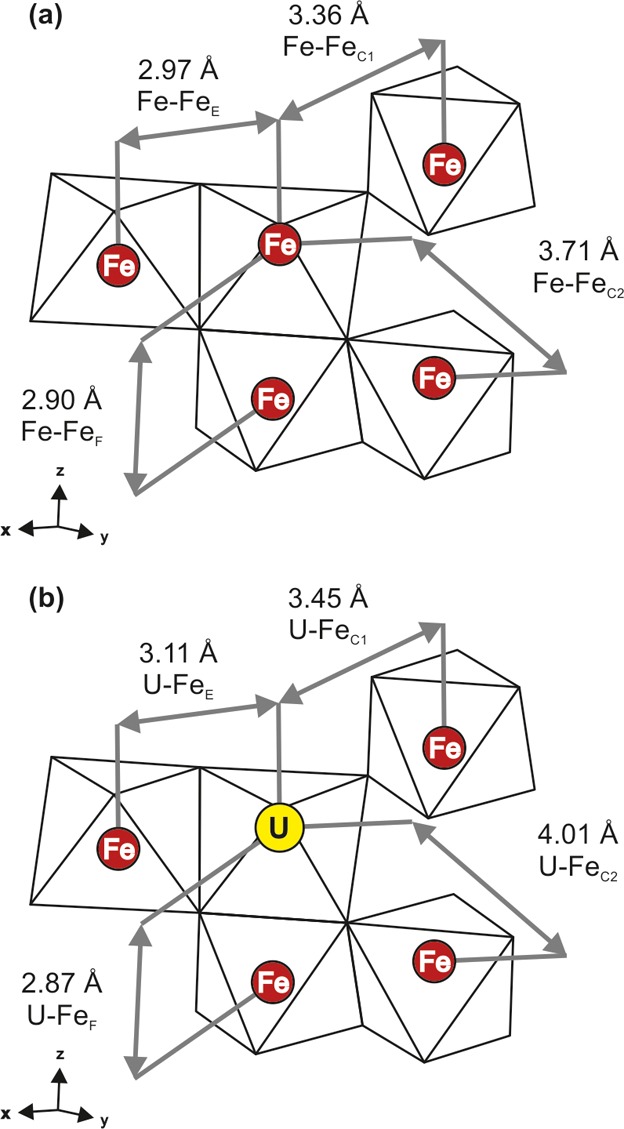
(a) Hematite structure showing Fe—Fe distances of Blake et al.40 (b) Uranium incorporated hematite showing U—Fe distances obtained from EXAFS fitting of the 105 °C data. Subscript notation indicates the polygon sharing relationship: F = face; E = edge; C = corner. Redrawn after Cornell and Schwertmann.28
The usual shell-by-shell approach to EXAFS fitting was not possible here because the Fe scatterers mostly contribute to the EXAFS spectrum in the low to middle k range (4–10 Å–1) (Supporting Information Figure SI-2); hence, any model that excludes these contributions will be unsatisfactory. Additionally, we found that having data with good signal-to-noise ratio in the high k range (>10 Å–1) was essential to adequately fit the U—O shells (Supporting Information Figure SI-2). Therefore, with our incorporation hypothesis in mind, we iteratively refined the U—O shells and U—Fe shells simultaneously. Once the U—O shells had been satisfactorily resolved, we then constructed a model from a single Fe shell to the full model including four Fe shells refining the model each time and assessing the statistical relevance of each additional shell by way of an F-test (Supporting Information Table SI-7).43 The F-test results confirmed that the addition of each subsequent Fe shell significantly improved the fit of the model to the data and was statistically valid.
The Debye–Waller factor of the shortest U—O distance, the axial oxygens, is the highest of the three oxygen shells, when normally it would be expected that they would be the tightest bound and, thus, have the lowest Debye–Waller factors. This may be due to static disorder, possibly related to the averaged nature of the EXAFS spectrum and the complexity of the structure with U—O octahedra potentially in several different orientations, resulting in a range of U—O axial oxygen distances that are averaged in the fit. Additionally, the outermost Fe shell has a comparatively large Debye–Waller factor, ∼0.02 Å2. Again, this is likely to be due to the relative increase in static disorder in the spectrum as the distance from the central uranium atom increases. Overall, these data, coupled to TEM and QXRD show that the U(VI) within the 105 °C, 45 day aged samples was incorporated into the hematite structure by replacing Fe(III).
Application to the 60 °C Data
The fit model from the 105 °C data can be fitted to the 60 °C data but requires the removal of the outer two Fe shells from the model. This may be due to the reduced useable k range of the 60 °C data or to the heterogeneity of the uranium location (e.g., an adsorbed and incorporated component) in this sample compared to the high temperature experiment. The fit parameters for the two closest Fe shells (Table 1) were statistically valid and were essentially the same as the 105 °C fit. Quantitative analysis of the XRD revealed up to 45% goethite present in this sample (Supporting Information Table SI-1). The Fe—Fe distances in goethite44 are similar to those in hematite, although the hematite face-sharing octahedra at 2.90 Å is absent in goethite. However, amplitude at this distinctive distance was clearly present in our data, meaning that the majority of the U(VI) must reside within the hematite, although it was not possible to eliminate some fraction of uranium residing within the goethite. The fit parameters for the O shells in the 60 °C fit did not remain the same as in the 105 °C fit. The U—Oax bond distance (1.84 Å) in this sample was close to that of the adsorbed model (1.81 Å), and both the equatorial shells have longer U—O atomic distances than the 105 °C fit. Because the Fe—O octahedra in goethite and hematite are nearly identical, U(VI) present in goethite was unlikely to be the cause for the changed U—O environment between the 105 and 60 °C data sets. However, our chemical extraction data illuminate the differences between the U—O shell fit parameters between the two systems. Approximately 30% of the uranium in the 60 °C sample was acid leachable, indicating a significant proportion remains surface bound after 30 days aging. Hence, it seems the bulk EXAFS data contained a significant component of signal from U(VI) in a surface adsorption site, which caused the average U—O bond length in the EXAFS signal to be closer to those of surface bound U(VI). Additionally, the Debye–Waller factors for each U—O shell were >0.008 Å2, indicating significant disorder: our model did not account for the adsorbed component, and trying to fit two similar U—O environments simultaneously to the 60 °C data set resulted in large disorder in the U—O shells and, thus, was unjustifiable.
Linear combination fitting of the 60 °C, 30 day data, using the 0 h and 105 °C data as end members, indicated a contribution from the adsorbed U(VI) species of approximately 20% (see Supporting Information), which is in agreement with the chemical extraction data (Figure 2). Applying the same linear combination fitting to data taken after 24 h aging at 60 °C indicated approximately 40% U was adsorbed, whereas the chemical extraction suggested closer to 50% was adsorbed. This modest discrepancy may be partially due to an overestimate of the adsorbed fraction by the operationally defined chemical extraction. This is not uncommon for indirect techniques and suggests a small proportion of the partially crystalline iron oxyhydroxide was dissolved at pH 2.5.45
Uranium Incorporation into Hematite
For our end-member 105 °C experiment, the EXAFS analysis showed that during hematite crystallization and U(VI) incorporation, the uranyl axial bonds lengthen by 0.06 Å and the average equatorial bonds shorten by 0.17 Å. These changes to the U—O bond lengths are in agreement with our interpretation of the changes in energy of the resonance features in the XANES data. The EXAFS analysis is consistent with 6-fold coordinated U(VI) residing in a distorted uranate-like octahedral site within the hematite structure (Figure 5), although we accept there may be a contribution from minor amounts of U(VI) in goethite.
In earlier work, Duff et al.18 formed U(VI)-containing hematite via a coprecipitation method at pH 11 and interpreted their EXAFS as showing incorporation of uranium into the crystal structure, with an oxygen coordination of approximately 4 at radial distances of 2.19 Å (N = 1.4 ± 15%) and 2.36 Å (N = 2.1 ± 20%), with a single Fe atom (N = 1.12 ± 25%) at a distance of 3.19 Å. The implication is that uranium was incorporated into hematite with the loss of the axial U—O bonds. Latterly, it has been suggested that the Duff model had an unexpectedly low U—O coordination, suggesting that not all of the U—O bond distances were fully resolved from the EXAFS.20 The same approach to uranium incorporation into hematite was followed by Ilton et al.19 who reported a similar uranium environment to Duff et al.18 Our EXAFS data analysis showed that these models are incorrect and that U(VI) is fully coordinated by 6 oxygens within a distorted octahedral site in the hematite structure.
Our interpretation is supported by recent work on atomic simulations of uranium incorporation into hematite,20 which shows that incorporation of octahedrally coordinated U(VI), with reduction of Fe(III) as the charge compensation mechanism, maintains an average U—O bond distance of 2.06 Å. This is identical to the average U—O bond distance obtained from the fit to our data (2.06 ± 0.02 Å). However, the U—Fe atomic distances returned by the simulations were in excess of those obtained from our EXAFS fitting, in particular the calculated single U—Fe distance from the face sharing octahedra was reported at 3.37 Å.20 These differences may be due to the simulations assuming a single U—O bond length; this does not take into account the shape of the distorted UO6 octahedron that we propose. The corresponding simulation that considers incorporation of the U(VI) into an unoccupied interstitial site within the hematite structure returns similar average U—O and U—Fe bond distances to those of our EXAFS fit.20 However, the calculated Fe shells in the simulation are doubly overcoordinated compared to our fit and we were unable to reconcile this model with our data, leading us to discount U(VI) incorporation into a vacancy site. We can see no viable mechanism to achieve charge compensation by reduction of Fe(III) to Fe(II) in our fully oxidized system. Similarly, reduction of U(VI) to U(V) again seems to be improbable in the absence of a suitable reducing agent. Although distinction of U(V) from U(IV) and U(VI) has been shown to be possible with high resolution XAS techniques,46 it is not possible to do so for our samples at their low U-loadings. Furthermore, in crystalline materials, reportedly the U(V) cation may occur in octahedral or pentagonal bipyramidal coordination with a near linear O—U—O unit, but the U—O bond length is typically around 2 Å,47 which is in vast excess to the 1.87 Å we observed, giving confidence that U(V) was not present in our samples. Another potential charge compensation mechanism postulated by Kerisit et al.20 for U(VI) substituting for Fe(III) is via creation of an Fe vacancy in its vicinity. To test for this we refitted the model described above, but with the coordination number of each Fe shell reduced by one, sequentially (Supporting Information Tables SI-3 and SI-4). None of these fits gave a statistical improvement on that presented here, and in fact, the omission of the face-sharing Fe significantly worsened the fit (Supporting Information Table SI-7). This suggests that if the charge compensation is via creation of a Fe(III) vacancy, then the vacancy is (a) located in the edge-sharing or corner-sharing shells and (b) is randomly distributed relative to U(VI) or is undetectable within the constraints of the EXAFS measurements we made.
Overall, in this study, we present clear evidence for U(VI) incorporation into hematite in an octahedrally coordinated environment and via direct substitution for Fe(III). Our model requires retention of the uranyl bonds as evidenced by the XANES and EXAFS analyses, albeit elongated within the structure, which is in direct contrast to previous studies.18,19 Our data also evidence the importance of high quality spectroscopic data out to high k when attempting to model actinide incorporation into iron oxides.
Implications for Uranium in the Environment
Our work highlights that under conditions relevant to both geological disposal and contaminated land, a significant proportion of U(VI) adsorbed to ferrihydrite is incorporated into the hematite crystal structure during crystallization. In our experiments, hematite showed the ability to incorporate approximately 3000 ppm U(VI) (0.3 wt %) in the solid. This is relevant to a wide range of nuclear decommissioning and waste management scenarios where iron oxides are ubiquitous. Indeed, the incorporation of uranium into iron oxides, specifically hematite, has implications for reducing the long-term environmental mobility of U(VI), especially given the long-term stability of hematite, which is found in geological settings older than 1 Ga.48 It is also worth noting that elevated temperatures associated with disposal of heat-yielding radioactive wastes may enhance hematite formation and, thereby, U(VI) immobilization. In addition, under conditions where biogeochemical processes can occur, it is interesting to note that hematite is recalcitrant to microbial reduction due to its crystallinity, with only a thin surface layer of bioavailable Fe(III) present,49 again suggesting its stability may be significant in, for example, oxic-contaminated land environments. Fe(II)aq has been shown to enhance the release of iron oxide incorporated trace metals,50 although interestingly, natural iron oxides substituted with, for example, Al3+ are less susceptible to Fe(II)-activated recrystallization, and as such, trace metal release may be inhibited in these phases.51 In particular, the alkaline conditions used in this study show that these processes are directly relevant to the conditions expected around a cementitious disposal facility for radioactive watse27 as well as alkaline waste management scenarios (e.g., Hanford tanks52). Thus, our results show that substantial incorporation of U(VI) into hematite can occur, which is potentially a significant new pathway to immobilize U(VI) and has clear implications for the environmental mobility of this important radionuclide, especially in high pH conditions relevant to engineered waste environments.
Acknowledgments
This work has been funded as part of the U. K. Natural Environment Research Council (NERC) BIGRAD consortium through consortium grant NE/H007768/1. Diamond Light Source is thanked for providing Beamtime grants SP7367 and SP7593. We thank Dr. Steve Parry and Richard Doull for assistance at Diamond. We thank Dr. Mike Ward, Dr. John Waters, and Mr. Paul Lythgoe for assistance with TEM imaging, BET/XRD, and ICP-MS analyses, respectively.
Supporting Information Available
Additional information about materials, experimental set up, and results, including EXAFS fits. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Moyes L. N.; Parkman R. H.; Charnock J. M.; Vaughan D. J.; Livens F. R.; Hughes C. R.; Braithwaite A. Uranium Uptake from Aqueous Solution by Interaction with Goethite, Lepidocrocite, Muscovite, and Mackinawite: An X-Ray Absorption Spectroscopy Study. Environ. Sci. Technol. 2000, 34, 1062–1068. [Google Scholar]
- Dodge C. J.; Francis A. J.; Gillow J. B.; Halada G. P.; Eng C.; Clayton C. R. Association of Uranium with Iron Oxides Typically Formed on Corroding Steel Surfaces. Environ. Sci. Technol. 2002, 36, 3504–3511. [DOI] [PubMed] [Google Scholar]
- Lovley D. R.; Phillips E. J. P. Reduction of Uranium by Desulfovibrio-Desulfuricans. Appl. Environ. Microbiol. 1992, 58, 850–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovley D. R.; Phillips E. J. P. Bioremediation of Uranium Contamination with Enzymatic Uranium Reduction. Environ. Sci. Technol. 1992, 26, 2228–2234. [Google Scholar]
- Boyanov M. I.; Fletcher K. E.; Kwon M. J.; Rui X.; O’Loughlin E. J.; Loeffler F. E.; Kemner K. M. Solution and Microbial Controls on the Formation of Reduced U(IV) Species. Environ. Sci. Technol. 2011, 45, 8336–8344. [DOI] [PubMed] [Google Scholar]
- Renshaw J. C.; Butchins L. J. C.; Livens F. R.; May I.; Charnock J. M.; Lloyd J. R. Bioreduction of Uranium: Environmental Implications of a Pentavalent Intermediate. Environ. Sci. Technol. 2005, 39, 5657–5660. [DOI] [PubMed] [Google Scholar]
- O’Loughlin E. J.; Kemner K. M.; Burris D. R. Effects of Ag-I, Au-III, and Cu-II on the Reductive Dechlorination of Carbon Tetrachloride by Green Rust. Environ. Sci. Technol. 2003, 37, 2905–2912. [DOI] [PubMed] [Google Scholar]
- Ilton E. S.; Boily J. F.; Buck E. C.; Skomurski F. N.; Rosso K. M.; Cahill C. L.; Bargar J. R.; Felmy A. R. Influence of Dynamical Conditions on the Reduction of U(VI) at the Magnetite-Solution Interface. Environ. Sci. Technol. 2010, 44, 170–176. [DOI] [PubMed] [Google Scholar]
- Zhou P.; Gu B. H. Extraction of Oxidized and Reduced Forms of Uranium from Contaminated Soils: Effects of Carbonate Concentration and pH. Environ. Sci. Technol. 2005, 39, 4435–4440. [DOI] [PubMed] [Google Scholar]
- Wilkins M. J.; Livens F. R.; Vaughan D. J.; Beadle I.; Lloyd J. R. The Influence of Microbial Redox Cycling on Radionuclide Mobility in the Subsurface at a Low-Level Radioactive Waste Storage Site. Geobiology 2007, 5, 293–301. [Google Scholar]
- Law G. T. W.; Geissler A.; Burke I. T.; Livens F. R.; Lloyd J. R.; McBeth J. M.; Morris K. Uranium Redox Cycling in Sediment and Biomineral Systems. Geomicrobiol. J. 2011, 28, 497–506. [Google Scholar]
- Singh B.; Sherman D. M.; Gilkes R. J.; Wells M.; Mosselmans J. F. W. Structural Chemistry of Fe, Mn, and Ni in Synthetic Hematites as Determined by Extended X-Ray Absorption Fine Structure Spectroscopy. Clays Clay Miner. 2000, 48, 521–527. [Google Scholar]
- Liu J.; Liang C.; Zhang H.; Tian Z.; Zhang S. General Strategy for Doping Impurities (Ge, Si, Mn, Sn, Ti) in Hematite Nanocrystals. J. Phys. Chem. C 2012, 116, 4986–4992. [Google Scholar]
- Nico P. S.; Stewart B. D.; Fendorf S. Incorporation of Oxidized Uranium into Fe (Hydr)oxides during Fe(II) Catalyzed Remineralization. Environ. Sci. Technol. 2009, 43, 7391–7396. [DOI] [PubMed] [Google Scholar]
- Stewart B. D.; Nico P. S.; Fendorf S. Stability of Uranium Incorporated into Fe (Hydr)oxides under Fluctuating Redox Conditions. Environ. Sci. Technol. 2009, 43, 4922–4927. [DOI] [PubMed] [Google Scholar]
- Boland D. D.; Collins R. N.; Payne T. E.; Waite T. D. Effect of Amorphous Fe(III) Oxide Transformation on the Fe(II)-Mediated Reduction of U(VI). Environ. Sci. Technol. 2011, 45, 1327–1333. [DOI] [PubMed] [Google Scholar]
- Payne T. E.; Davis J. A.; Waite T. D. Uranium Retention by Weathered Schists–The Role of Iron Minerals. Radiochim. Acta 1994, 66/67, 297–303. [Google Scholar]
- Duff M. C.; Coughlin J. U.; Hunter D. B. Uranium Co-Precipitation with Iron Oxide Minerals. Geochim. Cosmochim. Acta 2002, 66, 3533–3547. [Google Scholar]
- Ilton E. S.; Pacheco J. S. L.; Bargar J. R.; Shi Z.; Liu J.; Kovarik L.; Engelhard M. H.; Felmy A. R. Reduction of U(VI) Incorporated in the Structure of Hematite. Environ. Sci. Technol. 2012, 46, 9428–9436. [DOI] [PubMed] [Google Scholar]
- Kerisit S.; Felmy A. R.; Ilton E. S. Atomistic Simulations of Uranium Incorporation into Iron (Hydr)Oxides. Environ. Sci. Technol. 2011, 45, 2770–2776. [DOI] [PubMed] [Google Scholar]
- Smith S. C.; Douglas M.; Moore D. A.; Kukkadapu R. K.; Arey B. W. Uranium Extraction From Laboratory-Synthesized, Uranium-Doped Hydrous Ferric Oxides. Environ. Sci. Technol. 2009, 43, 2341–2347. [DOI] [PubMed] [Google Scholar]
- Music S.; Gotic M.; Popovic S. X-Ray Diffraction and Fourier-Transform Infrared-Analysis of the Rust Formed by Corosion of Steel in Aqueous-Solutions. J. Mater. Sci. 1993, 28, 5744–5752. [Google Scholar]
- Collier N. C.; Milestone N. B.; Hill J.; Godfrey I. H. The Disposal of Radioactive Ferric Floc. Waste Manage. 2006, 26, 769–775. [DOI] [PubMed] [Google Scholar]
- The 2010 U. K. Radioactive Waste Inventory Main Report; Pöyry Energy Limited: West Sussex, U. K., 2011. [Google Scholar]
- Ferris F. G.; Hallberg R. O.; Lyven B.; Pedersen K. Retention of Strontium, Cesium, Lead and Uranium by Bacterial Iron Oxides from a Subterranean Environment. Appl. Geochem. 2000, 15, 1035–1042. [Google Scholar]
- Geological Disposal: Near-Field Evolution Status Report; Nuclear Decommissioning Authority: Didcot, U. K., 2011. [Google Scholar]
- Wallace S. H.; Shaw S.; Morris K.; Small J. S.; Burke I. T. Alteration of Sediments by Hyperalkaline K-Rich Cement Leachate: Implications for Strontium Adsorption and Incorporation. Environ. Sci. Technol. 2013, 47, 3694–700. [DOI] [PubMed] [Google Scholar]
- Cornell R. M.; Schwertmann U.. The Iron Oxides: Structure, Properties, Reactions, Occurences and Uses, 2nd ed.; Wiley-VCH: Weinham, 2003. [Google Scholar]
- Schwertmann U.; Murad E. Effect of pH on the Formation of Goethite and Hematite from Ferrihydrite. Clays Clay Miner. 1983, 31, 277–284. [Google Scholar]
- Fischer W. R.; Schwertmann U. Formation of Hematite from Amorphous Iron(III) Hydroxide. Clays Clay Miner. 1975, 23, 33–&. [Google Scholar]
- Combes J. M.; Manceau A.; Calas G. Formation of Ferric Oxides from Aqueous-Solutions–A Polyhedral Approach by X-Ray Absorption-spectroscopy. 2. Hematite Formation from Ferric Gels. Geochim. Cosmochim. Acta 1990, 54, 1083–1091. [Google Scholar]
- Shaw S. The Kinetics and Mechanisms of Goethite and Hematite Crystallization under Alkaline Conditions, and in the Presence of Phosphate. Am. Mineral. 2005, 90, 1852–1860. [Google Scholar]
- Vu H. P.; Shaw S.; Brinza L.; Benning L. G. Crystallization of Hematite (α-Fe2O3) under Alkaline Condition: The Effects of Pb. Cryst. Growth Des. 2010, 10, 1544–1551. [Google Scholar]
- Waite T. D.; Davis J. A.; Payne T. E.; Waychunas G. A.; Xu N. Uranium(VI) Adsorption to Ferrihydrite–Application of a Surface Complexation Model. Geochim. Cosmochim. Acta 1994, 58, 5465–5478. [Google Scholar]
- Dent A. J.; Cibin G.; Ramos S.; Smith a D.; Scott S. M.; Varandas L.; Pearson M. R.; Krumpa N. a; Jones C. P.; Robbins P. E. B18: A Core XAS Spectroscopy Beamline for Diamond. J. Phys.: Conf. Ser. 2009, 190, 012039. [Google Scholar]
- Ravel B.; Newville M. ATHENA, ARTEMIS, HEPHAESTUS: Data Analysis for X-Ray Absorption Spectroscopy Using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [DOI] [PubMed] [Google Scholar]
- Vu H. P.; Shaw S.; Brinza L.; Benning L. G. Partitioning of Pb(II) during Goethite and Hematite Crystallization: Implications for Pb Transport in Natural Systems. Appl. Geochem. 2013, 39, 119–128. [Google Scholar]
- Farges F.; Ponader C. W.; Calas G.; Brown G. E. Structural Enviroments of Incompatible Elements in Silicate Glass Melt Systems II. U(IV), U(V), AND U(VI). Geochim. Cosmochim. Acta 1992, 56, 4205–4220. [Google Scholar]
- Misra N. L.; Lahiri D.; Mudher K. D. S.; Olivi L.; Sharma S. M. XANES Study on Novel Mixed-Valent A(2)U(4)O(12)(A = K, Rb or Tl) Uranates. X-Ray Spectrom. 2008, 37, 215–218. [Google Scholar]
- Blake R. L.; Hessevic Re; Zoltai T.; Finger L. W. Refinement of the Hematite Structure. Am. Mineral. 1966, 51, 123–129. [Google Scholar]
- Loopstra B. O.; Rietveld H. M. The Structure of Some Alkaline-Earth Metal Uranates. Acta Crystallogr. 1969, B25, 787–791. [Google Scholar]
- Shannon R. D. Revised Effective Ionic-Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr. 1976, 32, 751–767. [Google Scholar]
- Downward L.; Booth C. H.; Lukens W. W.; Bridges F.. A Variation of the F-Test for Determining Statistical Relevance of Particular Parameters in EXAFS Fits. In X-Ray Absorption Fine Structure—XAFS13: 13th International Conference; Hedman B., Painetta P., Eds.; American Institute of Physics: College Park, MD, 2007; Vol. 882, pp 129–131. [Google Scholar]
- Szytula A.; Burewicz A.; Dimitrij Z.; Krasnick S.; Rzany H.; Todorovi J.; Wanic A.; Wolski W.; Dimitrijvic Z.; Krasnicki S.; Todorovic J. Neutron Diffraction Studies of Alpha-FeOOH. Phys. Status Solidi 1968, 429, 429–434. [Google Scholar]
- Reyes I.; Torrent J. Citrate-Ascorbate as a Highly Selective Extractant for Poorly Crystalline Iron Oxides. Soil Sci. Soc. Am. J. 1997, 61, 1647–1654. [Google Scholar]
- Vitova T.; Kvashnina K. O.; Nocton G.; Sukharina G.; Denecke M. A.; Butorin S. M.; Mazzanti M.; Caciuffo R.; Soldatov A.; Behrends T.; Geckeis H.. High Energy Resolution X-Ray Absorption Spectroscopy Study of Uranium in Varying Valence States. Phys. Rev. B 2010, 82. [Google Scholar]
- Burns P. C.; Ewing R. C.; Hawthorne F. C.; Polyhedra P. O. F. The Crystal Chemistry of Hexavalent Uranium: Polyhedron Geometries, Bond-Valence Parameters, and Polymerization of Polyhedra. Can. Mineral. 1997, 35, 1551–1570. [Google Scholar]
- Hitzman M. W.; Oreskes N.; Einaudi M. T. Geological Characteristics and Tectonic Setting of Proterozoic Iron Oxide (Cu-U-Au-REE) Deposits. Precambrian Res. 1992, 58, 241–287. [Google Scholar]
- Cutting R. S.; Coker V. S.; Fellowes J. W.; Lloyd J. R.; Vaughan D. J. Mineralogical and Morphological Constraints on the Reduction of Fe(III) Minerals by Geobacter Sulfurreducens. Geochim. Cosmochim. Acta 2009, 73, 4004–4022. [Google Scholar]
- Frierdich A. J.; Catalano J. G. Fe(II)-Mediated Reduction and Repartitioning of Structurally Incorporated Cu, Co, and Mn in Iron Oxides. Environ. Sci. Technol. 2012, 46, 11070–7. [DOI] [PubMed] [Google Scholar]
- Frierdich A. J.; Scherer M. M.; Bachman J. E.; Engelhard M. H.; Rapponotti B. W.; Catalano J. G. Inhibition of Trace Element Release During Fe(II)-Activated Recrystallization of Al-, Cr-, and Sn-Substituted Goethite and Hematite. Environ. Sci. Technol. 2012, 46, 10031–10039. [DOI] [PubMed] [Google Scholar]
- Um W.; Wang Z. M.; Serne R. J.; Williams B. D.; Brown C. F.; Dodge C. J.; Francis A. J. Uranium Phases in Contaminated Sediments below Hanford’s U Tank Farm. Environ. Sci. Technol. 2009, 43, 4280–4286. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.