

Abstract
Self-assembly of three-dimensional solid-state nanostructures containing approximately 33% by weight globular protein is demonstrated using a globular protein-polymer diblock copolymer, providing a route to direct nanopatterning of proteins for use in bioelectronic and biocatalytic materials. A mutant red fluorescent protein, mCherryS131C, was prepared by incorporation of a unique cysteine residue and site-specifically conjugated to end-functionalized poly(N-isopropylacrylamide) through thiol-maleimide coupling to form a well-defined model protein-polymer block copolymer. The block copolymer was self-assembled into bulk nanostructures by solvent evaporation from concentrated solutions. Small-angle X-ray scattering and transmission electron microscopy illustrated the formation of highly disordered lamellae or hexagonally perforated lamellae depending upon the selectivity of the solvent during evaporation. Solvent annealing of bulk samples resulted in a transition towards lamellar nanostructures with mCherry packed in a bilayer configuration and a large improvement in long range ordering. Wide-angle X-ray scattering indicated that mCherry did not crystallize within the block copolymer nanodomains and that the β-sheet spacing was not affected by self-assembly. Circular dichroism showed no change in protein secondary structure after self-assembly, while UV-vis spectroscopy indicated approximately 35% of the chromophore remained optically active.
Keywords: self-assembly, block copolymer, mCherry, PNIPAM, bioconjugation
Enzymes have garnered a great deal of attention for incorporation into a variety of biocatalytic and bioelectronic devices1–3 because of their substrate specificity, high catalytic rates, and ability to operate under mild reaction conditions. Enzymes have been incorporated into highly selective and sensitive biosensors for applications such as glucose detection and medical diagnostics.4–5 Biofuel cells have been prepared that operate on nontraditional fuels such as sugars, utilize low operating overpotentials, and do not require an electrolyte membrane.6–8 Enzymes also continue to be widely used to catalyze the synthesis of pharmaceutical compounds,9 and more recently, biocatalysts are being pursued for a wide variety of energy applications, including carbon sequestration,10 carbon dioxide reduction,11–13 and hydrogen production.14–16 Photodiodes or photovoltaics have been prepared from reaction center proteins,17 photosynthetic complexes,18 and fluorescent proteins19 in attempts to exploit the high absorption cross section and quantum efficiency of the proteins. All of these protein-based devices offer the added advantages of biodegradability and renewability, making them attractive as green chemistry techniques become increasingly prevalent.
Although proteins offer the aforementioned desirable advantages, the incorporation of these complex molecules into highly efficient and functional devices presents a large number of challenges. In order to increase the enzyme activity per unit area, a high density of enzyme should be achieved by structuring the material in three dimensions. Diffusion of substrate/product or charge carriers into and out of the material must be rapid to minimize transport limitations, and control over the orientation of the enzyme is desired to maintain access to the substrate and cofactor binding areas. Finally, the material should be engineered to improve the stability and lifetime of the protein. These engineering design goals have motivated the development of a number of strategies for immobilizing or nanopatterning enzymes.20 Operational stability, efficiency and lifetime of enzymes have been shown to increase through immobilization with multiple tethers on porous supports or within a crosslinked material.21
Nanopatterning of proteins is required to increase the density of enzymes at the surface and to ensure continuous pathways for transport of electrons, substrates, and products through the material. Nanopatterning may also be used as a method to enforce proper orientation and stabilize the fold of the protein. Layer-by-layer techniques have been used to pattern proteins and polyelectrolytes on surfaces by taking advantage of electrostatic, hydrophobic and hydrogen bond interactions between the materials.22–23 Alternately, a variety of lithographic methods including microcontact printing, photolithography and dip-pen lithography have been used to create templates for protein immobilization by patterning an attachment site where the protein selectively binds.24–26 Covalent binding using lysine-NHS or cysteine-maleimide chemistries,27 or non-covalent binding using biotin-avidin28 or His tag-NTA29 interactions are then employed to immobilize enzymes on the pre-formed patterns. In a third approach, nanostructured templates are self-assembled from amphiphilic molecules and the proteins are inserted into the templates. This type of template has been demonstrated with the use of block copolymer thin films,30 nanostructures formed from surfactants in solution,31 and through the self-assembly of lipid materials.32
Block copolymer self-assembly provides a simple, low cost method for fabricating nanopatterns in three dimensions that may be of high value for producing enzyme-based materials. Block copolymers prepared with two or more Gaussian coil polymer blocks are well-known to self-assemble into a wide variety of nanostructures with characteristic length scales from 5–100 nm.33–35 The multidomain structures formed by these block copolymers make them ideal candidates for building three-dimensional structures with efficient mass or charge carrier transport.36–37 However, the thermodynamics of coil-coil block copolymers do not generalize to more complex systems. Changes in the topology of the polymer chain and liquid crystalline or other specific interactions between polymers result in large changes in the phase behavior of these systems, as has been demonstrated for rod-coil block copolymers.38 Incorporating a globular protein as one block in a block copolymer to direct the protein’s self-assembly into nanostructured materials introduces a great deal of added complexity beyond both coil-coil and rod-coil systems due to the specific chain fold in the protein domain, the strong, directional interactions between proteins that lead to macromolecular crystallization, and the complex thermodynamics of interactions between proteins and synthetic polymers. It is unclear how the traditional concepts of block copolymer self-assembly will apply to globular protein-polymer block copolymers, particularly when the constraint of maintaining protein fold and function during processing and nanostructure self-assembly is imposed.
While significant effort has been directed towards the self-assembly of bulk and thin film block copolymers with α-helical, β-sheet, coiled-coil, and cyclical peptide blocks,39–41 studies of globular protein-polymer conjugates have been restricted primarily to self-assembly in solution. Giant amphiphiles consisting of globular proteins and synthetic polymers have been synthesized and their self-assembly has been studied in solution.42–44 The variety of observed morphologies including spherical, rod-like and toroid micelles is due primarily to changes in relative coil fraction as well as processing conditions which kinetically favor certain morphologies.45 Among these amphiphilic conjugates are PEGylated proteins which have been used to improve the stability and lifetime of proteins in serum.46–48 However, there is currently a lack of fundamental knowledge on the self-assembly of globular protein-polymer conjugates in concentrated solutions or solid-state materials, and these materials have not been evaluated as a means to nanopattern protein-based devices.
Here, we demonstrate block copolymer self-assembly as an effective technique for producing nanostructured plastics from globular proteins. A model globular protein-polymer diblock copolymer is synthesized based on the site-specific conjugation of poly(N-isopropylacrylamide) (PNIPAM) to the fluorescent protein mCherry. Self-assembly is induced by evaporation of water from concentrated solutions of the copolymers, and the kinetic effects of evaporation method and post-evaporation solvent annealing are shown to influence nanostructure formation. The fold and optical activity of mCherry within the nanostructured assembly is investigated, and the packing of mCherry within the self-assembled nanostructure is explored.
Results and Discussion
Globular Protein-Polymer Diblock Copolymer Synthesis
A model globular protein-polymer conjugate with a single well-defined bioconjugation site was synthesized using the red fluorescent protein mCherry and the thermoresponsive synthetic polymer PNIPAM. The protein mCherry was selected because the native sequence lacked cysteine residues, the high yield expression and purification of folded protein are well-established, and the fluorescent nature provided a simple and robust spectrophotometric method for conjugate characterization. The protein mCherry49–50 was mutated to introduce a unique thiol conjugation site into its sequence by replacing serine with cysteine at residue 131, yielding the mutant mCherryS131C. The mutation is located in a loop region on the end of the β-barrel structure opposite both the N and C termini, as illustrated in Scheme 1. A 6xHis-tagged variant of this protein was expressed in Escherichia coli (E. coli) and then purified using metal affinity chromatography under native reducing conditions to preserve the delicate chromophore bond and prevent thiol inactivation. The yield of purified protein, determined spectrophotometrically at 586 nm based on the known extinction coefficient of mCherry,49 was 121 mg/L culture.
Scheme 1.
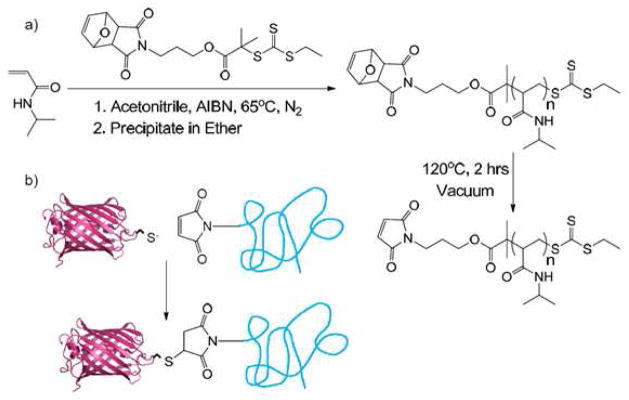
Synthesis of maleimide-functionalized poly(N-isopropylacrylamide) and its bioconjugation to mCherryS131C to create a protein-polymer diblock copolymer.
The diblock copolymer was synthesized by conjugating mCherryS131C to maleimide-functionalized PNIPAM. A reversible addition-fragmentation chain transfer (RAFT) agent containing a protected maleimide group was used to synthesize low polydispersity PNIPAM (Scheme 1). After polymerization, the maleimide group was thermally deprotected to yield maleimide end-functionalized PNIPAM with a molar mass of 51.3 kg/mol and a polydispersity of 1.24. Gel permeation chromatography (GPC) analysis is shown in Figure 1. The polydispersity in these samples originates in part from a small shoulder at twice the peak molecular weight in the GPC trace that represents 8% of the total polymer mass. This minor high molecular weight fraction is believed to originate from a slight reactivity of the double bond in the protected maleimide group on the RAFT agent during polymerization to conversions greater than 50%. When an identical RAFT agent is used without the protected maleimide functionality, no coupling is observed.
Figure 1.

Normalized gel permeation chromatography trace of deprotected poly(N-isopropylacrylamide) product with a poly(methyl methacrylate)-equivalent molecular weight of 51.3 kg/mol and a polydispersity of 1.24.
Bioconjugation was then performed at room temperature in 20 mM Tris buffer (pH 8) using a eight-fold excess of PNIPAM. SDS-PAGE of the crude conjugation product and purified fractions is shown in Figure 2. The band at approximately 70,000 g/mol corresponds to the mCherry-PNIPAM conjugate with a PNIPAM volume fraction of 0.66 and a weight fraction of 0.66. The band at 28,000 g/mol corresponds to unconjugated mCherry, while the bands at 19,000 g/mol and 9,000 g/mol correspond to the partial hydrolysis of the mCherry chromophore acylimine bond during SDS-PAGE analysis.50–53 Lane analysis of the crude reaction mixture revealed a conversion of approximately 78%, comparable to previously reported conversions for thiol-maleimide couplings to globular proteins.54–56 Although the eight-fold excess of PNIPAM used gave maximum conversion of the mCherryS131C, decreasing to a five-fold excess still yielded approximately 70% conversion. After conjugation, unreacted mCherry was removed using ammonium sulfate precipitation; SDS-PAGE showed that after the first precipitation no additional unreacted mCherry is removed. Unconjugated PNIPAM was subsequently removed by metal affinity chromatography; washing with 7 column volumes of buffer was used to ensure complete removal of the free PNIPAM. Analysis of the second elution lane (lane 8) suggests that the conjugate is > 90% pure, while the third elution lane (lane 9) demonstrates that at lower protein concentrations only the conjugate band is visible. The purified conjugate was obtained in a final yield of 30%. Circular dichroism and UV-vis spectroscopy (Supporting Information) confirmed that the protein structure and optical properties in the purified mCherry-PNIPAM diblock remained unchanged from that of the mCherryS131C in solution. Cloud point measurements (Supporting Information) show that the thermal transition of the conjugate increases by ~ 5 °C relative to that of the homopolymer due to addition of the large hydrophilic protein.
Figure 2.
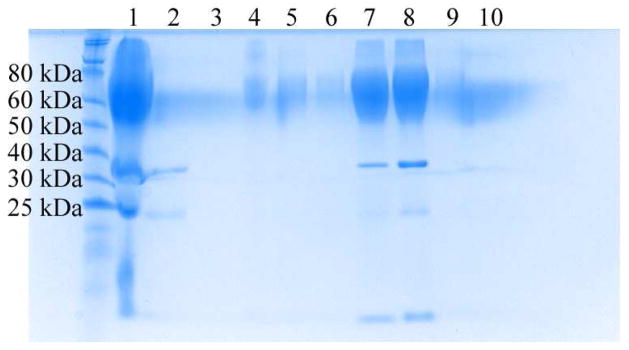
Denaturing SDS-PAGE gel showing purification of mCherryS131C-PNIPAM conjugate. The crude reaction mixture (lane 1) was purified by repeated precipitation of the conjugate using ammonium sulfate to remove unconjugated mCherryS131C. The discarded supernatants (lanes 2 and 3) contained most of the unconjugated mCherry. Next, metal affinity chromatography was used to remove excess PNIPAM. Minimal conjugate is lost from the column flow through (lane 4) and the two wash steps (lanes 5 and 6). The conjugate is then eluted in four fractions (lanes 7–10).
Nanostructure Formation
Self-assembly of mCherry-PNIPAM diblock copolymers was accomplished through evaporation of water from diblock copolymer solutions to form nanostructured bulk plastics. The formation of nanostructures strongly depends on the processing conditions used to prepare the material, as demonstrated by small-angle X-ray scattering (SAXS), shown in Figure 3, and transmission electron microscopy (TEM) images, shown in Figure 4. In the TEM images, the mCherry areas appear dark due to staining with ruthenium tetroxide which reacts with alcohols, amines and aromatics present on the protein surface.57 Two processing pathways were explored for water evaporation: a protein-selective solvent (40 °C water, above the lower critical solution temperature (LCST) of the PNIPAM homopolymer) and a nonselective solvent (room temperature water), as illustrated in Figure 5.
Figure 3.

Small-angle x-ray scattering (SAXS) data of as-cast and solvent annealed mCherry-PNIPAM block copolymers. Samples were cast from both protein-selective (40 °C water) and nonselective (room temperature water) solvents. The materials form long-range ordered nanostructures with weak order. After solvent annealing in water, the samples transition to lamellar nanostructures with improved long-range order. Traces have been offset for clarity.
Figure 4.

TEM images of mCherry-PNIPAM block copolymers demonstrate the formation of lamellae and hexagonally perforated lamellae in bulk samples cast at 40 °C (a) or room temperature (b), and samples cast at room temperature followed by solvent annealing in water at 4 °C (c) or room temperature (d). Panel e shows the 40 °C cast sample at a lower magnification.
Figure 5.

This schematic depicts two possible pathways towards self-assembly of mCherry-PNIPAM block copolymers. Room temperature water provides a nonselective solvent, whereas 40 °C water is a protein-selective solvent. Self-assembly is induced by increasing the block copolymer concentration through solvent evaporation.
Both as-cast samples contain multiple peaks in their SAXS patterns, indicating the formation of long-range ordered nanostructures. The nanostructures formed from a nonselective solvent exhibit two sets of peaks, a relatively intense set that can be indexed to a lamellar structure and a weaker set that may be indexed to a hexagonal lattice. The primary peak is asymmetric, composed of a 001 peak and a low q 100 shoulder. The first higher order reflections may be indexed to 110, 200, 002, and 003, corresponding to peaks from both a hexagonal and a lamellar lattice. These peak observations are consistent with a hexagonally packed lamellar (HPL) morphology with a lamellar domain spacing of 23.4 nm and a hexagonal domain spacing of 26.5 nm. The 002 and 003 scattering peaks, corresponding to the lamellar spacing, are much more intense than the 110 and 200 scattering peaks corresponding to the hexagonal perforations, as is typical for HPL structures.58 TEM images of the sample provide evidence for HPL formation and are consistent with previously reported images of HPL from coil-coil diblock copolymers.59–60 The micrograph in Figure 4b shows areas of lamellar structure with perforations parallel to the lamellar normal as well as regions of the sample that have a hexagonally packed structure. These two structures are consistent with different orientations of the HPL unit cell in the sample. The formation of an HPL morphology at a PNIPAM volume fraction of 0.66 is similar to that found in coil-coil block copolymer systems where HPL morphologies are typically seen as non-equilibrium structures in a narrow window between lamellae and cylinders.59 Even though the protein is the minority block in these copolymers, the protein domains perforate the coil regions. This suggests that the protein remains more highly hydrated than the PNIPAM during sample casting, increasing its effective volume fraction.
The assembled nanostructures from a protein-selective solvent show poorer ordering than those obtained from a nonselective solvent. The protein-selective solvent results in a relatively broad primary peak with a broad second order shoulder centered around a q value twice that of the primary peak. While the observed peak positions are consistent with the formation of lamellar nanostructures, the broad peaks indicate that the order in this sample is poorer than that in the sample cast from nonselective solvent. The TEM images in Figure 4a,e show the structurally heterogeneous nature of this sample containing small lamellar regions with hexagonally packed areas interspersed. These images show that the sample is composed of undulating lamellae along accompanied by regions of hexagonal structures with PNIPAM centers similar to the structure of the sample cast from a nonselective solvent. These structures may be the result of micelles which merged to form sheets. A lower magnification image (Figure 4e) reveals that these lamellar regions are interspersed with spherical protein aggregates, and that these domains are inverted from the observed HPL morphologies. Because this sample was prepared from a state where the protein and polymer are segregated due to immiscibility of the solvent and polymer, it is likely that kinetic barriers to nanoscale structural rearrangement during solvent evaporation result in the structural heterogeneity and a relatively low degree of order.
In comparison to the sample cast in the nonselective solvent, the sample cast from protein-selective solvent has a larger domain spacing of 32.1 nm, as measured by SAXS. However, TEM images suggest that the domain sizes for the more highly ordered (Figure 4a) and more disordered (Figure 4e) regions of the protein-selective solvent cast sample differ significantly. Fourier transforms of the images of well-ordered lamellar regions and the disordered regions indicate that the disordered regions are 40% larger than the lamellar regions. This increased domain spacing observed at larger length scales is consistent with the 37% larger domain spacing observed by SAXS of the protein-selective solvent compared to the lamellar sample from a nonselective solvent. The observed difference in domain spacing for disordered and lamellar regions of the protein-selective solvent condition suggests that the equilibrium lamellar spacing would be quite similar for both casting conditions; however, the processing-dependent effects that lead to the high degree of structural heterogeneity and kinetic trapping of nonequilibrium structures for the protein-selective solvent result in a large increase in the average domain spacing.
Solvent annealing of samples cast from the nonselective solvent condition was used to probe whether the HPL structure was at equilibrium. Annealed samples show enhanced ordering of the nanodomains with a clear lamellar symmetry and a domain spacing corresponding to the lamellar spacing observed in the as-cast sample, suggesting that the lamellar phase is closer to thermodynamic equilibrium for this sample. After annealing in water at either 4 °C or room temperature for 8 hours, the primary peak shifted to slightly lower q* than in the as-cast sample, all reflections in the SAXS pattern became sharper, and the two higher order reflections now occur at 2q* and 3q*, consistent with the formation of lamellae. TEM confirms the formation of lamellar nanostructures in the room temperature annealed sample while some hexagonally perforated lamellae are still present in the sample annealed at 4 °C. Both annealed samples show clear layered structures and larger grain sizes than the as-cast sample. In addition, dislocations typical of lamellar block copolymers may be observed in the annealed samples. The improvements in translational and orientational order are consistent with the sharpening of scattering peaks and the observation of the third order reflection. In addition, the lamellae are relatively straight as compared to the typical fingerprint patterns observed in coil-coil diblocks. The nanodomain persistence length appears to be longer than that of coil-coil block copolymer domains, but shorter than that of typical rod-coil block copolymer domains.61 This observation is likely due to the small, yet well-defined rigid shape of the mCherry protein which may introduce an enhanced bending rigidity within the nanodomains relative to that of a coil-coil diblock copolymer.
Although both solvent annealed samples show an evolution towards lamellar structures, the disappearance of the low q shoulder on the primary peak and the appearance of the third order peak are both more pronounced in the sample annealed at room temperature. In addition, the domain spacing of the sample annealed at room temperature (24.0 nm) is larger than the domain spacing of the sample annealed at 4 °C (23.1 nm). While both annealing conditions were chosen to occur in the nonselective solvent regime, both the chemical potential of water and the water-PNIPAM interactions are strongly temperature dependent. The PNIPAM block is anticipated to have a more favorable interaction with water at lower temperature, but the chemical potential of water vapor during solvent annealing decreases as a function of temperature. Both the slightly larger domain spacing and the increased degree of ordering observed in the room temperature annealed sample suggest that the increased chemical potential of water at higher temperature dominates the annealing behavior, resulting in increased swelling of the lamellar nanostructures and accounting for the increase in domain spacing. In addition, both higher temperature and higher water content will increase the mobility of the polymer, consistent with the observation of more intense higher order peaks and a stronger depletion of the hexagonally perforated lamellar phase in the sample annealed at room temperature.
Using scaling relationships for domain spacing as a function of molecular weight, the proteins may be inferred to pack in a bilayer structure within the lamellae. Based on the crystallographic structure of mCherry,50 the protein has a length of approximately 4.2 nm in the folded state. Because the folded proteins are rigid, the same scaling analysis for domain spacing in rod-coil diblock copolymers61 is expected to apply. For the mCherry-PNIPAM diblocks, this would yield an expected domain spacing of ~12.4 nm in the monolayer configuration. Because this value is approximately half that of the experimentally observed domain spacing, it is most likely that the mCherry is packed in a bilayer configuration within the lamellar nanodomains.
Protein Assembly Within the Nanodomain Structure
To enable the fabrication of nanostructured protein-based materials, the protein in these self-assembled block copolymers must remain properly folded and functional. The functionality of mCherry in solid-state mCherry-PNIPAM materials was quantified using both UV-vis spectroscopy and circular dichroism, demonstrating that the self-assembly process preserves a large fraction of the protein structure and optical activity. UV-vis spectra were measured for solid-state samples (Figure 6a) and for rehydrated diblock copolymers (Figure 6b). In the solid state, the peak absorption of the mCherry chromophore at 586 nm remains unchanged, but the shoulder near 550 nm increases in intensity relative to the peak, suggesting a change in absorbance in the solid state. This change is reversed when the materials are rehydrated. The diminishing of the absorbance peak at 586 nm and the enhancement of the shoulder at 510 nm and the peak at 390 nm in both solid-state and rehydrated samples indicates that the chromophore for some fraction of the material has been disrupted. A peak at 386 nm has been observed previously in similar red fluorescent proteins and has been attributed to the addition of water across the acylimine chromophore bond.52
Figure 6.

a) Solid state UV-vis spectra of mCherry-PNIPAM conjugate normalized by A280 b) Rehydrated solid state UV-vis spectra of mCherry-PNIPAM conjugate normalized by A280 showing retention of the characteristic absorbance peak shape of mCherry at 586 nm c) Rehydrated solid-state circular dichroism spectra of conjugate showing protein fold is not disturbed by self-assembly
Because quantum overlap in the solid state may influence the spectrum, a quantitative measure of the functionality of the materials was performed by measuring their absorbance spectrum upon rehydration in milli-Q water. Compared to the UV-vis data for as-synthesized protein-polymer conjugates in solution (Supporting Information), the rehydrated samples show a decrease in the ratio of A586:A280 from approximately 1.3 to 0.5. This indicates that for all four samples 30–40% of the protein remains active when compared to the conjugate in solution. Consistent with preservation of approximately 35% of the protein’s optical activity, the bulk material appears to have a deep red color (Supporting Information). Rehydration also results in a decrease in the peak absorbance at 390 nm to some level intermediate between the as-synthesized and the solid-state materials. This suggests the presence of three types of mCherry chromophore in the solid state: active, irreversibly inactive, and spectrally shifted. The active fraction is responsible for the major absorbance peak at 586 nm. The peak at 390 nm increases in the solid-state material then decreases upon rehydration, suggesting that a significant contribution to this peak comes from the spectrally shifted material. The shoulder at 510 nm also increases upon rehydration, suggesting that there is a fraction of irreversibly inactivated chromophore that absorbs at this wavelength only after rehydration. Some of the irreversibly inactivated chromophore may also have no absorption within the visible region.
In the dehydrated samples, it is observed that the extent of processing correlates with a loss of optical function in the protein. The relatively rapid casting process at 40 °C tends to promote a higher fraction of active chromophore compared to other samples by about 10%. Because the sample cast at 40 °C was never exposed to high vacuum, it is likely that it contains residual water that enhances the stability of the protein through hydrogen bonding. The room temperature cast sample has the next highest fraction of active chromophore, followed by the two solvent annealed samples which show a further decrease in absorbance at 586 nm and an increase in absorbance at 390 nm. By comparison, a control sample of lyophilized mCherry retains less than 15% of its optical activity at 586 nm, worse than any of the self-assembled materials. Upon rehydration, the relative order of peak intensities in the solid-state and rehydrated spectra changes. While all samples show an increase in the absorbance at 586 nm after rehydration, the sample solvent annealed at 4 °C shows the largest increase, indicating that it contains the largest fraction of reversibly inactive material.
Circular dichroism spectra of the rehydrated conjugates show very little change from the conjugate before self-assembly. This indicates that the majority of the protein retains identical secondary structure as the native protein, even after having been dehydrated and rehydrated. Quantitative analysis of the spectra showed that the mCherry alone was composed of 50% β-sheet, as compared to a theoretically predicted 57% β-sheet content from the crystal structure. The discrepancy between the measurement and prediction is accounted for by the presence of the 6xHis tag region in the experimental protein but not in the crystal structure. The mCherry-PNIPAM block copolymer in solution after purification was 43% β-sheet, and the rehydrated samples contained 41–48% β-sheet, indicating no loss of secondary structure upon PNIPAM conjugation within the resolution of the measurement. Taken together, the circular dichroism and UV-vis data illustrate that it is possible to maintain a substantial degree of globular protein fold and function in a solid-state self-assembled block copolymer. Because the fold is maintained to a much higher degree than the chromophore activity, it is likely that the activity of the sensitive chromophore is lost during self-assembly without large changes in the protein structure.
The packing and crystallinity of the mCherry within the block copolymer nanodomains was investigated using wide-angle X-ray scattering (WAXS), shown in Figure 7. The scattering patterns of self-assembled conjugates contain the same two peaks as a bulk sample composed of solid mCherry prepared by evaporation from solution. This demonstrates that the WAXS peaks seen for the block copolymer samples are a result of the mCherry structure and not affected by the PNIPAM. No peaks are present corresponding to the mCherry crystal structure, indicating that the mCherry is in an amorphous state. This is consistent with both kinetic limitations to crystallization during the self-assembly process, which occurs on a timescale of ~ 2 hrs, and with UV-vis results that suggest a portion of the protein may be structurally perturbed in the solid state and therefore unable to crystallize within the mCherry unit cell.
Figure 7.

Wide-angle x-ray scattering data of as-cast and solvent annealed mCherry-PNIPAM block copolymers shows scattering due to the inter-sheet and inter-strand spacing of β-strands. The spacing of the β strands of mCherry is not altered in conjugate materials.
WAXS data for the mCherry protein indicate that the material retains a predominantly β-barrel structure in the solid state. All of the block copolymers contain two peaks, one at approximately 6.23 nm−1 and the other at 14.32 nm−1 corresponding to 1.0 and 0.45 nm, respectively, in real space. The larger domain spacing peak is attributed to either inter-sheet spacing or inter-helix spacing for proteins containing β-sheets or α-helices, respectively.62 The smaller domain spacing peak arises from inter-strand hydrogen bonding in β-sheet proteins, or backbone hydrogen bonding in α-helices.62 Given that circular dichroism and the known crystal structure of mCherry suggest a predominantly β-sheet secondary structure, these peaks are inferred to result from inter-sheet spacing and inter-strand hydrogen bonding between β-strands. Because the q values of these peaks do not change between samples, it is concluded that the inter-sheet and inter-strand spacing is not changed significantly by the processing method used to induce self-assembly. The block copolymer samples all show similar intensities for both peaks with the ratio of the 6.23 nm−1 to the 14.32 nm−1 peak equal to approximately 0.98. In contrast, mCherry shows a higher intensity for the second peak with a ratio of 0.84. An increase in this ratio has previously been correlated with a decrease in β-sheet content,62 suggesting that the β-sheet content is slightly lower in the self-assembled block copolymers than in the bulk mCherry. This result is consistent with the minor decrease in β-sheet content upon bioconjugation observed by CD.
Conclusions
Protein-polymer diblock copolymers composed of mCherryS131C and PNIPAM were synthesized and self-assembled into nanostructured materials, demonstrating an attractive route towards high density three-dimensional protein nanopatterning with precise control over protein orientation and placement. Self-assembly was induced by solvent evaporation, and the selectivity of the solvent during the evaporation process was shown to have a large effect on the nanostructure formed, resulting in a heterogeneous nanodomain structure for a protein-selective solvent and a hexagonally perforated lamellar phase for a nonselective solvent. Subsequent solvent annealing resulted in an evolution towards well-ordered lamellar structures, suggesting that this lamellar structure may be closer to thermodynamic equilibrium. After annealing at room temperature, SAXS indicated a domain spacing for the material of 24.0 nm, suggesting that the mCherry packs in bilayers within the lamellae. The mCherry structure within the lamellar domains was largely amorphous, with the only observed WAXS peaks assigned to inter-sheet and inter-strand spacing of β-strands. While circular dichroism indicated no irreversible change in protein secondary structure after self-assembly, UV-vis spectroscopy showed that one third of the protein chromophores remained active in the solid-state material.
Methods
Synthesis of 2-ethylsulfanylthiocarbonylsulfanyl-2-methyl propionic acid (EMP)
This procedure was adapted from the work of You and Oupický.63 First, ethanethiol (7.21 mL, 0.1 mol), acetone (73 mL), and tricaprylylmethylammonium chloride (1.0 g, 2.5 mmol) were combined and the mixture was cooled on ice under nitrogen. Next, 9 mL of 50% (w/v) of sodium hydroxide was added dropwise. After an additional 20 minutes, carbon disulfide (6.03 mL, 0.1 mol) and acetone (12.6 mL) were combined and added dropwise. Chloroform (12 mL, 0.15 mol) was added, followed by the addition of 80 mL of 50% (w/v) sodium hydroxide over 10 minutes. The yellow-orange mixture was stirred overnight. Water (200 mL) was added, followed by concentrated hydrochloric acid (80 mL) to drop the pH below 1. The mixture was extracted three times into diethyl ether and concentrated to a dark red oil. Crude product was purified via silica gel chromatography (1:1 hexanes:ether) and then distilled to yield 11.0 g of a bright orange, viscous liquid (49% yield). 1H NMR (CDCl3, δ): 1.33 (t, 3H, -CH2CH3), 1.72 (s, 6H, -C(CH3)2COOH), 3.30 (q, 2H, -CH2CH3).
Synthesis of exo-3a,4,7,7a-tetrahydro-2-(3-hydroxypropyl)-4,7-epoxy-14-isoindole-1,3(2H)-dione (1)
Following the work of Neubert and Snider,64 3-amino-1-propanol (4.08 g, 54.2 mmol) was added dropwise to a solution of exo-3,6-epoxy-1,2,3,6-tetrahydrophthalic anhydride (9.0 g, 54.2 mmol) in 500 mL methanol. The reaction was stirred at 56 °C for 3 days, after which the solvent was removed by rotary evaporation to give a clear yellow oil. 100 mL dichloromethane was added and washed three times with 100 mL brine. The organic fraction was dried over sodium sulfate and the solvent removed under reduced pressure to give 2.71 g of a white solid (22% yield). 1H NMR (CDCl3, δ): 1.75 (tt, 2H, -CH2(CH2)CH2-), 2.88 (s, 2H, -NC(O)CH-), 3.52 (t, 2H, -NCH2-), 3.65 (t, 2H, -CH2O-), 5.27 (s, 2H, -CH(O)-), 6.52 (s, 2H, -CHCH-).
Synthesis of RAFT agent
Functional RAFT agent was prepared by carbodiimide coupling.54 EMP (1.88 g, 8.40 mmol), 1 (1.50 g, 6.72 mmol), 4-dimethylaminopyridine (103.8 mg, 0.84 mmol), and N,N′-dicyclohexylcarbodiimide (3.46 g, 16.8 mmol) were combined in 58 mL dry tetrahydrofuran and stirred under nitrogen at room temperature overnight. The reaction mixture was filtered and concentrated, and the product was purified by silica gel chromatography (1:1 hexanes:ethyl acetate) to yield 1.23 g of a bright yellow solid (34% yield). 1H NMR (CDCl3, δ): 1.29 (t, 3H, -S-CH2CH3), 1.69 (s, 6H, -C(CH3)2-), 1.85–2.00 (tt, 2H, -CH2(CH2)CH2-), 2.83 (s, 2H, -NC(O)CH-), 3.26 (q, 2H, -CH2CH3), 3.55 (t, 2H, -NCH2-), 4.05 (t, 2H, -CH2O-), 5.24 (s, 2H, -CH(O)-), 6.49 (s, 2H, -CHCH-).
Polymerization
The RAFT agent and azobisisobutyronitrile (AIBN) (recrystallized twice from methanol) were added to a 2.0 M solution of NIPAM (sublimated) in acetonitrile in the ratio 600:1:0.2 (monomer:RAFT agent:initiator). The solution was degassed by three freeze-pump-thaw cycles. The polymerization was carried out in a sealed flask at 65 °C and terminated after 75 minutes by removal of heat and exposure to oxygen. The polymer was then precipitated in cold diethyl ether and dried under vacuum. The maleimide was deprotected by heating to 120 °C under vacuum for 2 hours. The molecular weight and polydispersity were determined by gel permeation chromatography using a Waters Breeze 1525 HPLC system with a series 2414 refractive index detector, calibrated with poly(methyl methacrylate) standards, and N,N-dimethylformamide with 0.01 M LiBr as the mobile phase.
Cloning and Protein Expression
The gene for mCherry, optimized for prokaryotic codon usage, was subcloned into the pQE9 vector (Qiagen) which encodes for an N-terminal His tag. Site directed mutagenesis was used to create the mutant mCherryS131C by replacing a serine with a cysteine at residue 131, located in a loop region on the end of the β-barrel opposite the N and C termini. The protein was expressed in the E. coli strain SG13009 containing the pREP4 repressor plasmid, grown in Terrific Broth at 37 °C, and induced with 1 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) at OD600 = 1. The cells were cultured for 4.5 hours after induction and were then harvested. The cell were resuspended in lysis buffer (50 mM NaH2PO4, 300 mM sodium chloride, 10 mM imidazole, 10 mM β-mercaptoethanol (BME), pH 8.0), incubated with 1 mg/mL lysozyme at 4 °C for 30 minutes and sonicated. The lysate was clarified, and the protein was purified using Ni-NTA metal affinity chromatography. Throughout the purification, 10 mM BME was used in all buffers. Elution fractions containing purified protein were dialyzed into 20 mM Tris buffer, pH = 8. The yield in the elution fractions was determined spectrophotometrically using the absorbance peak at 586 nm (extinction coefficient of 72,000 M−1cm−1).49, 65
The purity of the protein was confirmed by denaturing gel electrophoresis (SDS-PAGE), native state gel electrophoresis, and matrix assisted laser desorption ionization mass spectrometry (MALDI-MS). SDS-PAGE (Supporting Information) shows three bands near 28 kg/mol, 19 kg/mol and 9 kg/mol due to partial cleavage of the protein during sample boiling before loading the gel.51 The highest molecular weight band corresponds to the expected molar mass of the whole mCherry molecule, and the sum of the molar masses of the two lower mass bands also corresponds to the mass of the whole protein, consistent with hydrolysis of the sensitive acylimine bond within the mCherry chromophore.50 The native gel (Supporting Information), run at a high protein concentration, shows only a single prominent protein band corresponding to the mCherry and a higher molecular weight band corresponding to dimerization through the formation of a disulfide bond. Consistent with previously reported expressions of mCherry,53, 66 it is noted that the lower molar mass fragments are not observed by SDS-PAGE in more dilute samples. The observation of a single dominant band in the native gel indicates that cleavage occurs only during SDS-PAGE sample preparation. The purity of the protein was further confirmed by MALDI-TOF mass spectrometry; the measured molar mass of the protein was 28,201 g/mol compared to the expected mass of 28,134.48 g/mol. No peak was observed for the higher molar mass hydrolysis product (18,896 g/mol calculated molar mass), although a small peak was observed for the low molecular weight fragment (9,256 g/mol calculated molar mass), consistent with the preferential observation of low molecular weight species by MALDI.
Bioconjugation
The coupling reaction between mCherryS131C and maleimide end-functionalized PNIPAM was performed in 20 mM Tris buffer, pH 8.0. Tris(2-carboxyethyl)phosphine (TCEP) (0.0185g, 0.0646 mmol) was added to the mCherryS131C solution (0.173g, 0.00646 mmol, 1.8 mg/mL) one hour prior to PNIPAM addition to reduce all thiol groups. Maleimide functionalized PNIPAM (2.61201g, 0.0451 mmol) was added to the solution and allowed to react overnight. The conjugate was purified by precipitation in 1.0 M ammonium sulfate solution followed by centrifugation at room temperature. The pellet containing conjugate and unreacted PNIPAM homopolymer was then resuspended in 20 mM Tris buffer (pH 8) and precipitated a second time. The excess PNIPAM was removed from the second pellet using Ni-NTA affinity chromatography to yield purified conjugate. After collecting the flowthrough, the column was washed with approximately 7 column volumes of wash buffer before elution to completely remove unconjugated PNIPAM. The purified conjugate was dialyzed into pure water. Purity was analyzed using SDS-PAGE and the yield was determined spectrophotometrically.
Sample Preparation and Characterization
Conjugate solution was concentrated to approximately 44 mg/mL conjugate using Millipore Ultra-15 centrifugal filters with a molecular weight cutoff of 3 kDa. Bulk samples were prepared by evaporation of water either at 40 °C under ambient pressure or at room temperature under vacuum. Solvent annealing was performed at 4 °C or room temperature in sealed jars using nanopure water as the solvent. UV-vis spectra were collected at ambient temperature on a Cary 50 UV-vis spectrophotometer using a quartz cuvette and normalized to the absorbance at 280 nm. CD spectra were obtained using an Aviv Model 202 circular dichroism spectrometer operating at 25 °C and converted into molar ellipticity by correcting for the water background and using the concentration determined by the A280. CDPro was used to analyze spectra to determine the secondary structure content using CONTINLL, SELCON3 and CDSSTR methods. Disk-shaped samples for x-ray scattering were cast on kapton tape using 7 mm diameter washers as a mold. SAXS and WAXS data were collected using a Molecular Metrology ASSY 610-004378 system, and corrected for empty cell and dark field scattering. Bulk samples were cryo-microtomed using a Leica EM UC6 at −100 °C to a thickness of 50–60 nm for TEM analysis. Samples were stained with ruthenium tetroxide vapors from a 0.5% aqueous solution for 20–40 minutes. Due to the greater number of alcohol, amine and aromatic functional groups on the protein compared with the polymer, the protein domains were selectively stained and appear dark in images. A JEOL 2000FX Transmission Electron Microscope was used to obtain bright field images using an accelerating voltage of 120 kV and a LaB6 filament. Images were captured using an ORCA camera in a fixed bottom mount configuration.
Supplementary Material
Acknowledgments
This work was supported by the MIT Energy Initiative (award number 015728-066). GPC, SAXS, WAXS, microtome, and TEM experiments were performed at the Institute for Soldier Nanotechnology. NMR experiments were performed at the MIT Department of Chemistry Instrumentation Facility, and CD experiments were performed at the Biophysical Instrumentation Facility for the Study of Complex Macromolecular Systems (NSF-0070319 and NIH GM68762).
Footnotes
Supporting Information Available: Protein sequence for mCherryS131C, native and denaturing gels for protein purification, UV-vis and circular dichroism spectra of mCherryS131C and the mCherry-PNIPAM conjugate, cloud point data, a photograph of the solid-state nanostructured plastic, analysis of CD data to quantitatively determine protein secondary structure, and the method for estimating PNIPAM volume fraction in the block copolymer. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Willner I, Katz E. Integration of Layered Redox Proteins and Conductive Supports for Bioelectronic Applications. Angew Chem, Int Ed. 2000;39:1180–1218. doi: 10.1002/(sici)1521-3773(20000403)39:7<1180::aid-anie1180>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 2.LaVan DA, Cha JN. Approaches for Biological and Biomimetic Energy Conversion. Proc Natl Acad Sci US A. 2006;103:5251–5255. doi: 10.1073/pnas.0506694103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benkovic SJ, Hammes-Schiffer S. A Perspective on Enzyme Catalysis. Science. 2003;301:1196–1202. doi: 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
- 4.Rahman MM, Ahammad AJS, Jin JH, Ahn SJ, Lee JJ. A Comprehensive Review of Glucose Biosensors Based on Nanostructured Metal-Oxides. Sensors. 2010;10:4855–4886. doi: 10.3390/s100504855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rapp BE, Gruhl FJ, Länge K. Biosensors with Label-Free Detection Designed for Diagnostic Applications. Anal Bioanal Chem. 2010;398:2403–2412. doi: 10.1007/s00216-010-3906-2. [DOI] [PubMed] [Google Scholar]
- 6.Cracknell JA, Vincent KA, Armstrong FA. Enzymes as Working or Inspirational Electrocatalysts for Fuel Cells and Electrolysis. Chem Rev. 2008;108:2439–2461. doi: 10.1021/cr0680639. [DOI] [PubMed] [Google Scholar]
- 7.Yu EH, Scott K. Enzymatic Biofuel Cells-Fabrication of Enzyme Electrodes. Energies. 2010;3:23–42. [Google Scholar]
- 8.Minteer SD, Liaw BY, Cooney MJ. Enzyme-Based Biofuel Cells. Curr Opin Biotechnol. 2007;18:228–234. doi: 10.1016/j.copbio.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 9.Patel RN. Synthesis of Chiral Pharmaceutical Intermediates by Biocatalysis. Coord Chem Rev. 2008;252:659–701. [Google Scholar]
- 10.Favre N, Christ ML, Pierre AC. Biocatalytic Capture of CO2 with Carbonic Anhydrase and its Transformation to Solid Carbonate. J Mol Catal B: Enzym. 2009;60:163–170. [Google Scholar]
- 11.Reda T, Plugge CM, Abram NJ, Hirst J. Reversible Interconversion of Carbon Dioxide and Formate by an Electroactive Enzyme. Proc Natl Acad Sci U S A. 2008;105:10654–10658. doi: 10.1073/pnas.0801290105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parkinson BA, Weaver PF. Photoelectrochemical Pumping of Enzymatic CO2 Reduction. Nature. 1984;309:148–149. [Google Scholar]
- 13.Kuwabata S, Tsuda R, Yoneyama H. Electrochemical Conversion of Carbon Dioxide to Methanol with the Assistance of Formate Dehydrogenase and Methanol Dehydrogenase as Biocatalysts. J Am Chem Soc. 1994;116:5437–5443. [Google Scholar]
- 14.Karyakin AA, Morozov SV, Karyakina EE, Varfolomeyev SD, Zorin NA, Cosnier S. Hydrogen Fuel Electrode based on Bioelectrocatalysis by the Enzyme Hydrogenase. Electrochem Commun. 2002;4:417–420. [Google Scholar]
- 15.Hambourger M, Gervaldo M, Svedruzic D, King PW, Gust D, Ghirardi M, Moore AL, Moore TA. [FeFe]-Hydrogenase-Catalyzed H2 Production in a Photoelectrochemical Biofuel Cell. J Am Chem Soc. 2008;130:2015–2022. doi: 10.1021/ja077691k. [DOI] [PubMed] [Google Scholar]
- 16.Krassen H, Schwarze A, Friedrich B, Ataka K, Lenz O, Heberle J. Photosynthetic Hydrogen Production by a Hybrid Complex of Photosystem I and [NiFe]-Hydrogenase. ACS Nano. 2009;3:4055–4061. doi: 10.1021/nn900748j. [DOI] [PubMed] [Google Scholar]
- 17.Cogdell RJ, Gall A, Köhler J. The Architecture and Function of the Light-Harvesting Apparatus of Purple Bacteria: From Single Molecules to in vivo Membranes. Q Rev Biophys. 2006;39:227–324. doi: 10.1017/S0033583506004434. [DOI] [PubMed] [Google Scholar]
- 18.Das R, Kiley PJ, Segal M, Norville J, Yu AA, Wang LY, Trammell SA, Reddick LE, Kumar R, Stellacci F, et al. Integration of Photosynthetic Protein Molecular Complexes in Solid-State Electronic Devices. Nano Lett. 2004;4:1079–1083. [Google Scholar]
- 19.Choi JW, Fujihira M. Molecular-Scale Biophotodiode Consisting of a Green Fluorescent Protein/cytochrome c Self-Assembled Heterolayer. Appl Phys Lett. 2004;84:2187–2189. [Google Scholar]
- 20.Minteer SD. Enzyme Stabilization and Immobilization. Springer; New York: 2010. [Google Scholar]
- 21.Mateo C, Palomo JM, Fernandez-Lorente G, Guisan JM, Fernandez-Lafuente R. Improvement of Enzyme Activity, Stability and Selectivity via Immobilization Techniques. Enzyme Microb Technol. 2007;40:1451–1463. [Google Scholar]
- 22.Caruso F, Schüler C. Enzyme Multilayers on Colloid Particles: Assembly, Stability, and Enzymatic Activity. Langmuir. 2000;16:9595–9603. [Google Scholar]
- 23.Wang Y, Angelatos AS, Caruso F. Template Synthesis of Nanostructured Materials via Layer-by-Layer Assembly. Chem Mater. 2007;20:848–858. [Google Scholar]
- 24.Christman KL, Enriquez-Rios VD, Maynard HD. Nanopatterning Proteins and Peptides. Soft Matter. 2006;2:928–939. doi: 10.1039/b611000b. [DOI] [PubMed] [Google Scholar]
- 25.Kwak SK, Lee GS, Ahn DJ, Choi JW. Pattern Formation of cytochrome c by Microcontact Printing and Dip-Pen Nanolithography. Mater Sci Eng C-Biomimetic Supramol Syst. 2004;24:151–155. [Google Scholar]
- 26.Alang Ahmad SA, Wong LS, ul-Haq E, Hobbs JK, Leggett GJ, Micklefield J. Protein Micro- and Nanopatterning Using Aminosilanes with Protein-Resistant Photolabile Protecting Groups. J Am Chem Soc. 2011;133:2749–2759. doi: 10.1021/ja1103662. [DOI] [PubMed] [Google Scholar]
- 27.Kalia J, Raines RT. Advances in Bioconjugation. Curr Org Chem. 2010;14:138–147. doi: 10.2174/138527210790069839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim DC, Sohn JI, Zhou DJ, Duke TAJ, Kang DJ. Controlled Assembly for Well-Defined 3D Bioarchitecture Using Two Active Enzymes. ACS Nano. 2010;4:1580–1586. doi: 10.1021/nn900610u. [DOI] [PubMed] [Google Scholar]
- 29.Kim KH, Kim JD, Kim YJ, Kang SH, Jung SY, Jung H. Protein Immobilization Without Purification via Dip-Pen Nanolithography. Small. 2008;4:1089–1094. doi: 10.1002/smll.200700519. [DOI] [PubMed] [Google Scholar]
- 30.Presley AD, Chang JJ, Xu T. Directed Co-Assembly of Heme Proteins with Amphiphilic Block Copolymers Toward Functional Biomolecular Materials. Soft Matter. 2011;7:172–179. [Google Scholar]
- 31.Cardoso MB, Smolensky D, Heller WT, Hong K, O’Neill H. Supramolecular Assembly of Biohybrid Photoconversion Systems. Energy Environ Sci. 2011;4:181–188. [Google Scholar]
- 32.Laible PD, Kelley RF, Wasielewski MR, Firestone MA. Electron-Transfer Dynamics of Photosynthetic Reaction Centers in Thermoresponsive Soft Materials. J Phys Chem B. 2005;109:23679–23686. doi: 10.1021/jp053884n. [DOI] [PubMed] [Google Scholar]
- 33.Bates FS, Fredrickson GH. Block Copolymers - Designer Soft Materials. Phys Today. 1999;52:32–38. [Google Scholar]
- 34.Hamley IW. The Physics of Block Copolymers. Oxford University Press; New York: 1998. [Google Scholar]
- 35.Park C, Yoon J, Thomas EL. Enabling Nanotechnology with Self Assembled Block Copolymer Patterns. Polymer. 2003;44:6725–6760. [Google Scholar]
- 36.Gomez ED, Panday A, Feng EH, Chen V, Stone GM, Minor AM, Kisielowski C, Downing KH, Borodin O, Smith GD, et al. Effect of Ion Distribution on Conductivity of Block Copolymer Electrolytes. Nano Lett. 2009;9:1212–1216. doi: 10.1021/nl900091n. [DOI] [PubMed] [Google Scholar]
- 37.Yang X, Loos J. Toward High-Performance Polymer Solar Cells: The Importance of Morphology Control. Macromolecules. 2007;40:1353–1362. [Google Scholar]
- 38.Olsen BD, Segalman RA. Self-Assembly of Rod-Coil Block Copolymers. Mater Sci Eng R-Rep. 2008;62:37–66. [Google Scholar]
- 39.Canalle LA, Lowik DWPM, van Hest JCM. Polypeptide-Polymer Bioconjugates. Chem Soc Rev. 2010;39:329–353. doi: 10.1039/b807871h. [DOI] [PubMed] [Google Scholar]
- 40.Krishna OD, Kiick KL. Protein- and Peptide-Modified Synthetic Polymeric Biomaterials. Pept Sci. 2010;94:32–48. doi: 10.1002/bip.21333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rabotyagova OS, Cebe P, Kaplan DL. Protein-Based Block Copolymers. Biomacromolecules. 2011;12:269–289. doi: 10.1021/bm100928x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lavigueur C, García JG, Hendriks L, Hoogenboom R, Cornelissen JJLM, Nolte RJM. Thermoresponsive Giant Biohybrid Amphiphiles. Polym Chem. 2011;2:333–340. [Google Scholar]
- 43.Duncan R. Polymer Conjugates as Anticancer Nanomedicines. Nat Rev Cancer. 2006;6:688–701. doi: 10.1038/nrc1958. [DOI] [PubMed] [Google Scholar]
- 44.Gil ES, Hudson SM. Stimuli-Reponsive Polymers and their Bioconjugates. Prog Polym Sci. 2004;29:1173–1222. [Google Scholar]
- 45.Reynhout IC, Cornelissen JLM, Nolte RJM. Self-Assembled Architectures from Biohybrid Triblock Copolymers. J Am Chem Soc. 2007;129:2327–2332. doi: 10.1021/ja066790f. [DOI] [PubMed] [Google Scholar]
- 46.Harris JM, Chess RB. Effect of Pegylation on Pharmaceuticals. Nat Rev Drug Discovery. 2003;2:214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 47.Kim MY, Kwon JS, Kim HJ, Lee EK. In vitro Refolding of PEGylated Lipase. J Biotechnol. 2007;131:177–179. doi: 10.1016/j.jbiotec.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 48.Veronese FM. Peptide and Protein PEGylation: A Review of Problems and Solutions. Biomaterials. 2001;22:405–417. doi: 10.1016/s0142-9612(00)00193-9. [DOI] [PubMed] [Google Scholar]
- 49.Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, Tsien RY. Improved Monomeric Red, Orange and Yellow Fluorescent Proteins Derived from Discosoma sp. Red Fluorescent Protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 50.Shu XK, Shaner NC, Yarbrough CA, Tsien RY, Remington SJ. Novel Chromophores and Buried Charges Control Color in mFruits. Biochemistry. 2006;45:9639–9647. doi: 10.1021/bi060773l. [DOI] [PubMed] [Google Scholar]
- 51.Gross LA, Baird GS, Hoffman RC, Baldridge KK, Tsien RY. The Structure of the Chromophore within DsRed, a Red Fluorescent Protein from Coral. Proc Natl Acad Sci U S A. 2000;97:11990–11995. doi: 10.1073/pnas.97.22.11990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pakhomov AA, Pletneva NV, Balashova TA, Martynov VI. Structure and Reactivity of the Chromophore of a GFP-like Chromoprotein from Condylactis gigantea. Biochemistry. 2006;45:7256–7264. doi: 10.1021/bi060207q. [DOI] [PubMed] [Google Scholar]
- 53.Chung JA, Wollack JW, Hovlid ML, Okesli A, Chen Y, Mueller JD, Distefano MD, Taton TA. Purification of Prenylated Proteins by Affinity Chromatography on Cyclodextrin-Modified Agarose. Anal Biochem. 2009;386:1–8. doi: 10.1016/j.ab.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bays E, Tao L, Chang CW, Maynard HD. Synthesis of Semitelechelic Maleimide Poly(PEGA) for Protein Conjugation By RAFT Polymerization. Biomacromolecules. 2009;10:1777–1781. doi: 10.1021/bm9001987. [DOI] [PubMed] [Google Scholar]
- 55.Lapiene V, Kukolka F, Kiko K, Arndt A, Niemeyer CM. Conjugation of Fluorescent Proteins with DNA Oligonucleotides. Bioconjugate Chem. 2010;21:921–927. doi: 10.1021/bc900471q. [DOI] [PubMed] [Google Scholar]
- 56.Kukolka F, Niemeyer CM. Synthesis of Fluorescent Oligonucleotide-EYFP Conjugate: Towards Supramolecular Construction of Semisynthetic Biomolecular Antennae. Org Biomol Chem. 2004;2:2203–2206. doi: 10.1039/b406492e. [DOI] [PubMed] [Google Scholar]
- 57.Sawyer LC, Grubb DT, Meyers GF. Polymer Microscopy. Springer; New York: 2008. [Google Scholar]
- 58.Lai C, Loo YL, Register RA, Adamson DH. Dynamics of a Thermoreversible Transition between Cylindrical and Hexagonally Perforated Lamellar Mesophases. Macromolecules. 2005;38:7098–7104. [Google Scholar]
- 59.Khandpur AK, Forster S, Bates FS, Hamley IW, Ryan AJ, Bras W, Almdal K, Mortensen K. Polyisoprene-Polystyrene Diblock Copolymer Phase Diagram Near the Order-Disorder Transition. Macromolecules. 1995;28:8796–8806. [Google Scholar]
- 60.Loo YL, Register RA, Adamson DH, Ryan AJ. A Highly Regular Hexagonally Perforated Lamellar Structure in a Quiescent Diblock Copolymer. Macromolecules. 2005;38:4947–4949. [Google Scholar]
- 61.Olsen BD, Segalman RA. Nonlamellar Phases in Asymmetric Rod-Coil Block Copolymers at Increased Segregation Strengths. Macromolecules. 2007;40:6922–6929. [Google Scholar]
- 62.Elshemey WM, Elfiky AA, Gawad WA. Correlation to Protein Conformation of Wide-Angle X-ray Scatter Parameters. Protein J. 2010;29:545–550. doi: 10.1007/s10930-010-9291-z. [DOI] [PubMed] [Google Scholar]
- 63.You YZ, Oupický D. Synthesis of Temperature-Responsive Heterobifunctional Block Copolymers of Poly(ethylene glycol) and Poly(N-isopropylacrylamide) Biomacromolecules. 2007;8:98–105. doi: 10.1021/bm060635b. [DOI] [PubMed] [Google Scholar]
- 64.Neubert BJ, Snider BB. Synthesis of (±)-Phloeodictine A1. Org Lett. 2003;5:765–768. doi: 10.1021/ol034042e. [DOI] [PubMed] [Google Scholar]
- 65.Dennis AM, Bao G. Quantum Dot-Fluorescent Protein Pairs as Novel Fluorescence Resonance Energy Transfer Probes. Nano Lett. 2008;8:1439–1445. doi: 10.1021/nl080358+. [DOI] [PubMed] [Google Scholar]
- 66.Lee BC, Zuckermann RN. Templated Display of Biomolecules and Inorganic Nanoparticles by Metal Ion-Induced Peptide Nanofibers. Chem Commun. 2010;46:1634–1636. doi: 10.1039/b925395e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.