

Abstract
Due to their specialized location, stem and progenitor cells are often exposed to oxidative stress. Although ATP-binding cassette transporter subfamily G member 2 (Abcg2)-expressing cells have been implicated in cardiac protective mechanisms involving oxidative stress, there remains a lack of understanding regarding the behavior of cardiac Abcg2-expressing cells when exposed to ROS. The aim of the present study was to characterize the response of the cardiac Abcg2 lineage to oxidative stress. In vitro analysis demonstrated that the antioxidant program regulated by Abcg2 is dependent on a functional transporter. Delivery of paraquat dichloride (PQ), a systemic oxidative stress-inducing agent, to mice confirmed that Abcg2 provides a survival benefit. When exposed to PQ, reporter mice showed an increase in the Abcg2 lineage. Transcriptional and immunohistochemical analysis of Abcg2 lineage-positive cells revealed an enhanced vascular commitment after stress. Finally, preconditioning with PQ demonstrated a reduction in scar size and an increase in angiogenesis after permanent left coronary artery ligation. In conclusion, the data suggest that Abcg2 plays a cytoprotective role in response to in vivo oxidative stress. The contribution of the Abcg2 lineage to the vasculature in the heart is increased after PQ delivery.
Keywords: ATP-binding cassette transporter subfamily G member 2, oxidative stress, cardioprotection, lineage tracing, preconditioning, cardiac vasculature
atp-binding cassette transporter subfamily G member 2 (Abcg2) is an energy-dependent transport protein located in the plasma membrane and is responsible for the efflux of a variety of substrates. Originally described in a drug-resistant human breast cancer cell line (9), it has been identified in numerous mammalian tissues and used as a stem cell marker (15, 17, 23, 30, 37, 39). Side population (SP) cells, which exhibit Hoechst DNA-binding dye efflux capability primarily due to high Abcg2 expression, demonstrate clonality and multipotency when isolated from various adult tissue types (23, 24, 30, 37, 39). We (25) have previously demonstrated that cardiac Abcg2 expression as well as resident cardiac SP cell numbers increase after myocardial injury, highlighting a role in response to tissue damage. A number of previous studies (18, 23, 30, 39) have also implicated SP cells as a suitable candidate for tissue renewal. However, the reported contribution toward cardiac regeneration has been minimal, and a significant functional improvement has not been recognized.
Due to its quiescent nature and heterogeneous character (19, 24, 36), following the SP in vivo has been difficult, resulting in an incomplete understanding of cell function. To more efficiently target the SP, specific markers have necessitated identification. We (24) have previously established that Abcg2 is the molecular determinant for stem cell character in adult cardiac SP cells. Recent Abcg2-specific analysis in vivo has helped to illuminate the functional role of Abcg2 beyond the xenobiotic transport for which it was originally discovered. Studies using Abcg2-deficient mice have allowed organ-specific regenerative analysis. In response to cardiac injury, Abcg2-null mice suffer increased mortality as well as decreased cardiac function, associating Abcg2 with angiogenesis, macrophage recruitment, and redox regulation (18, 19). In skeletal muscle, tissue regeneration is obstructed after injury, with a small level of Abcg2 contribution toward novel myofibers (10). Despite the importance of these findings, there remains a lack of understanding regarding the fate of Abcg2-expressing cells in the heart.
ROS are powerful agents of oxidative stress that originate from multiple sources, including extensive tissue injury, such as myocardial infarction (11). The relationship between Abcg2 and oxidative stress in vitro has been previously described (17–19, 23, 30, 37, 39). We (25) have previously shown that Abcg2 is a direct downstream target of hypoxia-inducible factor (HIF)-2α, the master regulator of oxidative stress, suggesting that Abcg2 is activated by elevated ROS levels. When exposed to oxidant culture conditions, Abcg2-overexpressing cells demonstrated a survival advantage compared with non-Abcg2-expressing cells. Although the protective effects against high-level ROS have been alluded to in a small number of animal injury models (10, 18, 19, 23, 30), the nature of injuries used included an extensive degree of adverse cellular events such as ischemia, inflammation, and apoptosis. To date, an exclusively oxidant stress-specific in vivo injury model has not been used. We therefore sought to characterize the Abcg2 response to oxidative stress by using paraquat dichloride (PQ), a well-known herbicide that generates excessive superoxide radicals in the cytosol and thus creates an imbalance in redox homeostasis (34). Taking advantage of a recently established tamoxifen-inducible Abcg2 lineage-tracing mouse model (14), we used multiple techniques to determine the fate of cardiac Abcg2 lineage after PQ challenge. Furthermore, we showed that a protective antioxidant transcriptional program is dependent on functional Abcg2 and that preconditioning with PQ attenuates ventricular remodeling and increases vascular density after myocardial infarction. Characterizing the response to oxidative stress allows for further insights into cardiac Abcg2 biology.
METHODS
Experimental animals.
All experiments were performed in accordance with the Guide for the Care and Use Laboratory Animals, and the experimental protocol was submitted to and approved by the Institutional Animal Care and Use Committee of the University of Minnesota. All experiments used adult mice aged 2–6 mo. Abcg2-null and Abcg2CreERT2 mice were obtained from Dr. Brian P. Sorrentino (St. Jude Children's Research Hospital, Memphis, TN). C57Bl/6J wild-type (000664), RosaLacZ (003474), and RosaCAGZsGreen (007906) mice were purchased from The Jackson Laboratories (Bar Harbor, ME).
Abcg2 overexpression in C2C12 cells.
C2C12 myoblast cells were transfected with empty IRES-GFP plasmid (PSAM control), wild-type Abcg2-IRES plasmid, or mutant Abcg2-IRES plasmid as previously described (17). Cells were collected for H2O2 consumption assay or quantitative RT-PCR analysis as described below.
H2O2 consumption assay.
One hundred thousand C2C12 myoblast cells transfected with wild-type Abcg2, mutant Abcg2, or PSAM control were sorted into a 48-well plate. H2O2 (100 μM) was added to each well, and samples were incubated for 5 min at 37°C. Culture medium (100 μl) was removed at specific time points and added to 2.0 ml of indicator mix, and fluorescence was measured.
Quantitative RT-PCR.
Cells from a single cell suspension were sorted with a MoFlo cytometer into TRIzol (Invitrogen). RNA was extracted with chloroform, and cDNA was made according to the manufacturer's instructions (Invitrogen). Real-time RT-PCR was performed with TaqMan MasterMix kit (Applied Biosystems). All measurements were taken on an Applied Biosystems 7900 Fast Real-time PCR System instrument. For Abcg2-overexpressing C2C12 cells, the transcript level was normalized to the PSAM control empty vector. For adult cardiac Abcg2 lineage cells, the transcript level was normalized to GAPDH.
Immunofluorescence microscopy.
Sections (10 μm) were stained overnight at 4°C for the following primary antibodies: α-actinin (Sigma), CD31 (BD Pharmigen), smooth muscle myosin (Biomedical Technologies), and von Willebrand factor (vWF; Novocastra). Secondary staining was performed for 1 h at room temperature for the following antibodies: goat anti-rabbit AF594 (Molecular Probes) for smooth muscle myosin and vWF, goat anti-mouse AF594 (Molecular Probes) for α-actinin, and rabbit anti-rat (Molecular Probes) for CD31. All images were taken using a Zeiss LSM 510 meta confocal microscope.
Western blot analysis and SOD assay.
Tissue samples were homogenized and quantified with a BCA protein assay kit (Thermo Scientific). Fifty micrograms of protein were loaded per well. Blots were stained with the following primary antibodies overnight at 4°C: GAPDH (Abcam), glutathione reductase (Abcam), and SOD (Calbiochem). Secondary staining was performed for 1 h at room temperature with the following antibodies: goat anti-mouse horseradish peroxidase (Jackson ImmunoResearch Laboratories) for GAPDH, (Jackson ImmunoResearch Laboratories) goat anti-rabbit horseradish peroxidase for glutathione reductase, and donkey anti-sheep horseradish peroxidase for SOD (R&D Systems). SOD activity was quantified from homogenized cardiac samples by a SOD assay kit according to the manufacturer's instructions (Cayman Chemical).
Chemical reagents for intraperitoneal injection.
Each adult mouse received 60 mg/kg body wt tamoxifen (Sigma), dissolved in corn oil, for 12 consecutive days. Consecutive doses were given 24 h apart. The vehicle control was corn oil. For the toxicity experiment, mice received 20, 40, 60, 80, or 100 mg/kg PQ (Sigma) dissolved in sterile 1× PBS. The vehicle control was sterile 1× PBS. For oxidative stress experiments, each mouse received a single dose of 20 mg/kg PQ or PBS 24 h after the final tamoxifen dose. For the wild-type versus Abcg2-null survival experiment, each mouse received a single dose of 70 mg/kg PQ or volume-matched PBS. For the preconditioning experiment, each mouse received two doses, 1 wk apart, of 20 mg/kg PQ or PBS beginning 2 wk before cardiac injury. All injections were intraperitoneal.
LacZ analysis.
Samples were fixed and stained overnight at 37°C while being rotated with LacZ stain solution. Tissue was sectioned at 10 μm thickness with a cryostat. The counterstain was nuclear fast red, and images were taken using a Zeiss Axio Imager M1 upright microscope with Axiovision software. Heart and bone marrow single cell suspensions were obtained with the SP cell preparation as previously described (9). After being resuspended, the cell solutions were stained with a FluoReporter lacz Flow Cytometry kit according to the manufacturer's instructions (Invitrogen). Cells were analyzed on a MoFlo flow cytometer (Cytomation).
Cell expression quantification.
All histological and immunohistochemical images were quantified using Image Pro Plus 6.0 software analysis. For all analyses, five random fields of view were chosen per mouse from at least three mice per group.
Myocardial injury.
Mice underwent permanent ligation of the left anterior coronary artery. Briefly, mice were anesthetized with 1.5% isofluorane, intubated to the level of the carina, placed on a heating pad, and put on a ventilator to aid respiration. A thoracotomy was performed, the heart was exposed, and the proximal left anterior coronary artery was ligated below the atrial appendage using a 9-0 silk suture. The thoracic wall was approximated using 3-0 silk sutures, and extubation was achieved. Mice were kept on a heating pad and observed until fully recovered.
Echocardiography.
Echocardiography was obtained under 1.5% isuflurane anesthesia using a Vevo 770 (VisualSonics) high-frequency ultrasound biomicroscope using a 30-MHz probe operating at a range of 100–120 frames/second. Echocardiography was performed at baseline and 1, 4, and 12 wk after myocardial injury. Ejection fraction (EF; in %) and shortening fraction (FS; in %) values were calculated from M- and B-mode images of the left ventricle (LV) in standard long- and short-axis views, respectively. EF and FS were calculated using the following formulas: EF (in %) = [(LVEDV − LVESV)/LVEDV] × 100 and FS (in %) = [(LVIDd − LVIDs)/LVIDd] × 100, where LVEDV is LV end-diastolic volume, LVESV is LV end-systolic volume, LVIDd is LV internal diameter at diastole, and LVIDs is LV internal diameter at systole.
Scar size assessment.
Hearts were perfused with cold PBS and cut transversely into three 3-mm rings (the base, middle, and apex). Sections (10 μm) at the landmark level of the papillary muscles were stained with Sirius red. Tiled images were taken on a Zeiss AxioObserver Z1 inverted microscope with Zeiss AxioVision software, and scar size, indicated by red-stained collagen, was calculated as a percent of the entire LV using ImageJ software.
Survival analysis.
Age-matched mice (17 mice/group) were used for the wild-type compared with Abcg2-null survival experiment after 70 mg/kg ip PQ injection. Age-matched wild-type mice (at least 8 mice/group) were used for the PQ toxicity study. Survival data were analyzed with GraphPad Prism software (version 5.02).
Statistical analysis.
All results are means ± SD. Survival significance (with a 95% confidence interval) was established by a log-rank (Mantel-Cox) test for two groups or one-way ANOVA with Tukey's post hoc test for correction for five groups. All other significance was established by Student's t-test. P values of <0.05 were considered significant.
RESULTS
Cytoprotection is dependent on functionally active Abcg2.
C2C12 myoblast cells were transfected with empty vector, wild-type Abcg2, or mutant Abcg2, and Abcg2 transport was verified by elimination of Hoechst efflux activity (27.66% SP for wild-type Abcg2, 0.71% SP for mutant Abcg2, and 0.33% SP for the empty control, n = 3, P < 0.05; Fig. 1A). The antioxidative activity of transfected C2C12 cells was then characterized. Wild-type Abcg2-overexpressing cells were able to scavenge a greater amount of H2O2 from the culture medium compared with mutant Abcg2-overexpressing cells (33.97 ± 0.98% remaining H2O2 for wild-type Abcg2 vs. 44.69 ± 0.52% for mutant Abcg2 and 44.22 ± 3.49% for the empty control, n = 3, P < 0.05; Fig. 1B). Furthermore, mRNA analysis confirmed that antioxidative transcript levels of glutathione-S-transferase A, N-myc downstream-regulated gene 1, activating transcription factor 3, and p21 were upregulated in wild-type Abcg2-expressing C2C12 cells, but this induction of antioxidation was absent in mutant Abcg2-expressing cells (wild-type Abcg2 and mutant Abcg2 normalized to the empty vector control; Fig. 1C).
Fig. 1.

Oxidative stress activates a cytoprotective program in ATP-binding cassette transporter subfamily G member 2 (Abcg2)-expressing cells in vitro. A: Mutation of the Abcg2 ATP-binding site. Hoechst expression profiles of C2C12 cells overexpressing wild-type Abcg2 (Abcg2 wt), mutant Abcg2 (Abcg2 mut), or empty vector (control) are shown. B: percentage of extracellular H2O2 consumed from the culture medium. C: fold change of antioxidative gene expression in C2C12 cells overexpressing either mutant or wild-type Abcg2 normalized to the empty vector control. Gsta, glutathione-S-transferase A; Ndr1, N-myc downstream-regulated gene 1; Atf3, activating transcription factor 3. *P < 0.05.
Abcg2 protects against ROS in vivo.
PQ was administered to wild-type mice at multiple concentrations, and mortality was assessed. PQ was toxic, with an estimated 50% lethal dose of 50 mg/kg (n = 8 mice/group, P < 0.05 between all groups; Fig. 2A). The PBS vehicle control was nontoxic to all mice used in the study (data not shown). To confirm elevated ROS generation in the heart after nonlethal PQ, cardiac samples were taken from wild-type mice administered PQ (20 mg/kg) or the saline control. Glutathione reductase and SOD protein levels were measured by Western blot analysis. Both antioxidant enzyme levels were significantly higher in animals that received PQ compared with the vehicle control (Fig. 2B). SOD activity was similarly increased in cardiac samples after PQ compared with control treatment (4.07 ± 1.81 U/mL PQ vs. 3.00 ± 0.84 PBS, n = 3, P < 0.05; Fig. 2C), confirming the induction of oxidative stress.
Fig. 2.
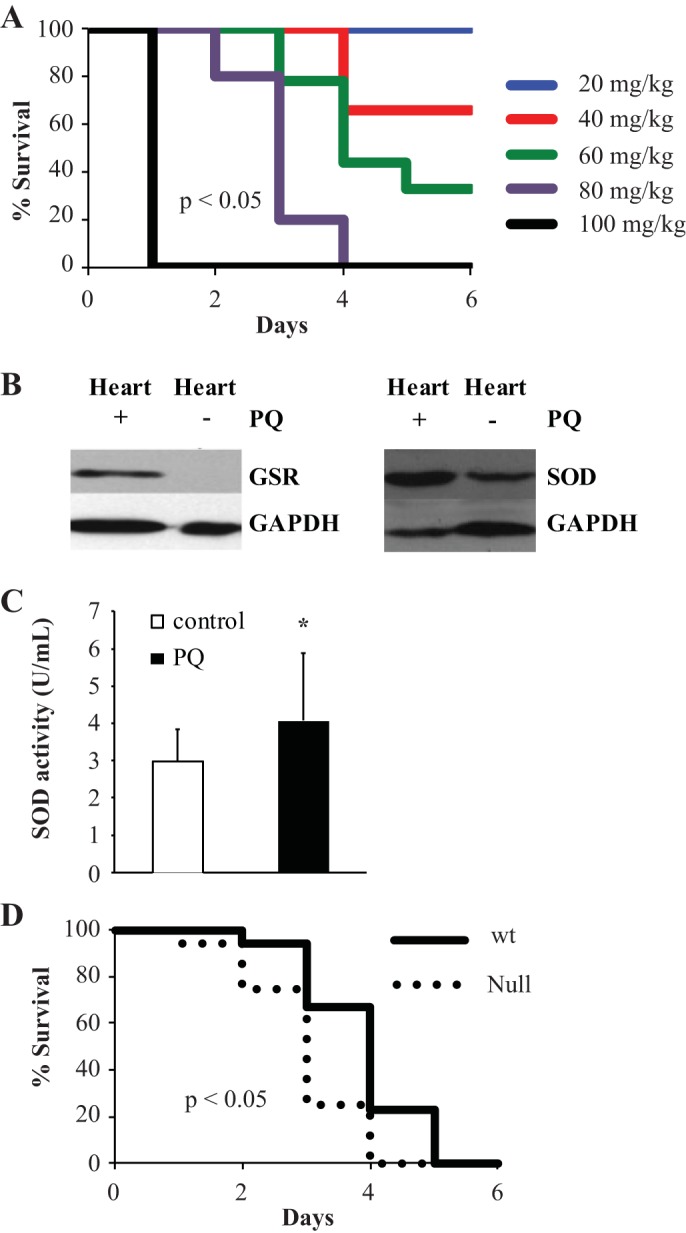
Paraquat dichloride (PQ) generates ROS in vivo and is toxic. A: Kaplan-Meier survival curve for wild-type mice treated with referenced concentrations of PQ (n = 8 mice/group). B: Western blot analysis demonstrating the expression levels of glutathione reductase (GSR) and SOD (SOD) proteins in control or PQ-treated wild-type cardiac tissue. C: SOD activity measured by a SOD assay kit for control or PQ-treated wild-type cardiac samples. D: wild-type (solid line) and Abcg2-null (dotted line) survival after 70 mg/kg PQ treatment (n = 17 mice/group). *P < 0.05.
We next sought to determine whether genetic deletion of Abcg2 has an impact on animal survival upon high-level ROS challenge. Mortality after PQ treatment (70 mg/kg) was assessed in adult mice lacking functional protein (Abcg2-null mice) compared with wild-type mice. Abcg2-null mice demonstrated a significant increase in the rate of mortality, supporting that the presence of Abcg2 affords greater survivability after ROS insult (n = 17 mice/group, P < 0.05; Fig. 2D).
Cardiac Abcg2 lineage increases after oxidative stress.
To further characterize the response of Abcg2-expressing cells to oxidant stress, we crossed tamoxifen-inducible Abcg2CreERT2 mice with R26R and RosaCAGZsGreen reporter strains (Fig. 3). Tamoxifen was administered, and histological analysis was performed to assess cardiac expression of the Abcg2 lineage in the uninjured mouse. The majority of lineage+ cells were observed in the interstitial space, residing between and extralaminar to cardiomyocytes (Fig. 4B,g and h). Reporter cells 10–20 μm in size, characterized by a spindle shape when viewed longitudinally, were observed in a continuous pattern similar to the microcirculation. A small amount of expression was also present in the luminal surface of small veins and arteries. Interestingly, the extent and size of lineage+ cells were qualitatively greater in the atria compared with the ventricles.
Fig. 3.

The Abcg2 lineage is expressed throughout the heart and increases after PQ. (A) ZsGreen expression in the heart after 20 mg/kg PQ treatment compared with control. 4′,6-Diamidino-2-phenylindole staining is shown in blue. B and C: quantification of the percent lineage+ cells (B) and average lineage+ cell size (C) per field of view. ZsG, ZsGreen; AU, arbitrary units. D: FACS quantification of reporter cells in the digested whole heart after cardiomyocyte enrichment. Scale bars = 50 μm. *P < 0.05.
Fig. 4.

The Abcg2-lineage increases after oxidative stress and is vascular. A: gene expression of lineage-sorted cardiac cells treated with PQ compared with control. eNOS3, endothelial nitric oxide synthase (NOS3); vWF, von Willebrand factor; Tagln, transgelin; Tnnt2, troponin T type 2. B: immunohistochemical comparison of endothelial markers [CD31 (a and b) and vWF (c and d)], smooth muscle marker [smooth muscle myosin (SMM); e and f], and cardiac muscle [α-sarcomeric actinin (a-actinin); g and h]. Arrowheads denote coexpression of ZsGreen and labeled marker. C: quantification of coexpression normalized to total nuclei. Scale bars = 50 μm. *P < 0.05.
Establishing baseline expression of the cardiac Abcg2 lineage permitted the characterization of this population's response to in vivo oxidant stress. Mice were treated with 20 mg/kg PQ or saline (control) 24 h after tamoxifen recombination and euthanized 1 mo later. PQ-treated mice demonstrated increased reporter expression throughout the heart compared with control mice (Fig. 3A). Quantification of cardiac sections revealed that in addition to the average cell number, the average size of Abcg2 lineage+ cells per field of view was significantly increased (57.52 ± 14.10% ZsGreen+ cells/total nuclei for PQ vs. 45.56 ± 8.70% ZsGreen+ cells/total nuclei for PBS, n = 3, P < 0.05, Fig. 3B; 213.72 ± 62.69 ZsGreen pixels/cell for PQ vs. 160.89 ± 42.68 ZsGreen pixels/cell for PBS, n = 3, P < 0.05, Fig. 3C). One month after treatment, tissue samples were also harvested and subjected to FACS analysis for quantitative assessment. In the heart as well as bone marrow, Abcg2 reporter expression was significantly increased in mice that received PQ treatment (28.92 ± 0.17% ZsGreen+ cells/whole heart for PQ vs. 25.14 ± 0.36% ZsGreen+ cells/whole heart for PBS, n = 3, P < 0.05; Fig. 3D). This confirms that an exogenous oxidant stress-inducing insult is capable of stimulating cardiac Abcg2 lineage expression in vivo, which parallels the proliferative response seen in vitro.
Abcg2 lineage contributes to the vasculature after ROS treatment.
We next sought to determine the fate commitment of Abcg2 lineage+ cells. Using the PQ treatment and tamoxifen regimen discussed above, cardiac samples were FACS sorted and subjected to real-time quantitative PCR. Treatment with oxidative stress resulted in a significant upregulation of endothelial (CD31 and vWF) and antioxidant transcripts (endothelial nitric oxide synthase and Abcg2, n = 3, P < 0.01; Fig. 4A) in lineage+ cells compared with saline-treated lineage+ cells. In contrast, there were no significant differences observed between treatment groups in smooth and cardiomyocyte-specific muscle expression (transgelin and troponin T type 2, respectively), indicating that an oxidative stress insult compels the Abcg2 lineage to commit to a vascular program.
Cardiac samples were also sectioned and characterized by immunohistochemistry for lineage identification. The majority of Abcg2 lineage+ cells in both treatment groups were CD31 posititive and localized to the endothelium (Fig. 4B,a and b). Quantification of CD31-ZsGreen coexpression demonstrated an increase in lineage commitment to the endothelium after stress (11.64 ± 4.64% double-positive pixels/total nuclei for PBS vs. 36.93 ± 7.36% double-positive pixels/total nuclei for PQ, n = 3, P < 0.05; Fig. 4C). Staining for the endothelial marker vWF supported this result (1.25 ± 0.49% double-positive pixels/total nuclei for PBS vs. 5.98 ± 1.66% double-positive pixels/total nuclei for PQ, n = 3, P < 0.05; Fig. 4, B,c and d, and C). Surprisingly, reporter expression was also observed in conjunction with smooth muscle. Colabeling with smooth muscle myosin was identified in small amounts in control tissue compared with more frequent observation in the treated group (1.66 ± 0.37% double-positive pixels/total nuclei for PBS vs. 4.10 ± 0.75% double-positive pixels/total nuclei for PQ, n = 3, P < 0.05; Fig. 4B,e and f, and C). Finally, there was a complete absence of cardiomyocyte labeling in both groups (α-actinin; Fig. 4B,g and h). Collectively, these findings support an enhanced contribution to the cardiac vasculature after oxidant insult.
Preconditioning with PQ ameliorates ventricular remodeling after myocardial infarction.
Wild-type mice were divided into two groups and subjected to a pretreatment regimen followed by permanent ligation of the left anterior coronary artery. Mice received either PQ (20 mg/kg) or the saline control at 7 and 14 days before injury. Echocardiography was performed at multiple times to assess cardiac function, with baseline analysis after the pretreatment regimen. As shown in Fig. 5A, preconditioning with PQ indicated a trend toward an improvement in FS and EF, yet the differences were not significant (3 mo postinjury, FS: 11.47 ± 5.02% for PBS vs. 15.18 ± 6.47% for PQ, n = 26, not significant; EF: 16.41 ± 11.13% for PBS vs. 18.68 ± 9.28% for PQ, n = 26, not significant). Quantification of the collagen volume fraction in cardiac sections did, however, demonstrate that oxidant stress treatment mitigated the size of scarring (collagen volume fraction: 16.72 ± 4.30% for PBS vs. 13.81 ± 2.66% for PQ, n = 26, P ≤ 0.05; Fig. 5B). Furthermore, CD31 staining in the peri-infarct region established that the capillary density was significantly increased with stress treatment (344.00 ± 112.24 capillaries/mm2 for PBS vs. 486.40 ± 165.25 capillaries/mm2 for PQ, n = 10, P < 0.05; Fig. 5C). Taken together, these results indicate a cardioprotective mechanism that reverses detrimental remodeling through improved vascularization.
Fig. 5.

Oxidative stress pretreatment reverses ventricular remodeling after injury. A: Fractional shortening (FS) and ejection fraction (EF) of mice preconditioned with PQ (n = 13) compared with control (n = 13). B: quantification of myocardial infarct size, as measured by collagen volume fraction (CVF; n = 13 mice/group). C: capillary density in the peri-infarct region in control and treated tissue determined by CD31 staining and quantification (n = 5 mice/group). Scale bars = 50 μm. *P < 0.05.
DISCUSSION
Oxidative stress, typically defined as the imbalance in redox homeostasis toward elevated ROS, is a common feature of heart failure and is highly upregulated after tissue-damaging events, such as myocardial infarction (11). Abcg2 has been implicated in the oxidative stress pathway in numerous studies and is usually characterized as a protective agent that reduces harmful substrate levels and influences other prosurvival mechanisms through unidentified signaling mechanisms (10, 18, 19, 23, 30, 36, 37). In a previous study (25), we demonstrated that in vitro Abcg2 overexpression confers a survival advantage in the presence of H2O2. In the present study, we confirmed this interaction with elevated ROS, demonstrating that functional Abcg2 has a role in redox cycling between the cell and its environment. Our data suggest that oxidative stress stimulates a change in Abcg2-expressing cells toward proliferation and cytoprotection. After stress, cardiac and bone marrow-derived SP cells demonstrated increased proliferation in culture, whereas the total cardiac Abcg2 lineage increased in mice. An increase in the antioxidative transcriptional program after stress was also demonstrated both in vitro and in vivo by transcriptional analysis. The cell cycle regulator p21 and enzyme glutathione-S-transferase A were upregulated after oxidative stress and serve to reduce potential oxidative damage (5, 7, 21, 26), whereas the proteins N-myc downstream-regulated gene 1 and activating transcription factor 3 have been implicated in the oxidative stress response (27, 28). These data support the hypothesis that Abcg2 has a vital role in the oxidative stress pathway and that elevated ROS influences Abcg2 expression to initiate a protective mechanism. This relationship is probable given that we previously demonstrated that HIF-2α binds and transactivates Abcg2 (25). Necessary for the transcriptional regulation of antioxidant genes after oxidative stress, HIF-2α activity has been identified as especially prevalent in the endothelium of multiple tissues (2, 4, 22, 33, 35). In a study associated with oxidative stress injury, Kojima et al. (22) reported an increased susceptibility of HIF-2α knockdown mice to ischemia-reperfusion injury in the kidney, an event particularly damaging to renal endothelial cells. Furthermore, Skuli et al. (35) reported disrupted arteriogenesis in skeletal muscle after hindlimb ischemia after deletion of HIF-2α in vascular endothelial cells. Collectively, these data suggest an increased antioxidative capacity in Abcg2-expressing cells that is activated by HIF-2α in response to elevated ROS.
Stimulation of Abcg2-expressing cells with oxidative stress was achieved with Paraquat dichloride, a stable electron carrier which elevates superoxide radical levels and influences further ROS production to the point of oxidant stress (13, 34). This chemical was chosen as a simple and effective agent for generating cellular oxidative stress levels without concomitant tissue injury. To date, there has not been a suitable alternative for an in vivo oxidative stress model without tissue injury (1, 3, 6, 8, 12, 29, 32, 38). As reported in previous studies (2, 4) using PQ to generate high-level ROS to mimic disease systems, we observed a significant increase in oxidative stress markers in cardiac tissue within hours of treatment. Importantly, mice possessing Abcg2 experienced a survival advantage compared with Abcg2-null mice after PQ insult. Because all mice survived the 20 mg/kg PQ treatment used for systemic oxidant insult and preconditioning experiments, a higher concentration (70 mg/kg) was required to detect a significant difference in survival. This trend of protection supports past studies (18, 19) demonstrating increased mortality of Abcg2-null mice after cardiac injury. Due to the interaction with elevated ROS described above, it is likely that the presence of Abcg2 is necessary to maintain the redox balance in the cellular compartment.
Lineage tracing via an inducible Cre-lox system allows for temporally controlled cell labeling, thus providing a reliable method to follow a specific population and the cells they daughter. Importantly, reporter expression can be initiated at virtually any stage of the animal's life. Given the inherent difficulties with marking the SP, including poor antibody reliability and downregulation of Abcg2 expression with SP cell differentiation (24, 30), this method of fate mapping was especially appropriate for our interests in targeting adult Abcg2 function. Use of the Cre-lox reporter system with inducible activation allowed labeling of not only Abcg2-expressing SP cells but also any progeny derived within the 1-mo study period. This is an important advancement as determination of the SP cell fate was not previously possible. Previous studies (10, 14) using Abcg2CreERT2 transgenic animals have confirmed the stem cell activity of Abcg2-expressing cells; however, this population has not been characterized after cardiovascular injury. Our results mark the first in vivo characterization of the cardiac Abcg2 lineage in response to an exclusively oxidative event. Before injury, we identified an interstitial cell population that was present throughout the adult heart, supporting the original cardiac description for this specific Abcg2-CreERT2 mouse (14). The location of the Abcg2 lineage mirrored the previously described cardiac SP cell staining pattern in the adult heart (19). However, the extent of expression was qualitatively greater than previous descriptions. Most cells were small in size (diameter: 2–4 μm, length: 10–40 μm) and located between cardiac myofibers. A low level of lineage expression was observed in the endothelial lining of small veins and arterioles. Although the pattern was different in size and orientation, atrial expression was similarly observed in endothelial locations. A major finding of this study was the difference observed in Abcg2 lineage expression after ROS insult. Quantification revealed an increase of 12% by histological counting of cardiac sections. FACS analysis after cardiomyocyte enrichment revealed a 3% increase in the Abcg2 lineage. This discrepancy suggested that the response to ROS-induced injury was more pronounced in noncardiac lineages. Expansive reporter expression was observed in the macrovasculature, broadly defined as blood vessels possessing a diameter of >100 μm (20). ZsGreen and LacZ were often observed throughout the entire luminal circumference of large blood vessels after PQ treatment. Previous Abcg2 localization within macrovascular structures had been described exclusively in the luminal layer and adjacent to smooth muscle (39). We revealed coexpression with smooth muscle in the radial portions of the vessel as well as the lumen, highlighting that Abcg2-expressing cells and their progeny are not confined to the endothelium. In contradiction to previous reports on cardiac SP cell differentiation (23, 30), we were unable to find any contribution of the Abcg2 lineage to striated, cardiomyoctye-like structures. There is a possibility that the difference in cardiac muscle differentiation we observed compared with previous reports is due to disparities in the experimental method. Studies by Oyama et al. (30) and Liang et al. (23) used transplantation of cardiac SP cells into the cryoinjured and infarcted heart, respectively, whereas the present study targeted an endogenous cell population using a systemic oxidative insult. Therefore, we cannot exclude the ability of resident cells to contribute to contractile muscle in response to a severe damaging event involving cardiomyocyte death. In fact, TUNEL staining revealed a total absence of apoptosis in cardiac tissue after 20 mg/kg PQ treatment (data not shown).
Due to the increase in the Abcg2 lineage after oxidative stress insult, we hypothesized that a preconditioning regimen of an exogenously administered ROS-inducing agent before myocardial infarction would preserve cardiac function and limit the extent of injury. Pretreatment with PQ did, in fact, demonstrate protection in the form of reduced scar size. Capillary density in the peri-infarct region also increased, indicating that collagen deposition was limited by improved vascularization. The postoxidative stress increase we observed in the Abcg2 lineage, combined with increased antioxidative transcriptional activity, suggests that the attenuation of injury was influenced by the cardiac Abcg2 lineage. Although we were unable to reveal a significant difference in EF and FS, there was an overall trend toward improved function with stress treatment. These data indicate that Abcg2-expressing cells have the capacity to influence contractile health postinjury. In fact, Higashikuni et al. (18) demonstrated that Abcg2-null mice posses significantly deteriorated cardiac function compared with wild-type mice 1 mo after myocardial infarction. Their finding was believed to be in large part due to elevated oxidative species in microvascular endothelial cells, causing obstructed angiogenesis and eventual heart failure. These abnormalities coincide with the findings asserted in this study, yet likely emphasize the more severe physiological consequence of Abcg2 lineage knockout, as opposed to upregulation. The capacity for functional improvement via oxidative stress pretreatment may have been precluded by the extensive degree of injury obtained with permanent ligation of the left coronary artery. Additional studies, possibly involving injury models with attenuated myocardial cell death, will be necessary to comprehensively determine the impact of Abcg2-expressing cells in the regeneration of cardiac performance after injury.
In summary, through the introduction of a systemic oxidative stress agent, we have uncovered novel behaviors of Abcg2-expressing cells and their progeny in the adult heart, further illuminating the function and regulation of this critical transport protein. The cardiac Abcg2 lineage increased in number and size after delivery of PQ. Specifically, we have shown by lineage tracing that Abcg2-expressing cells and their progeny contribute to the cardiac vasculature, including the endothelium and smooth muscle. We have also demonstrated that the presence of Abcg2 is necessary for protection against high-level ROS and that preconditioning with PQ mitigates ventricular remodeling. These data collectively suggest that Abcg2 is highly involved in the antioxidant response in the adult heart and is a suitable target for future cardioprotective and regenerative studies.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant K08-HL-102157.
REFERENCES
- 1.Bivalacqua TJ, Champion HC, Hyman AL, McNamara DB, Kadowitz PJ. Analysis of responses to angiotensin II in the mouse. J Renin-Angioten-Aldost Sys 2: S48–S53, 2001 [DOI] [PubMed] [Google Scholar]
- 2.Brooks AI, Chadwick CA, Gelbard HA, Cory-Slechta DA, Federoff HJ. Paraquat elicited neurobehavioral syndrome caused by dopaminergic neuron loss. Brain Res 823: 1–10, 1999 [DOI] [PubMed] [Google Scholar]
- 3.Carll AP, Willis MS, Lust RM, Costa DL, Farraj AK. Merits of non-invasive rat models of left ventricular heart failure. Cardiovasc Tox 11: 91–112, 2011 [DOI] [PubMed] [Google Scholar]
- 4.Chen Q, Niu Y, Zhang R, Guo H, Gao Y, Li Y, Liu R. The toxic influence of paraquat on hippocampus of mice: Involvement of oxidative stress. Neurotoxicol 31: 310–316, 2010 [DOI] [PubMed] [Google Scholar]
- 5.Chen W, Sun Z, Xiao-Jun W, Tao J, Zheping H, Deyu F, Zhang DD. Direct interaction between Nrf2 and p21Cip1/WAF-1 upregulates the Nrf2-mediated antioxidant response. Mol Cell 34: 663–673, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Communal C, Singh K, Pimentel DR, Colucci WS. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the β-adrenergic pathway. Circulation 98: 1329–34, 1998 [DOI] [PubMed] [Google Scholar]
- 7.Curtis JM, Grimsrud PA, Wright WS, Xu X, Foncea RE, Graham DW, Bretoff JR, Wiczer BM, Ilkayeva O, Cianflone K, Muoio DE, Arriaga EA, Bernlohr DA. Downregulation of adipose glutathione S-transferase A4 leads to increased protein carbonylation, oxidative stress, and mitochondrial dysfunction. Diabetes 59: 1132–1142, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davel AP, Brum PC, Rossoni LV. Isoproterenol induces vascular oxidative stress and endothelial dysfunction via a Giα-coupled β2-adrenoceptor signaling pathway. PLoS One 9: e918771–10, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doyle LA, Yang W, Abruzzo LV, Krogmann T, Gao Y, Rishi AK, Ross DD. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci USA 26: 15665–70, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doyle M, Zhou S, Tanaka KK, Pisconti A, Farina NH, Sorrentino BP, Olwin BB. Abcg2 labels multiple cell types in skeletal muscle and participates in muscle regeneration. J Cell Bio 195: 147–162, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Droge W. Free radicals in the physiological control of cell function. Physiol Rev 82: 47–95, 2002 [DOI] [PubMed] [Google Scholar]
- 12.Engle SK, Jordan WH, Pritt ML, Chiang AY, Davis MA, Zimmermann JL, Rudmann DG, Heinz-Taheny KM, Irizarry AR, Yamamoto Y, Mendel D, Schultze AE, Cornwell PD, Watson DE. Qualification of cardiac troponin I concentration in mouse serum using isoproterenol and implementation in pharmacology studies to accelerate drug development. Toxicol Path 37: 617–28, 2009 [DOI] [PubMed] [Google Scholar]
- 13.Farrington JA, Ebert M, Land EJ, Fletcher K. Bipyridylium quaternary salts and related compounds. V. Pulse radiolysis studies of the reaction of paraquat radical with oxygen Implications for the mode of action of bipyridyl herbicides. Biochim Biophys Acta 314: 372–381, 1973 [DOI] [PubMed] [Google Scholar]
- 14.Fatima S, Zhou S, Sorrentino BP. Abcg2 expression marks tissue-specific stem cells in multiple organs in a mouse progeny tracking model. Stem Cells 30: 210–21, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. Isolation and functional properties of hematopoietic stem cells that are replicating in-vivo. J Exp Med 183: 1797–1806, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Bio 244: 305–318, 2002 [DOI] [PubMed] [Google Scholar]
- 17.Hierlihy AM, Seale P, Lobe CG, Rudnicki MA, Megeney LA. The post-natal heart contains a myocardial stem cell population. FEBS 530: 239–243, 2002 [DOI] [PubMed] [Google Scholar]
- 18.Higashikuni Y, Sainz J, Nakamura K, Takaoka M, Enomoto S, Iwata H, Sahara M, Tanaka K, Koibuchi N, Ito S, Kusuhara H, Sugiyama Y, Hirata Y, Nagai R, Sata M. The ATP-binding cassette transporter BCRP1/ABCG2 plays a pivotal role in cardiac repair after myocardial infarction via modulation of microvascular endothelial cell survival and function. Arter Throm Vasc Bio 30: 2128–2135, 2010 [DOI] [PubMed] [Google Scholar]
- 19.Higashikuni Y, Sainz J, Nakamura K, Takaoka M, Enomoto S, Iwata H, Tanaka K, Sahara M, Hirata Y, Nagai R, Sata M. The ATP-binding cassette transporter ABCG2 protects against pressure overload-induced cardiac hypertrophy and heart failure by promoting angiogenesis and antioxidant response. Arter Throm Vasc Bio 32: 654–661, 2012 [DOI] [PubMed] [Google Scholar]
- 20.Junqueira LC, Carneiro J. The circulatory system. In: Basic Histology: Text and Atlas (10th ed.). New York: McGraw-Hill, 2003, p. 23–31 [Google Scholar]
- 21.Kfouri F, de Castro I, Testagrossa L, Delle H, Da Silva AM, Bastos AP, Vieira JM, Jr, Yu L. Role of p21 and oxidative stress on renal tubular resistance after acute ischaemic injury. Nephrol Dial Transplant 25: 1795–1803, 2010 [DOI] [PubMed] [Google Scholar]
- 22.Kojima I, Tanaka T, Inagi R, Kato H, Yamashita T, Sakiyama A, Ohneda O, Takeda N, Sata M, Miyata T, Fujita T, Nangaku M. Protective role of hypoxia-inducible factor-2 against ischemic damage and oxidative stress in the kidney. J Am Soc Nephrol 18: 1218–1226, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Liang SX, Tan TY, Gaudry L, Chong B. Differentiation and migration of Sca1+/CD31− cardiac side population cells in a murine myocardial ischemic model. Int J Card 138: 40–9, 2010 [DOI] [PubMed] [Google Scholar]
- 24.Martin CM, Meeson AP, Robertson SM, Hawke TJ, Richardson JA, Bates S, Goetsch SC, Gallardo TD, Garry DJ. Persistent expression of the ATP-binding cassette transporter, Abcg2, identifies cardiac SP cells in the developing and adult heart. Dev Bio 265: 262–275, 2004 [DOI] [PubMed] [Google Scholar]
- 25.Martin CM, Ferdous A, Gallardo T, Humphries C, Sadek H, Caprioli A, Garcia JA, Szweda LI, Garry MG, Garry DJ. Hypoxia-inducible factor-2a transactivates Abcg2 and promotes cytoprotection in cardiac side population cells. Circ Res 102: 1075–1081, 2008 [DOI] [PubMed] [Google Scholar]
- 26.McElhannon KE, Bose C, Sharma R, Wu L, Awasthi YC, Singh SP. Gsta4 null mouse embryonic fibroblasts exhibit enhanced sensitivity to oxidants: role of 4-hydroxynonenal in oxidant toxicity. Open J Apoptosis 2: 1–22, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Melotte V, Qu X, Mate O, van Criekinge W, de Bruine AP, Baldwin HS, van Engeland M. The N-myc downstream regulated gene (NDRG) family: diverse functions, multiple applications. FASEB J 24: 4153–4166, 2010 [DOI] [PubMed] [Google Scholar]
- 28.Miyamoto N, Izumi H, Miyamoto R, Bin H, Kondo H, Tawara A, Sasaguri Y, Kohno K. Transcriptional regulation of activating transcription factor 4 under oxidative stress in retinal pigment epithelial arpe-19/hpv-16 cells. IOVS 52: 1226–1234, 2011 [DOI] [PubMed] [Google Scholar]
- 29.Murray DR, Prabhu SD, Chandrasekar B. Chronic β-adrenergic stimulation induces myocardial proinflammatory cytokine expression. Circulation 101: 2338–2341, 2000 [DOI] [PubMed] [Google Scholar]
- 30.Oyama T, Nagai T, Wada H, Naito AT, Matsuura K, Iwanaga K, Takahashi T, Goto M, Mikami Y, Yasuda N, Akazawa H, Uezumi A, Takeda S, Komuro I. Cardiac side population cells have a potential to migrate and differentiate into cardiomyocytes in vitro and in vivo. J Cell Biol 176: 329–341, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peng J, Zhang L, Drysdale L, Fong GH. The transcription factor EPAS-1/hypoxia-inducible factor 2α plays an important role in vascular remodeling. Proc Natl Acad Sci USA 18: 8386–8391, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reil JC, Hohl M, Oberhofer M, Kazakov A, Kaestner L, Mueller P, Adam O, Maack C, Lipp P, Mewis C, Allessie M, Laufs U, Bohm M, Neuberger H. Cardiac Rac1 overexpression in mice creates a substrate for atrial arrhythmias characterized by structural remodeling. Cardiovas Res 87: 485–93, 2010 [DOI] [PubMed] [Google Scholar]
- 33.Scortegagna M, Ding K, Oktay Y, Gaur A, Thurmond F, Yan LJ, Marck BT, Matsumoto AM, Shelton JM, Richardson JA, Michael Bennett MJ J, Garcia JA. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1−/− mice. Nat Gen 35: 331–340, 2003 [DOI] [PubMed] [Google Scholar]
- 34.Sharp CW, Ottolenghi A, Posner HS. Correlation of paraquat toxicity with tissue concentrations and weight loss of the rat. Tox Appl Pharm 22: 241–251, 1972 [DOI] [PubMed] [Google Scholar]
- 35.Skuli N, Majmundar AJ, Krock BL, Mesquita RC, Mathew LK, Quinn ZL, Runge A, Liu L, Kim MN, Liang J, Schenkel S, Yodh AG, Keith B, Simon MC. Endothelial HIF-2α regulates murine pathological angiogenesis and revascularization processes. J Clin Invest 122: 1427–1443, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamahara K, Fukushima S, Coppen SR, Felkin LE, Varela-Carver A, Barton P, Yacoub MH, Suzuki K. Heterogeneic nature of adult cardiac side population cells. Biochem Biophys Res Comm 371: 615–620, 2008 [DOI] [PubMed] [Google Scholar]
- 37.Yoon J, Choi SC, Park CY, Shim WJ, Lim DS. Cardiac side population cells exhibit endothelial differentiation potential. Exp Mol Med 39: 653–662, 2007 [DOI] [PubMed] [Google Scholar]
- 38.Zhang G, Kimura S, Nishiyama A, Shokoji T, Rahman M, Yao L, Naga Y, Fujisawa Y, Miyatake A, Abe Y. Cardiac oxidative stress in acute and chronic isoproterenol-infused rats. Cardiovas Res 65: 230–8, 2005 [DOI] [PubMed] [Google Scholar]
- 39.Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, Lagutina I, Grosveld GC, Osawa M, Nakauchi H, Sorrentino BP. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med 7: 1028–1034, 2001 [DOI] [PubMed] [Google Scholar]