

Abstract
The current study examined the role of Na/K-ATPase α1-subunit in animals subjected to 5/6th partial nephrectomy (PNx) using Na/K-ATPase α1-heterozygous (α1+/−) mice and their wild-type (WT) littermates. After PNx, both WT and α1+/− animals displayed diastolic dimension increases, increased blood pressure, and increased cardiac hypertrophy. However, in the α1+/− animals we detected significant increases in cardiac cell death in PNx animals. Given that reduction of α1 elicited increased cardiac cell death with PNx, while at the same time these animals developed cardiac hypertrophy, an examination of cardiac cell number, and proliferative capabilities of those cells was carried out. Cardiac tissues were probed for the progenitor cell marker c-kit and the proliferation marker ki-67. The results revealed that α1+/− mice had significantly higher numbers of c-kit-positive and ki-67-positive cells, especially in the PNx group. We also found that α1+/− mice express higher levels of stem cell factor, a c-kit ligand, in their heart tissue and had higher circulating levels of stem cell factor than WT animals. In addition, PNx induced significant enlargement of cardiac myocytes in WT mice but has much less effect in α1+/− mice. However, the total cell number determined by nuclear counting is higher in α1+/− mice with PNx compared with WT mice. We conclude that PNx induces hypertrophic growth and high blood pressure regardless of Na/K-ATPase content change. However, total cardiac cell number as well as c-kit-positive cell number is increased in α1+/− mice with PNx.
Keywords: stem cells, cardiac hypertrophy, apoptosis, ATPases, caspase, cell proliferation, uremic cardiomyopathy, cardiac progenitor cells, cardiotonic steroids
studies indicate that renal insufficiency worsens congestive heart failure and is directly associated with morbidity and mortality in congestive heart failure patients, described as uremic cardiomyopathy (UrC) (6, 9, 37, 44). Mortality due to cardiovascular disease was 10 to 30 times higher in patients receiving dialysis than in the general population (31). However, direct mechanisms by which these cardiovascular changes are incurred are not well understood.
One of the major cardiovascular changes observed in patients and experimental models of UrC is cardiac remodeling, including cardiac hypertrophy and dilatation. Increased gravimetric weight of the heart as well as increased blood pressure occurs in both experimental models and patients with UrC (2, 5, 11, 38, 46). Cardiac hypertrophy typically occurs in two phases. First is the compensated phase where cardiomyocytes enlarge and the walls of the heart thicken to preserve cardiac ejection performance in response to systemic pressure or volume overload (5). The second phase called “decompensated” hypertrophy occurs in response to prolonged pressure or volume overload or in response to an illness or chronic disease condition (5). Decompensated hypertrophy is characterized by cardiac myocyte death, increased ventricular dimension, thinning of the walls of the heart, and significant declines in cardiac output (5). More recently, studies found that regeneration of myocardium via stem cell differentiation is also involved in the process of cardiac remodeling (21, 52, 53). The concept that the adult heart is a self-renewing organ has evolved during the past two decades, with reports showing that stem cells can differentiate into cardiac myocytes (3, 21, 30, 52, 53). Several biomarkers have been discovered that identify the residential cardiac stem cells or progenitor cells (3, 22). Among these, c-kit-positive cells are the most studied cardiac stem cells and have been shown to improve cardiac function in clinical trials (4, 36).
We have previously reported that cardiotonic steroids (CTS) are elevated and contribute to cardiac hypertrophy and fibrosis in rat and mouse models of UrC (2, 46). Conversely, we found that CTS such as marinobufagenin (MBG) induce cardiac myocyte apoptosis when the Na/K-ATPase α1-subunit is reduced (33). Decreased Na/K-ATPase and increased endogenous CTS have been found in patients with congestive heart failure and chronic kidney diseases (18, 25, 29, 40, 47). Additionally, infusion of MBG into mice with reduced Na/K-ATPase α1-subunit levels (α1+/−) resulted in increased myocyte apoptosis, ventricular dilation, decreased cardiac function, and cardiac wall thickness in α1+/− mice versus their wild-type (WT) littermates treated in a similar manner (33). These studies illustrate the causative nature of CTS in these deleterious physiological events. Surgical implementation of partial nephrectomy (PNx) in rats and mice has been previously found to emulate the clinical presentation of cardiovascular decline that patients with chronic kidney disease typically have, i.e., high blood pressure, increased diastolic dimension, and ventricular dysfunction (7, 17, 26, 43). To determine the importance of Na/K-ATPase α1-subunit receptor levels in the development of UrC and survival/regenerative pathways in cardiovascular tissues, we adapted and used a method of segmental infarction via arterial ligation to induce 5/6th PNx in mice expressing reduced levels of Na/K-ATPase α1-subunit and their WT littermates.
MATERIALS AND METHODS
Animals.
Animal experiments were conducted in accordance with the National Institutes of Health's Guide for the Care and Use of Laboratory Animals under protocols approved by the Institutional Animal Care and Use Committee at the University of Toledo. Na/K-ATPase α1+/− mice and their WT littermates were generated as previously described (39). Genomic DNA was obtained from tail biopsies and used for PCR-based genotyping. Adult male mice, which were ∼2 to 3 mo of age and weighing between 25–27 g, were used for this study. All mice were reared under a 12-h:12-h light-dark cycle, fed standard chow, and were provided water ad libitum. These conditions were used for the entire duration of the experiment.
The two genetic groups (i.e., α1+/− and WT littermates) were each divided into two groups based on surgical intervention: the first group consisted of sham-operated animals that served as controls, whereas, the second group consisted of animals receiving PNx. Surgery was performed on mice anesthetized with 2% isoflurane (mixed with oxygen). An incision was made in the left flank, through which the left kidney was extirpated, and the artery(ies) supplying the upper pole of the kidney were observed under a high-power dissecting microscope and subsequently ligated with 6-0 silk sutures. After ligation, the kidney was observed for a characteristic color change over approximately two-thirds of the kidney tissue, indicating successful interruption of blood flow to the discolored portion of the kidney. Once the color change was observed, the kidney was reinserted to the body cavity and the incision closed. After 1 wk, the right kidney, exposed through the right flank, was decapsulated to avoid removal of the adrenal gland, and subsequently the renal artery, vein, and ureter were ligated and the kidney was removed. In sham surgeries, incisions were made as in both steps of the PNx, and the kidneys were isolated; however, neither ligation nor decapsulation and removal of the opposing kidney were performed. After visualization of sham-operated kidneys, they were returned to the body cavity and the muscle and skin closed with suture. Weekly measurements of body weight ensured the overall health of the animals.
Echocardiographic imaging was performed (Acuson Sequoia C512) just before the first step of the operations listed above (baseline) and at 8 wk after the completion of the second step of the operation, just before euthanasia. Transthoracic echocardiography was accomplished on animals anesthetized with 2% isoflurane delivered on oxygen. Animals were secured to a heated metal platform in a supine position with medical tape on all four extremities. The chests of the animals were shaved, and the remaining hair was removed using a depiliatory cream. After removal of excess depiliatory cream, prewarmed echo gel was applied to the chest and a 15-mHz linear transducer 15L8 (Siemens) was used to acquire images in a shallow left-side position on the heated pad. Applying the probe to the chest of the animal just over the left ventricle (LV) perpendicular to the direction of the sternum, short-axis m-mode views of the heart were acquired to determine various echocardiographic parameters. End-diastolic and end-systolic dimensions were obtained via measuring the transverse internal distance between the interventricular septal wall and posterior wall at diastole and systole for three consecutive beats. These m-mode plots were also used for determining the thickness of the interventricular septal and posterior walls at systole and diastole using onboard calipers available with the echo machine. Fractional shortening and relative wall thickness were measured using standard formulas (14).
Additionally, every 2 wk during the postoperative window till the time of euthanasia, blood pressure measurement via tail cuff was performed using a CODA high through-put 8 channel non-invasive blood pressure system (Kent Scientific, Torrington, CT) as previously described (26, 27).
Mice were euthanized at the end of the 8th wk after surgical manipulation. After euthanasia, the heart was collected and weighed. Once weighed, the atria were removed and the LV isolated and cut in half. Half of the LV was immediately submerged in 4% formaldehyde solution for fixation, whereas the other half was flash frozen in liquid nitrogen and subsequently stored at −80°C for later use in biochemical analysis as previously described (17, 26, 27).
Measurement of plasma MBG concentrations.
After euthanasia (i.e., 8 wk after PNx), plasma MBG was measured using a competitive enzyme-linked immunosorbent assay (ELISA) method previously described (26, 33). Briefly, 100 μl of mouse plasma was extracted and resuspended in TBST solution consisting of 150 mM NaCl, 50 mM Tris, and 0.05% Tween-20 (pH 7.6). The resuspended solution (50 μl/well) was incubated with anti-MBG antibody (50 μl/well) in an MBG-BSA-coated plate for 1 h. A secondary horseradish peroxidase (HRP)-conjugated anti-mouse antibody was added after washing. 3,3',5,5'-Tetramethylbenzidine was used to measure color development, and optical density at 450 nm was measured after addition of 1 N H2SO4 to stop the reaction. MBG concentration was quantified against a standard curve.
Western blot analysis.
Tissue homogenates were prepared by submersion in ice-cold radioimmunoprecipitation assay lysis buffer (pH 7.0) from Santa Cruz Biotechnology (SC-24948; Santa Cruz, CA), followed by homogenization. Aliquots were then made and frozen at −80°C for future use. For Western blot analysis, the protein from homogenates were separated by SDS-polyacrylamide gel electrophoresis. The resolved proteins were then electrotransferred to a nitrocellulose membrane (Fisher Scientific, Hanover Park, IL) for immunoblotting. The following antibody was purchased from Abcam (Boston, MA) and used to probe the membranes for the protein of interest: rabbit anti-stem cell factor (SCF) polyclonal antibody (ab83866). Antibodies used to probe the blots for total-ribosomal S6 protein (tS6; Cat. No. 2217S) and phospho-ribosomal S6 protein recognizing phosphorylation at serine 235/236 (pS6; Cat. No. 2211S) as well as biotinylated antibodies to probe for Akt at phosphorylation sites threonine-308 (Cat. No. 5056S) and serine-473 (Cat. No. 5012S) and streptavidin-conjugated with HRP (Cat. No. 3999S) were purchased from Cell Signaling Technologies (Boston, MA). Loading conditions were controlled using a goat anti-actin polyclonal antibody from Santa Cruz Biotechnology (SC-1616). All appropriate HRP-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology, and α1-subunit levels were probed using the α6F antibody from the Developmental Studies Hybridoma Bank at the University of Iowa.
Histology.
LV sections were immediately fixed in 4% formaldehyde buffer solution (pH 7.2) and was paraffin embedded after 48 h of fixation. The tissues were then cut to a thickness of 4 μm and mounted onto microscopy slides. Masson's trichrome stain for cardiac fibrosis was conducted and computer-aided morphometry used to quantify the percentage of fibrosis in the tissue as previously described (7, 17, 26, 27). For immunostaining, slides were subjected to coimmunofluorescence staining for c-kit (CD117, marker of progenitor cells, Cat. No. AB25022, Abcam) and ki-67 (marker of proliferation, Cat. No. AB16667, Abcam) or c-kit and CD45 (marker for cells of hematopoietic origin, Cat. No. AB64100, Abcam). To count cardiac cell number, slides were stained for cardiac troponin I (cTnI, a cardiomyocyte specific marker, Cat. No. AB47003, Abcam) and 4′,6-diamidino-2-phenylindole (DAPI, a nuclear marker, Cat. No. D1306, Life Technologies). The protocol for these staining procedures is as follows: the mounted paraffin-embedded tissue sections were first deparaffinized with xylene and rehydrated by sequential incubations in ethanol and water. After rehydration, antigen retrieval was performed by boiling the slides in trisodium citrate buffer (pH 6.0; 10 mM sodium citrate, 0.05% Tween-20) for 15 min. After boiling, the slides were allowed to cool for 30 min before being washed twice for 3 min in Tris-buffered saline supplemented with Tween-20, TBST, consisting of 0.05% Tween-20 (pH 7.5), 20 mM Tris, and 500 mM NaCl. The tissue sections were then blocked for 30 min with 1% bovine serum albumin in TBST. Subsequently, the slides were incubated overnight at 4°C, with primary antibody solution containing the primary antibodies for c-kit and ki-67, or c-kit and CD45 in 1% BSA-TBST. After primary antibody incubation, the slides were washed three times for 5 min each in TBST. The appropriate secondary antibodies were applied, and the slides were incubated for 2 h at room temperature. The concentrations of the secondary antibodies were as follows: 1:100 for streptavidin Alexa Fluor-594 (for c-kit), and 1:50 dilution in 1% BSA-TBST for anti-rabbit Alexa Fluor-488 (for ki-67), and/or 1:50 dilution in 1% BSA-TBST for anti-rat Alexa Fluor-488 (CD45). After incubation with secondary antibody, the slides were washed three times in TBST and mounted with Antifade Gold from Life Technologies. Immunofluorescence was then visualized on a confocal microscope (TCS SP5 LCSM, Lecia, Buffalo Grove, IL).
Active caspase 3 was determined by immunohistochemistry. Preservation, mounting, deparaffinization, and antigen retrieval were similarly performed as above, but the level of active caspase 3 was determined using the HRP/3,3′-diaminobenzidine (DAB) detection kit from Abcam, according to the manufacturer's protocol. Computer-aided morphometry was used to quantify active caspase-3 staining. Additionally, slides were stained with hematoxalin alone to allow visualization of nuclei in ×20 images of each tissue section.
Terminal deoxynucleotidyl transferase dUTP nick end labeling assay.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was performed using a commercially available kit from Trevigen (Gaithersburg, MD) according to the company's protocol. Briefly, paraffin-embedded LV tissue was deparaffinized with xylene and rehydrated with sequential incubation with ethanol as detailed above. Rehydrated tissue sections were washed with PBS and incubated with proteinase K for 15 min, washed, and quenched before labeling with biotin-labeled dUTP. The labeling reaction was stopped by adding stop buffer as provided by the kit. The tissue sections were then incubated with HRP-conjugated streptavidin for 10 min, washed, and immersed in DAB solution for color development. These sections were also counterstained with methyl green before mounting. Apoptotic events are presented as the ratio of positive cells (stained with dark brown color) and the total nuclei (stained with green).
Wheat germ agglutinin staining.
Briefly, LV tissue sections were deparaffinized and rehydrated in four changes of xylene, two changes of 100% ethanol, two changes of 95% ethanol, and two changes of 70% ethanol. After slides were rinsed in running tap water for 5 min, the tissue sections were incubated with solution containing 5 μg/ml of Oregon Green 488 wheat germ agglutinin (WGA) stain (Life Technologies, Eugene, OR), at 4°C overnight. After incubation with WGA, slides were washed three times with PBS. The slides were allowed to air dry and then mounted with Prolong Gold antifade reagent from Life Technologies. Subsequently, eight ×20 photomicrographs were taken in random areas of each of the tissue sections, and the area of 20 cells in each image was quantified using ImageJ (National Institutes of Health).
Measurement of plasma levels of SCF.
After euthanasia (i.e., 8 wk after PNx), blood was collected from the animals in EDTA plasma tubes (Fisher, Chicago, IL). Fresh whole blood was centrifuged as 2,000 rpm for 10 min to allow for separation of the blood and collection of the plasma fraction. Plasma samples were flash frozen and stored at −80°C until used in analysis. Circulating levels of SCF were determined using the mouse anti-SCF ELISA from Abcam. Plasma samples isolated from whole blood by centrifugation were diluted four times in dilution buffer provided in the kit, and then portions of the diluted plasma were assayed for SCF content according to the manufacturer's protocol.
Statistical analyses.
Data are presented as means ± SE. Data obtained were analyzed by t-test or by two-way ANOVA followed by pairwise comparisons with Bonferroni's corrections conducted post hoc, where appropriate. All statistical analyses were performed using Prism 5 (GraphPad).
RESULTS
PNx induces increases in circulating MBG concentrations, high blood pressure, cardiac hypertrophy, and cardiac fibrosis in both WT and α1+/− mice.
Echocardiographic data were obtained at baseline (1 day before surgery) and 8 wk after surgery as summarized in Table 1. These data showed that PNx caused enlargement of the LV in both WT and α1+/− mice as indicated by the end-diastolic dimension (EDD) and end-systolic dimension. PNx also slightly decreased the body weight gain in the α1+/− mice compared with their sham-operated controls or to PNx WT mice, but this decrease did not reach statistical significance. Other echo parameters such as posterior wall thickness (PWT), septal wall thickness (SWT), relative wall thickness [calculated by (PWT + SWT)/EDD], and fractional shortening showed no difference between sham-operated and PNx or between WT and α1+/− mice.
Table 1.
Doppler image study in WT and α1+/− mice
| WT |
α1+/− |
|||||||
|---|---|---|---|---|---|---|---|---|
| Sham |
PNx |
Sham |
PNx |
|||||
| Variables | Baseline | 8 wk | Baseline | 8 wk | Baseline | 4 wk | Baseline | 4 wk |
| BW, g | 30.3 ± 0.7 | 36.7 ± 1.0 | 31.4 ± 0.6 | 36.7 ± 1.1 | 29.7 ± 1.1 | 36.2 ± 1.1 | 31.3 ± 1.1 | 34.6 ± 1.4 |
| HR, beats/min | 479 ± 8 | 523 ± 14 | 466 ± 7 | 480 ± 13 | 449 ± 11 | 501 ± 22 | 430 ± 13 | 479 ± 19 |
| EDD, mm | 3.4 ± 0.1 | 3.8 ± 0.2 | 3.4 ± 0.1 | 4.3 ± 0.2* | 3.4 ± 0.1 | 3.9 ± 0.1 | 3.6 ± 0.1 | 4.2 ± 0.1* |
| ESD, mm | 2.3 ± 0.1 | 2.9 ± 0.2 | 2.3 ± 0.1 | 3.3 ± 0.2* | 2.0 ± 0.1 | 2.9 ± 0.1 | 2.3 ± 0.1 | 3.2 ± 0.1* |
| PWT, mm | 1.03 ± 0.04 | 0.99 ± 0.05 | 1.12 ± 0.07 | 0.98 ± 0.05 | 1.01 ± 0.04 | 1.07 ± 0.06 | 0.94 ± 0.03 | 1.04 ± 0.04 |
| SWT, mm | 1.01 ± 0.06 | 0.87 ± 0.04 | 1.05 ± 0.07 | 0.88 ± 0.04 | 0.89 ± 0.04 | 0.77 ± 0.05 | 0.99 ± 0.08 | 0.78 ± 0.04* |
| RWT | 6.2 ± 0.4 | 5.0 ± 0.3 | 6.4 ± 0.4 | 4.4 ± 0.3 | 5.7 ± 0.2 | 4.7 ± 0.3 | 5.5 ± 0.4 | 4.4 ± 0.2 |
| FS, % | 32.1 ± 3.1 | 25.4 ± 1.8 | 32.5 ± 2.4 | 24.6 ± 1.5 | 40.3 ± 2.4 | 26.0 ± 1.1 | 31.5 ± 2.9 | 24.4 ± 1.3 |
Values are means ± SE. WT, wild-type; α1+/−, Na/K-ATPase α1-heterozygous mice; BW, body weight; HR, heart rate; EDD, end diastolic dimension; ESD, end systolic dimension; PWT, posterior wall thickness; SWT, anterior wall thickness; RWT, relative wall thickness = (PWT + SWT)/EDD; FS, fraction shortening (%) = (EDD − ESD)/EDD × 100.
P < 0.05, PNx vs. sham at 8 wk.
In accord with previous studies (17, 27), both WT and α1+/− animals display a significant increase in systolic (Fig. 1, A and B) and mean blood pressure (Fig. 1, C and D) over sham-operated mice, starting 2 wk after PNx and persisting throughout the rest of the experiment. At 8 wk post-PNx, the WT animals displayed systolic blood pressures that were 20.9 mmHg higher than sham-operated animals, whereas the α1+/− animals displayed systolic blood pressures that were 19.0 mmHg higher than sham-operated animals (Fig. 1). The WT animals displayed slightly higher levels of end-systolic and mean blood pressure in PNx animals than in α1+/− animals subjected to the same surgery, these differences were not significant (P > 0.05).
Fig. 1.

Partial nephrectomy (PNx) induces blood pressure (BP) and marinobufagenin (MBG) concentration increases in both wild-type (WT) and Na/K-ATPase α1 heterozygous knockout (α1+/−) mice. BP was measured by tail cuff as described in materials and methods. Both systolic BP (A and B) and mean BP (C and D) showed significant increases after PNx vs. sham, but there were no significant differences between the WT and α1+/− animals. MBG concentration increases significantly with PNx in both WT and α1+/− animals. E: means ± SE of plasma [MBG] determined by immunoassay. *P < 0.05, significantly different than sham.
Additionally, using a competitive ELISA assay, we measured circulating plasma MBG levels in mice of both genetic groups subjected to either sham surgery or PNx. Plasma MBG concentrations were significantly elevated with PNx in both genetic groups versus sham-operated animals (P < 0.05). The level of PNx-induced increase in MBG did not significantly differ between WT and α1+/− animals (Fig. 1E).
To test whether PNx also increased cardiac hypertrophy and fibrosis as we previously observed in rats, the hearts of experimental animals were collected and weighed. LV tissue was also fixed and stained with trichrome to detect fibrosis. As shown in Fig. 2A, hearts collected from PNx-operated animals of both genotypes were significantly larger (P < 0.01) than those collected from sham-operated animals, though the heart weight of α1+/− mice subjected to PNx was smaller than that of the WT mice (159 ± 6 vs. 179 ± 9 mg). Cardiac hypertrophy, as measured by heart weight (HW)-to-body weight (BW) ratio, was significantly increased (P < 0.05) with PNx in both WT (4.91 ± 0.27 vs. 3.9 ± 0.10) and α1+/− (4.64 ± 0.26 vs. 3.8 ± 0.06) mice versus sham-operated controls (Fig. 2B). Mice in both genotypes also displayed significantly (P < 0.01) more LV tissue, with WT PNx-operated animals having 34.1% more LV tissue by weight than sham-operated controls and α1+/− mice having 22.1% more LV tissue versus sham-operated controls (Fig. 2C). Both WT and α1+/− mice receiving PNx exhibit significant (P < 0.05) LV hypertrophy compared with sham-operated controls of the same genotype as indicated by the LVW-to-BW ratio (Fig. 2D). PNx also causes significant increase in fibrosis in both WT and α1+/− mice (Fig. 2E).
Fig. 2.

PNx induces cardiac hypertrophy and fibrosis in WT and α1+/− mice. Heart weight (HW) and left ventricle weight (LVW) (A and C) were measured after mice were euthanized, and the heart weight/body weight ratio (HW/BW, C) and LVW/BW ratio (D) were calculated based on the final body weight before euthanasia. Both HW/BW and LVW/BW data indicate a significant increase in cardiac hypertrophy. E: Masson's trichrome staining in left ventricle tissue from experimental mice: representative images from each group taken by an Olympus FSX100 microscope with a ×20 lens (left) and quantification data (n = 8 in each group; right). *P < 0.05, significantly different than sham of same genotype; **P < 0.01, significantly different than sham of same genotype; !!P < 0.01, significantly different than PNx of WT.
PNx induces hypertrophy of cardiac myocytes in WT animals while inducing hyperplasia of cardiac cells in α1+/− mice.
Using WGA-based immunofluorescence to show the borders of individual cardiomyocytes in these tissues, we evaluated the cross-sectional area of individual myocytes in LV tissue sections of both WT and α1+/−, sham-operated and PNx-operated animals. After PNx, the mean area of cardiac myocytes in WT animals significantly increases (P < 0.001) by more than 76% versus sham-operated controls, whereas only 16% increase was observed in α1+/− mice (Fig. 3A). Of particular interest is the finding that the average myocyte size in WT animals and α1+/− mice receiving PNx are significantly different. WT cardiomyocytes average at 509.9 μm2 after PNx, whereas those from α1+/− mice are 379.6 μm2 (P < 0.001). A comparison of the distribution of the cell sizes for the WT and α1+/− animals reveals that the center of the distribution is shifted in the direction of larger cells with PNx WT animals (Fig. 3B), wherein the α1+/− distribution of the myocytes does not vary from that of sham-operated controls (Fig. 3C).
Fig. 3.

PNx promotes cardiomyocyte hypertrophy in WT animals and hyperplasia in α1+/− mice. A: cardiomyocyte cross-sectional area was determined by wheat germ agglutinin staining as illustrated in materials and methods: representative images from each group (left) and quantification data analyzed by ImageJ from 5 animals in each group (right). Scale bar = 25 μm. B and C: representative frequency distribution curves illustrating the frequency of cells of a given size as a percentage of the total measured cells in the WT (B) and α1+/− (C). ***P < 0.001, significantly different than sham of shared genotype; !P < 0.001, significantly different than WT receiving PNx.
Since both WT and α1+/− mice exhibit significant cardiac hypertrophy, whereas only WT PNx-operated animals showed a dramatic increase in cell size, we also measured the cell number by counting the number of nuclei in tissue sections from the animals in this experiment. LV sections from these animals were costained with DAPI and cTnI. As shown in Fig. 4A, most DAPI-stained nuclei overlay with cTnI, whereas a small fraction are outside the cTnI staining area. Interestingly, both the nuclei that overlaid with cTnI (Fig. 4B) and those not costained with cTnI (Fig. 4C) are increased in α1+/− mice with PNx compared with their sham-operated controls. However, there is no significant difference between the WT PNx and sham-operated animals. These results indicate that both myocytes (indicated by nuclei overlaying with cTnI, Fig. 4B) and other types of cells (nuclei outside of cTnI staining area, Fig. 4C) may contribute to increased cell number in the heart tissue from α1+/− mice with PNx.
Fig. 4.

PNx induces increased nuclei numbers in α1+/− mice. Left ventricle sections from each experimental group were stained with 4′,6-diamidino-2-phenylindole (DAPI; blue) and cardiac troponin I (cTnI, red) as described in materials and methods. A: representative images of DAPI and cTnI staining. Scale bar = 25 μm. B: quantification data of nuclei number by counting the DAPI-stained particles that overlaid with cTnI staining. C: quantification data for nuclei that are outside of the cTnI staining area. *P < 0.05, PNx vs. sham in α1+/− animals.
PNx induces myocyte apoptosis that fails to activate Akt/mammalian target of rapamycin (mTOR) survival signaling in α1+/− mice.
Previous work by our laboratory and others has shown that reduction of Na/K-ATPase potentiates cell death in cardiac myocytes and in epithelial cells treated with CTS (33, 51). Because of the fact that Na/K-ATPase reduction has been reported in heart failure patients and that PNx induces significant increases in circulating CTS concentrations (2, 7, 17, 27), we sought to determine if PNx elicited increased cell death in α1+/− mice versus WT littermates subjected to the operation. To evaluate the role of cardiac cell death and the level of apoptotic activity in these animals, we used immunohistochemistry to measure the levels of active caspase 3 in heart tissues as previously described (33). Our analysis revealed that both WT and α1+/− mice receiving sham surgery similarly displayed low levels of active caspase-3 expression, but the PNx surgery in α1+/− mice induced significantly higher levels of active caspase 3 (Fig. 5A), indicating that reduction of α1 potentiates cardiac cell death.
Fig. 5.

PNx induces apoptosis in α1+/− mice but not in WT mice. Active caspase-3 levels in mouse heart tissue were measured using immunohistochemical methods as described in materials and methods. A, left: representative ×20 photomicrographs of active caspase 3 in sham- or PNx-operated animals. A, right: quantification data from 5 to 6 animals in each group. B, left, representative terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) images from sham- and PNx-operated animals. B, right: quantification of TUNEL-positive nuclei. Red arrows indicate TUNEL-positive nuclei. WT sham, n = 6; WT PNx, n = 5; α1+/− sham, n = 6; PNx+/−, n = 6. *P < 0.05, significantly different than sham; !significantly different than WT PNx. Scale bar is 60 μm.
Additional evidence for increased cardiac cell death was observed in LV tissue sections by TUNEL assay. As shown in Fig. 5B, PNx increased TUNEL-positive nuclei in α1+/− animals but not in WT animals, which is consistent with the current caspase-3 data and our previous publication (33).
To further study the mechanism of cardiac cell death, we examined Na/K-ATPase content and Akt/mTOR signaling in these mice. The Akt/mTOR pathway has been demonstrated to regulate survival signaling pathways that protect cells from death. We also illustrated in our previous study that reduction of Na/K-ATPase attenuates this pathway in cells or animals treated with MBG (33). As shown in Fig. 6A, the Na/K-ATPase α1-expression levels were about 40% less in α1+/− mice hearts compared with the WT hearts, which is consistent with previous reports (33, 39). PNx further decreased the Na/K-ATPase α1-levels in α1+/− mice. To determine if cardioprotective signaling as mediated through the Akt/mTOR pathway was regulated in these animals, we carried out Western blot analyses of the levels of phosphorylation at residue serine-235/236 of ribosomal S6 protein (S6). S6 protein is a downstream member of the Akt/mTOR pathway and can be activated through phosphorylation by S6 kinase (19). As shown in Fig. 6B, we found that PNx elicits significantly increased levels of phosphorylated S6 protein in WT animals, whereas in α1+/− animals there was no induction of S6 phosphorylation when the animals were subjected to PNx. Additionally, we assessed the activation of Akt, an upstream signaling component of S6, by examining the phosphorylation of the protein at two known phosphorylation sites, i.e., threonine-308 and serine-473. We found that in WT animals, PNx induces significant increases in phosphorylation of Akt at both sites (P < 0.01). In α1+/− sham-operated animals, the level of Akt phosphorylation at these two sites is significantly higher than that observed in sham-operated WT animals. Interestingly, Akt phosphorylation was significantly downregulated after PNx versus sham-operated controls in α1+/− mice (Fig. 6, C and D). These findings are consistent with our previous publication (33), which indicated that Na/K-ATPase reduction attenuates Akt/mTOR survival signaling. Reduction of Na/K-ATPase increases c-kit cell abundance and proliferation in heart tissue. Our previous work (33, 51) and the above-mentioned data indicate that reductions in Na/K-ATPase α1-subunit elicit significant increases in cardiac cell death, whereas the results from the current report show little difference in hypertrophy and diastolic dimension obtained with PNx in these mice. We postulated that the α1+/− mice must have a counteracting cellular program which compensates for the increase in cells lost to apoptosis. A pathway that could counteract cell death is the activation of proliferative/regenerative processes; as such, we examined whether reduction of α1 resulted in an increase in cardiac regenerative activity of the heart. Cardiovascular tissues were probed with an antibody against a specific surface marker for cardiac progenitor cells, i.e., CD117 (c-kit). We found a significantly higher number of c-kit-positive cells in heart tissues isolated from α1+/− mice than those from WT animals (Fig. 7). Additionally, the number of proliferating (i.e., ki-67-positive) c-kit-positive cells also increased significantly with PNx versus sham surgery in animals of both genotypes, with α1+/− animals having a significantly higher number of proliferating cells than WT animals subjected to either sham or PNx surgery (Fig. 7). We further confirmed that the c-kit-positive cell population in the heart was composed primarily of cardiac progenitor cells by dual staining with c-kit and CD45. As shown in Fig. 8, >90% of the c-kit-positive cells were CD45 negative, indicating that they are not of hematopoietic origin.
Fig. 6.

PNx activates the mammalian target of rapamycin pathway in WT mice but not in α1+/− mice. Heart tissue homogenates from experimental mice were analyzed for Na/K-ATPase α1 (NKA)-content, phosphorylation of Akt (pAkt) at threonine-308, pAkt at serine-473, and phosphorylation of S6 ribosomal protein (pS6) using Western blot analysis. Top, right: Western blots. tAkt, total Akt. A–D: respective quantification data of Na/K-ATPase α1 (A), pS6 (B), pAkt Thr308 (C), and pAkt Ser473 (D) taken from 8 animals in each group. *P < 0.05, significant difference vs. WT control; !P < 0.05, PNx α1+/− significantly different than sham α1+/−; #P < 0.01, significant difference between PNx WT and PNx α1+/−.
Fig. 7.

Heart tissues exhibit more c-kit-positive and ki-67-positive cells in α1+/− mice vs. WT mice. Left ventricle tissues fixed in 4% formaldehyde solution were used for immunofluorescent staining with anti-c-kit antibody and anti-ki-67 antibody as described in materials and methods. Top: representative images of c-kit (red) and ki-67 (green) staining. DIC, differential interference contrast. Bottom: quantification data from 5–7 animals from each group. *P < 0.05 and **P < 0.01 vs. WT sham. Scale bar is 25 μm.
Fig. 8.

Coimmunostaining of CD45 and c-kit in heart tissue. Left ventricle tissues fixed in 4% formaldehyde solution were used for immunofluorescent staining with anti-c-kit antibody and anti-CD45 antibody as described in materials and methods. Data show that over 90% of c-kit-positive cells are CD45 negative, indicating that most of the c-kit cells are not of hematopoietic origin. Scale bar is 25 μm.
The cytokine SCF binds to and initiates c-kit cell expansion and movement (35, 50). To evaluate whether the reduction in Na/K-ATPase α1-subunit and/or PNx altered heart tissue SCF levels, we used Western blot analysis to probe for SCF in cardiac tissues isolated from the experimental animals. As shown in Fig. 9A, cardiac tissue expression of SCF significantly increases with PNx in α1+/− animals but only slightly in WT animals. Additionally, tissue level expression of SCF is higher in the sham-operated α1+/− animals versus the WT sham-operated animals, but this higher level failed to reach the threshold for significance (Fig. 9A). Locally, the tissue level expression of SCF increased overall in the α1+/− animals with a significant increase occurring between PNx- and sham-operated animals (P < 0.01).
Fig. 9.
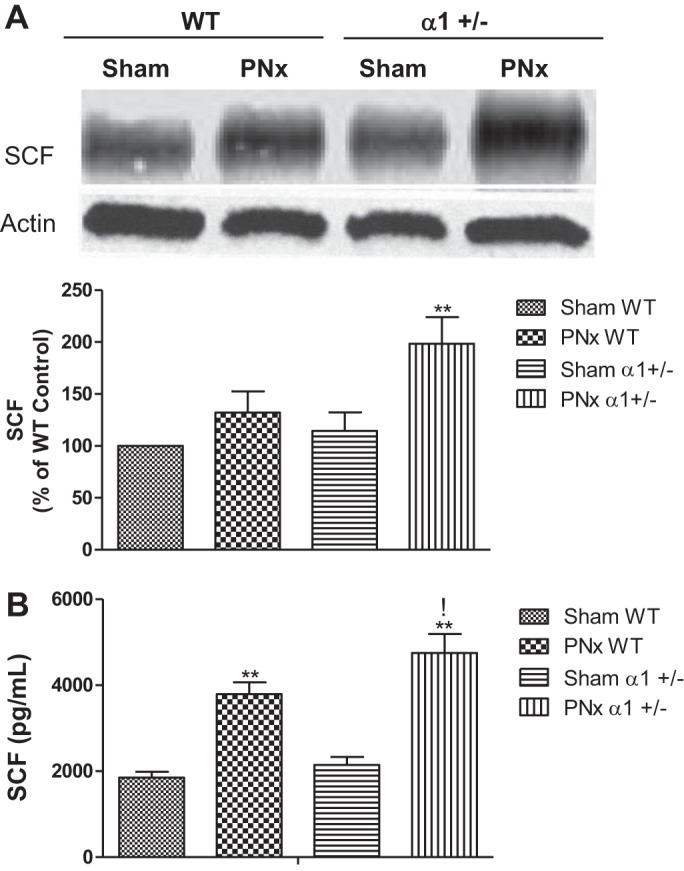
Stem cell factor (SCF) in heart tissue and in plasma. For SCF expression in heart tissue, left ventricle homogenates from experimental mice were analyzed using Western blot analysis, whereas the SCF level in plasma was measured using an ELISA kit from Abcam. A: representative Western blots of heart tissue SCF (top) and the quantification data (bottom). B: concentration of SCF in plasma collected after euthanasia from mice subjected to sham or PNx surgeries. **P < 0.01, PNx vs. sham; !P < 0.05, PNx α1+/− vs. PNx WT.
Circulating levels of SCF were also measured using a commercially available ELISA. As shown in Fig. 9B, there is a significant increase in the plasma levels of SCF with PNx in both WT and α1+/− animals (P < 0.01). Additionally, the level of SCF that is observed in α1+/− animals subjected to PNx was 80% higher than α1+/− sham-operated animals and 25% higher than the SCF levels observed in WT PNx-operated animals.
DISCUSSION
A major finding of this study is that reduction of the Na/K-ATPase α1-subunit results in increased cardiac cell death in PNx mice while also increasing cardiac progenitor cell abundance. Our data also suggest that reduction of this signaling subunit cause differential mechanisms of hypertrophy after PNx. In WT animals, hypertrophy is mostly attributed to the enlargement of individual cardiomyocytes, whereas when the α1-subunit of Na/K-ATPase is reduced, cardiac hypertrophy is more attributable to increases in cardiac cell number. Since Na/K-ATPase reduction is common in heart failure patients (2), it is clinically significant to further define the mechanisms regulating the balance between cardiac cell death and regeneration.
LV hypertrophy (23) and dilation (12, 34) are both prevalent in patients starting therapy for end-stage renal disease (ESRD), and both are markers of poor prognoses in dialysis patients (13, 41, 42, 49). We and others have demonstrated that Na/K-ATPase and its ligands are important regulators of cell survival and growth, which may contribute to hypertrophic growth or dilatation of the heart (20, 24, 51, 54). In our previous study using heterozygous knockout mice, we observed that genetic reduction of Na/K-ATPase stimulates the expression of proapoptotic proteins and potentiates CTS-induced cardiac cell death. It also caused decreased contractile function in these mice. More importantly, our results revealed that in normal cells, there exists a self-protective mechanism that preserves the membrane abundance of Na/K-ATPase and protects cells from death. Reduction of Na/K-ATPase attenuates the signaling function related with this self-protection (33). The current study further demonstrates that in chronic kidney disease induced by PNx, there was increased cardiac cell death and an attenuation of survival signaling when Na/K-ATPase is reduced. These data suggest that Na/K-ATPase amount and the receptor's related signaling are important regulators of organ health in UrC.
On the other hand, our data indicate that PNx induces apoptosis in heart tissue in α1+/− mice, but the heart size and weight in α1+/− mice after PNx surgery are increased in a degree close to their WT littermates. In terms of mechanisms, our current study suggests that cardiac hypertrophy may be attributable to different processes in WT and α1+/− mice, e.g., enlargement of cell size dominates in WT animals subjected to PNx, whereas hyperplasia is increased in α1+/− mice. However, because of technical limitations, it is difficult to identify whether increases in cell numbers result from myocyte proliferation exclusively and what portion of hyperplasia is the result of proliferation of other types of cells in the heart. Our data revealed that both the number of nuclei that colocalized with the myocyte marker cTnI and the number of those outside cTnI staining are increased in α1-knockdown mice after PNx. Since the border of individual myocytes was not very clear even with cTnI staining, the nuclei that overlaid with cTnI may contain other cell types (i.e., cells infiltrate in between muscle fibers) and does not represent an exact number of myocytes. In addition, we also found that PNx causes increased cardiac fibrosis, indicating an increase in fibroblast cells. Taken together, these results indicate that both myocytes and other types of cells such as fibroblasts are increased after PNx surgery and may contribute to the increase in cardiac hypertrophy, whereas the mechanism behind this merits further studies. These results also led us to reason that reduction of Na/K-ATPase in α1+/− mice may result in enhanced regeneration of the myocardium. Indeed, we found that α1+/− mice exhibit more cardiac stem cells, indicated by c-kit-positive and CD45-negative staining compared with the WT mice. PNx further increases the c-kit cell number in α1+/− mice but not in WT mice. PNx also increases c-kit cell proliferation in α1+/− mice, indicated by ki-67 expression. Since the c-kit marker only appears in early stage progenitor cells and disappears after differentiation, a reliable method tracing the fate of c-kit is still debated. Ellison et al. (8) demonstrated that c-kit cells are necessary and sufficient for functional cardiac regeneration and repair using rodent models of diffuse myocardial damage. However, Senyo et al. (48) used a pulse-chase method with stable isotope labeling and concluded that the division of preexisting cardiomyocytes rather than stem cells are the source for new heart cells. Our study observed increased cardiac cell numbers along with increased c-kit cells in Na/K-ATPase-deficient mice with PNx. Although no direct evidence showed that the increased c-kit cells differentiate into cardiac cells, it is conceivable that regenerative processes are activated in the α1+/− mice with PNx. This is further supported by increased total cell number in the heart tissue from these animals. Although our data did not directly demonstrate the increased cell number is originated from c-kit cells, it indicates that hyperplasia may be involved in cardiac hypertrophic growth in the α1+/− mice, which is different from the mechanism that causes hypertrophy in WT mice.
It is noted that Na/K-ATPase reduction potentiates myocyte death but promotes c-kit cell abundance. The mechanism of this differential regulation on different cell type is not clear. Since c-kit cells were reported to be surrounded by niches (10, 45), it is possible that the proliferation of c-kit cells was a second consequence which resulted from increased myocyte death. Previous reports have shown that acute myocardial infarction increases SCF expression in heart tissue, which induces c-kit cell proliferation and movement toward the injured area (32, 55). SCF is well documented to stimulate c-kit cells and regulate regenerative processes during heart tissue damage (22, 56). SCF is expressed in cardiac cells in two forms: the transmembrane form and soluble form found in the cytosol (1). It can also be secreted into blood and circulate to and penetrate other tissues. The molecular mechanism that regulates SCF secretion in injured tissue is not fully understood. However, hypoxic stress (28) and activation of the Akt pathway (15, 16) have been reported to increase SCF expression and secretion in mesenchymal stem cells. We measured SCF levels in LV tissue as well as in blood samples from these experimental mice. The results indicate that the level of SCF in heart tissue or in blood samples in α1+/− PNx mice is significantly higher than that of the sham-operated mice. It is also higher than the WT PNx mice (Fig. 9), which may explain the higher number of c-kit-positive cells in α1+/− mice. However, PNx surgery in the WT mice did not significantly increase the c-kit-positive cells despite the increase in SCF levels. These results indicate that SCF may not be the only factor that regulates c-kit cell abundance. It is also noted that c-kit cell number is more related with the tissue SCF level than the circulating levels. Taken together, the results show that Na/K-ATPase may be directly involved in the regulation of proliferative pathways in c-kit cells, while a determination of the specific mechanism(s) by which increased c-kit cell number and SCF levels are incurred, merits further studies.
In summary, we reason that the overall balance between survival signaling and apoptotic events in cardiac myocytes affects the fate of these cells in specific conditions of stress, while the balance of tissue injury and regenerative processes determine whether the affected organ can compensate and maintain relatively normal function in different disease states. It appears that reduction of Na/K-ATPase potentiates cell death but may also induce the activation of regenerative procedures in this organ as an adaptive mechanism. More study is needed to determine the specific pathways and signaling molecules that regulate this process.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants R01-HL-105649 (to J. Tian) and R01-HL-109015 (to Z. Xie and J. I. Shapiro).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
C.A.D., S.T.H., J.L., C.J.C., Z.X., J.I.S., and J.T. conception and design of research; C.A.D., M.S., K.L.E., H.S., X.W., S.T.H., and J.T. performed experiments; C.A.D., K.L.E., and J.T. analyzed data; C.A.D., C.J.C., Z.X., J.I.S., and J.T. interpreted results of experiments; C.A.D. and J.T. prepared figures; C.A.D. and J.T. drafted manuscript; C.A.D., S.T.H., J.L., C.J.C., Z.X., J.I.S., and J.T. edited and revised manuscript; C.A.D. and J.T. approved final version of manuscript.
REFERENCES
- 1.Anderson DM, Williams DE, Tushinski R, Gimpel S, Eisenman J, Cannizzaro LA, Aronson M, Croce CM, Huebner K, Cosman D, Lyman SD. Alternate splicing of mRNAs encoding human mast cell growth factor and localization of the gene to chromosome 12q22-q24. Cell Growth Differ 2: 373–378, 1991 [PubMed] [Google Scholar]
- 2.Bagrov AY, Shapiro JI, Fedorova OV. Endogenous cardiotonic steroids: physiology, pharmacology, and novel therapeutic targets. Pharmacol Rev 61: 9–38, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal-Ginard B, Anversa P. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 114: 763–776, 2003 [DOI] [PubMed] [Google Scholar]
- 4.Bolli R, Chugh AR, D'Amario D, Loughran JH, Stoddard MF, Ikram S, Beache GM, Wagner SG, Leri A, Hosoda T, Sanada F, Elmore JB, Goichberg P, Cappetta D, Solankhi NK, Fahsah I, Rokosh DG, Slaughter MS, Kajstura J, Anversa P. Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): initial results of a randomised phase 1 trial. Lancet 378: 1847–1857, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5.Diwan A, Dorn GW., 2nd Decompensation of cardiac hypertrophy: cellular mechanisms and novel therapeutic targets. Physiology (Bethesda) 22: 56–64, 2007 [DOI] [PubMed] [Google Scholar]
- 6.Dries DL, Exner DV, Domanski MJ, Greenberg B, Stevenson LW. The prognostic implications of renal insufficiency in asymptomatic and symptomatic patients with left ventricular systolic dysfunction. J Am Coll Cardiol 35: 681–689, 2000 [DOI] [PubMed] [Google Scholar]
- 7.Elkareh J, Kennedy DJ, Yashaswi B, Vetteth S, Shidyak A, Kim EG, Smaili S, Periyasamy SM, Hariri IM, Fedorova L, Liu J, Wu L, Kahaleh MB, Xie Z, Malhotra D, Fedorova OV, Kashkin VA, Bagrov AY, Shapiro JI. Marinobufagenin stimulates fibroblast collagen production and causes fibrosis in experimental uremic cardiomyopathy. Hypertension 49: 215–224, 2007 [DOI] [PubMed] [Google Scholar]
- 8.Ellison GM, Vicinanza C, Smith AJ, Aquila I, Leone A, Waring CD, Henning BJ, Stirparo GG, Papait R, Scarfo M, Agosti V, Viglietto G, Condorelli G, Indolfi C, Ottolenghi S, Torella D, Nadal-Ginard B. Adult c-kit(pos) cardiac stem cells are necessary and sufficient for functional cardiac regeneration and repair. Cell 154: 827–842, 2013 [DOI] [PubMed] [Google Scholar]
- 9.Ezekowitz J, McAlister FA, Humphries KH, Norris CM, Tonelli M, Ghali WA, Knudtson ML. The association among renal insufficiency, pharmacotherapy, and outcomes in 6,427 patients with heart failure and coronary artery disease. J Am Coll Cardiol 44: 1587–1592, 2004 [DOI] [PubMed] [Google Scholar]
- 10.Feng Y, Yu XY, Wang Y. Recent concepts for the roles of progenitor/stem cell niche in heart repair. Am J Cardiovasc Dis 2: 75–83, 2012 [PMC free article] [PubMed] [Google Scholar]
- 11.Foley RN. Clinical epidemiology of cardiac disease in dialysis patients: left ventricular hypertrophy, ischemic heart disease, and cardiac failure. Semin Dial 16: 111–117, 2003 [DOI] [PubMed] [Google Scholar]
- 12.Foley RN, Parfrey PS, Harnett JD, Kent GM, Martin CJ, Murray DC, Barre PE. Clinical and echocardiographic disease in patients starting end-stage renal disease therapy. Kidney Int 47: 186–192, 1995 [DOI] [PubMed] [Google Scholar]
- 13.Foley RN, Parfrey PS, Harnett JD, Kent GM, Murray DC, Barre PE. The prognostic importance of left ventricular geometry in uremic cardiomyopathy. J Am Soc Nephrol 5: 2024–2031, 1995 [DOI] [PubMed] [Google Scholar]
- 14.Gao S, Ho D, Vatner DE, Vatner SF. Echocardiography in mice. Curr Protoc Mouse Biol 1: 71–83, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gnecchi M, He H, Liang OD, Melo LG, Morello F, Mu H, Noiseux N, Zhang L, Pratt RE, Ingwall JS, Dzau VJ. Paracrine action accounts for marked protection of ischemic heart by Akt-modified mesenchymal stem cells. Nat Med 11: 367–368, 2005 [DOI] [PubMed] [Google Scholar]
- 16.Gnecchi M, He H, Noiseux N, Liang OD, Zhang L, Morello F, Mu H, Melo LG, Pratt RE, Ingwall JS, Dzau VJ. Evidence supporting paracrine hypothesis for Akt-modified mesenchymal stem cell-mediated cardiac protection and functional improvement. FASEB J 20: 661–669, 2006 [DOI] [PubMed] [Google Scholar]
- 17.Haller ST, Kennedy DJ, Shidyak A, Budny GV, Malhotra D, Fedorova OV, Shapiro JI, Bagrov AY. Monoclonal antibody against marinobufagenin reverses cardiac fibrosis in rats with chronic renal failure. Am J Hypertens 25: 690–696, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamlyn JM, Manunta P. Ouabain, digitalis-like factors and hypertension. J Hypertens Suppl 10: S99–S111, 1992 [PubMed] [Google Scholar]
- 19.Harada H, Andersen JS, Mann M, Terada N, Korsmeyer SJ. p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc Natl Acad Sci USA 98: 9666–9670, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haux J, Klepp O, Spigset O, Tretli S. Digitoxin medication and cancer; case control and internal dose-response studies. BMC Cancer 1: 11, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hierlihy AM, Seale P, Lobe CG, Rudnicki MA, Megeney LA. The post-natal heart contains a myocardial stem cell population. FEBS Lett 530: 239–243, 2002 [DOI] [PubMed] [Google Scholar]
- 22.Hosoda T. C-kit-positive cardiac stem cells and myocardial regeneration. Am J Cardiovasc Dis 2: 58–67, 2012 [PMC free article] [PubMed] [Google Scholar]
- 23.Huting J, Kramer W, Schutterle G, Wizemann V. Analysis of left-ventricular changes associated with chronic hemodialysis. A noninvasive follow-up study. Nephron 49: 284–290, 1988 [DOI] [PubMed] [Google Scholar]
- 24.Jing Y, Ohizumi H, Kawazoe N, Hashimoto S, Masuda Y, Nakajo S, Yoshida T, Kuroiwa Y, Nakaya K. Selective inhibitory effect of bufalin on growth of human tumor cells in vitro: association with the induction of apoptosis in leukemia HL-60 cells. Jpn J Cancer Res 85: 645–651, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kashkin VA, Bagrov AY, Fedorova OV, Bagrov YY, Agalakova NI, Patkina NA, Zvartau EE. Marinobufagenin (MBG) suppression of ethanol-seeking behavior is associated with inhibition of brain cortex Na/K-ATPase in mice. Eur Neuropsychopharmacol 12: 217–223, 2002 [DOI] [PubMed] [Google Scholar]
- 26.Kennedy DJ, Elkareh J, Shidyak A, Shapiro AP, Smaili S, Mutgi K, Gupta S, Tian J, Morgan E, Khouri S, Cooper CJ, Periyasamy SM, Xie Z, Malhotra D, Fedorova OV, Bagrov AY, Shapiro JI. Partial nephrectomy as a model for uremic cardiomyopathy in the mouse. Am J Physiol Renal Physiol 294: F450–F454, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kennedy DJ, Vetteth S, Periyasamy SM, Kanj M, Fedorova L, Khouri S, Kahaleh MB, Xie Z, Malhotra D, Kolodkin NI, Lakatta EG, Fedorova OV, Bagrov AY, Shapiro JI. Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension 47: 488–495, 2006 [DOI] [PubMed] [Google Scholar]
- 28.Kinnaird T, Stabile E, Burnett MS, Lee CW, Barr S, Fuchs S, Epstein SE. Marrow-derived stromal cells express genes encoding a broad spectrum of arteriogenic cytokines and promote in vitro and in vivo arteriogenesis through paracrine mechanisms. Circ Res 94: 678–685, 2004 [DOI] [PubMed] [Google Scholar]
- 29.Komiyama Y, Dong XH, Nishimura N, Masaki H, Yoshika M, Masuda M, Takahashi H. A novel endogenous digitalis, telocinobufagin, exhibits elevated plasma levels in patients with terminal renal failure. Clin Biochem 38: 36–45, 2005 [DOI] [PubMed] [Google Scholar]
- 30.Laugwitz KL, Moretti A, Lam J, Gruber P, Chen Y, Woodard S, Lin LZ, Cai CL, Lu MM, Reth M, Platoshyn O, Yuan JX, Evans S, Chien KR. Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature 433: 647–653, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levey AS, Beto JA, Coronado BE, Eknoyan G, Foley RN, Kasiske BL, Klag MJ, Mailloux LU, Manske CL, Meyer KB, Parfrey PS, Pfeffer MA, Wenger NK, Wilson PW, Wright JT., Jr Controlling the epidemic of cardiovascular disease in chronic renal disease: what do we know? What do we need to learn? Where do we go from here? National Kidney Foundation Task Force on Cardiovascular Disease. Am J Kidney Dis 32: 853–906, 1998 [DOI] [PubMed] [Google Scholar]
- 32.Limana F, Germani A, Zacheo A, Kajstura J, Di Carlo A, Borsellino G, Leoni O, Palumbo R, Battistini L, Rastaldo R, Muller S, Pompilio G, Anversa P, Bianchi ME, Capogrossi MC. Exogenous high-mobility group box 1 protein induces myocardial regeneration after infarction via enhanced cardiac C-kit+ cell proliferation and differentiation. Circ Res 97: e73–e83, 2005 [DOI] [PubMed] [Google Scholar]
- 33.Liu C, Bai Y, Chen Y, Wang Y, Sottejeau Y, Liu L, Li X, Lingrel JB, Malhotra D, Cooper CJ, Shapiro JI, Xie ZJ, Tian J. Reduction of Na/K-ATPase potentiates marinobufagenin-induced cardiac dysfunction and myocyte apoptosis. J Biol Chem 287: 16390–16398, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.London GM, Fabiani F, Marchais SJ, de Vernejoul MC, Guerin AP, Safar ME, Metivier F, Llach F. Uremic cardiomyopathy: an inadequate left ventricular hypertrophy. Kidney Int 31: 973–980, 1987 [DOI] [PubMed] [Google Scholar]
- 35.Magenta A, Avitabile D, Pompilio G, Capogrossi MC. c-kit-Positive cardiac progenitor cells: the heart of stemness. Circ Res 112: 1202–1204, 2013 [DOI] [PubMed] [Google Scholar]
- 36.Makkar RR, Smith RR, Cheng K, Malliaras K, Thomson LE, Berman D, Czer LS, Marban L, Mendizabal A, Johnston PV, Russell SD, Schuleri KH, Lardo AC, Gerstenblith G, Marban E. Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. Lancet 379: 895–904, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McClellan WM, Flanders WD, Langston RD, Jurkovitz C, Presley R. Anemia and renal insufficiency are independent risk factors for death among patients with congestive heart failure admitted to community hospitals: a population-based study. J Am Soc Nephrol 13: 1928–1936, 2002 [DOI] [PubMed] [Google Scholar]
- 38.Middleton RJ, Parfrey PS, Foley RN. Left ventricular hypertrophy in the renal patient. J Am Soc Nephrol 12: 1079–1084, 2001 [DOI] [PubMed] [Google Scholar]
- 39.Moseley AE, Cougnon MH, Grupp IL, El Schultz J, Lingrel JB. Attenuation of cardiac contractility in Na,K-ATPase alpha1 isoform-deficient hearts under reduced calcium conditions. J Mol Cell Cardiol 37: 913–919, 2004 [DOI] [PubMed] [Google Scholar]
- 40.Norgaard A, Bagger JP, Bjerregaard P, Baandrup U, Kjeldsen K, Thomsen PE. Relation of left ventricular function and Na,K-pump concentration in suspected idiopathic dilated cardiomyopathy. Am J Cardiol 61: 1312–1315, 1988 [DOI] [PubMed] [Google Scholar]
- 41.Parfrey PS, Griffiths SM, Harnett JD, Taylor R, King A, Hand J, Barre PE. Outcome of congestive heart failure, dilated cardiomyopathy, hypertrophic hyperkinetic disease, and ischemic heart disease in dialysis patients. Am J Nephrol 10: 213–221, 1990 [DOI] [PubMed] [Google Scholar]
- 42.Parfrey PS, Harnett JD, Griffiths SM, Taylor R, Hand J, King A, Barre PE. The clinical course of left ventricular hypertrophy in dialysis patients. Nephron 55: 114–120, 1990 [DOI] [PubMed] [Google Scholar]
- 43.Periyasamy SM, Liu J, Tanta F, Kabak B, Wakefield B, Malhotra D, Kennedy DJ, Nadoor A, Fedorova OV, Gunning W, Xie Z, Bagrov AY, Shapiro JI. Salt loading induces redistribution of the plasmalemmal Na/K-ATPase in proximal tubule cells. Kidney Int 67: 1868–1877, 2005 [DOI] [PubMed] [Google Scholar]
- 44.Saltzman HE, Sharma K, Mather PJ, Rubin S, Adams S, Whellan DJ. Renal dysfunction in heart failure patients: what is the evidence? Heart Fail Rev 12: 37–47, 2007 [DOI] [PubMed] [Google Scholar]
- 45.Sanada F, Kim J, Czarna A, Chan NY, Signore S, Ogorek B, Isobe K, Wybieralska EW, Borghetti G, Pesapane A, Sorrentino A, Mangano EN, Cappetta D, Mangiaracina CM, Ricciardi M, Cimini M, Ifedigbo E, Perrella MA, Goichberg P, Choi AM, Kajstura J, Hosoda T, Rota M, Anversa P, Leri A. c-Kit-positive cardiac stem cells nested in hypoxic niches are activated by stem cell factor reversing the aging myopathy. Circ Res 114: 41–55, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schoner W, Scheiner-Bobis G. Endogenous and exogenous cardiac glycosides: their roles in hypertension, salt metabolism, and cell growth. Am J Physiol Cell Physiol 293: C509–C536, 2007 [DOI] [PubMed] [Google Scholar]
- 47.Semb SO, Lunde PK, Holt E, Tonnessen T, Christensen G, Sejersted OM. Reduced myocardial Na+, K+-pump capacity in congestive heart failure following myocardial infarction in rats. J Mol Cell Cardiol 30: 1311–1328, 1998 [DOI] [PubMed] [Google Scholar]
- 48.Senyo SE, Steinhauser ML, Pizzimenti CL, Yang VK, Cai L, Wang M, Wu TD, Guerquin-Kern JL, Lechene CP, Lee RT. Mammalian heart renewal by pre-existing cardiomyocytes. Nature 493: 433–436, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Silberberg JS, Barre PE, Prichard SS, Sniderman AD. Impact of left ventricular hypertrophy on survival in end-stage renal disease. Kidney Int 36: 286–290, 1989 [DOI] [PubMed] [Google Scholar]
- 50.Sturzu AC, Wu SM. Developmental and regenerative biology of multipotent cardiovascular progenitor cells. Circ Res 108: 353–364, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tian J, Li X, Liang M, Liu L, Xie JX, Ye Q, Kometiani P, Tillekeratne M, Jin R, Xie Z. Changes in sodium pump expression dictate the effects of ouabain on cell growth. J Biol Chem 284: 14921–14929, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Urbanek K, Quaini F, Tasca G, Torella D, Castaldo C, Nadal-Ginard B, Leri A, Kajstura J, Quaini E, Anversa P. Intense myocyte formation from cardiac stem cells in human cardiac hypertrophy. Proc Natl Acad Sci USA 100: 10440–10445, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Urbanek K, Torella D, Sheikh F, De Angelis A, Nurzynska D, Silvestri F, Beltrami CA, Bussani R, Beltrami AP, Quaini F, Bolli R, Leri A, Kajstura J, Anversa P. Myocardial regeneration by activation of multipotent cardiac stem cells in ischemic heart failure. Proc Natl Acad Sci USA 102: 8692–8697, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamada K, Hino K, Tomoyasu S, Honma Y, Tsuruoka N. Enhancement by bufalin of retinoic acid-induced differentiation of acute promyelocytic leukemia cells in primary culture. Leuk Res 22: 589–595, 1998 [DOI] [PubMed] [Google Scholar]
- 55.Yaniz-Galende E, Chen J, Chemaly E, Liang L, Hulot JS, McCollum L, Arias T, Fuster V, Zsebo KM, Hajjar RJ. Stem cell factor gene transfer promotes cardiac repair after myocardial infarction via in situ recruitment and expansion of c-kit+ cells. Circ Res 111: 1434–1445, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao LR, Berra HH, Duan WM, Singhal S, Mehta J, Apkarian AV, Kessler JA. Beneficial effects of hematopoietic growth factor therapy in chronic ischemic stroke in rats. Stroke 38: 2804–2811, 2007 [DOI] [PubMed] [Google Scholar]