

Highlights
-
•
miR-34a was decreased in both glioma and glioma stem cell-lines as compared to normal brain tissues.
-
•
Glioma stem cell-lines HNGC-2 and NSG-K16 possess the mesenchymal glioblastoma phenotype.
-
•
miR-34a over-expression in these cell lines decreased their proliferative and migratory potential, and induced apoptosis.
-
•
Rictor, a part of the mTORC2 complex, is a novel target for miR-34a in glioma stem cells.
-
•
The tumor suppressive function of miR-34a is mediated via Rictor and affects the AKT/mTOR pathway and Wnt signaling.
Abbreviations: bFGF, basic fibroblast growth factor; CNS, central nervous system; EGF, epidermal growth factor; EMT, epithelial–mesenchymal transition; EV, empty vector; GBM, glioblastoma multiforme; GIC, glioma initiating cell; GSC, glioma stem cell; GSK-3β, glycogen synthase kinase 3β; NOD/SCID, nonobese diabetic/severe combined immunodeficiency; PARP, poly ADP-ribose polymerases; PDGFRA, platelet-derived growth factor receptor-α; qRT-PCR, quantitative real time PCR; TCGA, the cancer genome atlas database
Keywords: Glioblastoma, Mesenchymal, Beta-catenin, Rictor, Heterogeneity
Abstract
MiRNA-34a is considered as a potential prognostic marker for glioma, as studies suggest that its expression negatively correlates with patient survival in grade III and IV glial tumors. Here, we show that expression of miR-34a was decreased in a graded manner in glioma and glioma stem cell-lines as compared to normal brain tissues. Ectopic expression of miR-34a in glioma stem cell-lines HNGC-2 and NSG-K16 decreased the proliferative and migratory potential of these cells, induced cell cycle arrest and caused apoptosis. Notably, the miR-34a glioma cells formed significantly smaller xenografts in immuno-deficient mice as compared with control glioma stem cell-lines. Here, using a bioinformatics approach and various biological assays, we identify Rictor, as a novel target for miR-34a in glioma stem cells. Rictor, a defining component of mTORC2 complex, is involved in cell survival signaling. mTORC2 lays downstream of Akt, and thus is a direct activator of Akt. Our earlier studies have elaborated on role of Rictor in glioma invasion (Das et al., 2011). Here, we demonstrate that miR34a over-expression in glioma stem cells profoundly decreased levels of p-AKT (Ser473), increased GSK-3β levels and targeted for degradation β-catenin, an important mediator of Wnt signaling pathway. This led to diminished levels of the Wnt effectors cyclin D1 and c-myc. Collectively, we show that the tumor suppressive function of miR-34a in glioblastoma is mediated via Rictor, which through its effects on AKT/mTOR pathway and Wnt signaling causes pronounced effects on glioma malignancy.
1. Introduction
Gliomas are the most common tumors of central nervous system (CNS). Amongst the different grades of glioma, glioblastoma multiforme (GBM), classified by World Health Organization (WHO) as grade IV glioma is particularly aggressive, infiltrative and vascularized tumor with patients showing a one year median survival. Major challenges in GBM therapy are associated with location of the disease and high level of inter- and intra-tumoral heterogeneity, leading to accumulation of several genetic changes over time [1]. The heterogeneity possibly is an outcome of existence of hierarchy of subpopulations of tumorigenic cancer stem cells and their non-tumorigenic progeny [2]. The cell of origin for glioma is considered to be a neural stem cell with propensity to be transformed into a glioma stem cell (GSC) also referred to as a glioma initiating cell (GIC) [3]. This transformation to a glioma stem cell state involves aberrant activation of signaling pathways along with multiplicity of abnormal events at genetic and epigenetic levels including alterations in class of non-coding RNAs known as microRNAs.
MicroRNAs (miRNAs) are of 21–25 nucleotides size and are involved in post-transcriptional control of gene expression [4]. They regulate protein synthesis by base pairing to partially complementary sequences in the 3′ un-translated regions (UTRs) of target mRNAs leading to gene repression. Essentially, miRNAs execute their function in a dynamic and context-dependent manner by targeting diverse downstream target genes from transcriptional factors to epigenetic regulators. In GBM, several miRNAs have either oncogenes or tumor suppressor genes as their targets. A comprehensive overview on the expression profiles and functions of miRNAs in GBM specify that compared to normal brain tissues about 253 miRNAs are up-regulated, about 95 are down-regulated and 17 show disputed status. Importantly, about 85% of these miRNAs are not yet functionally characterized [5]. The overexpressed miRNAs include few like miR-17, miR-21, miR-93, and miRNAs – 221/222 that influence processes like proliferation, in vivo tumor growth, invasiveness and angiogenesis. The miRNAs with growth suppressive properties that are down-regulated in GBM include miR-7, miR-45, miR-29b, miR-101, miR-124, miR-145 and miR-34a [6–8].
MicroRNA-34a is mapped to a region of chromosome 1p36.23 in human and shows deviant expression in multiple cancer types like neuroblastoma [9,10], colon cancer [11], prostate [12] and pancreatic cancer [13]. It is shown to be a transcriptional target and validated component of the p53 tumor suppressor network and a legit tumor suppressor for glioma [14]. Studies showed that higher miR-34a levels were associated with wild-type p53 tumors possessing lower Bcl-2 expression levels than in tissues with lower miR-34a expression [15]. The role of miR-34a as a tumor suppressive RNA was demonstrated for glioma stem cells with Notch1/2 and c-Met as its functional targets. Recently, Musashi-1 and platelet-derived growth factor receptor-α (PDGFRA) [16,17] were identified as miR-34a targets and hence miR-34a loss in GBM was considered responsible for increased PDGF signaling.
The miRNA expression signatures both characterize and contribute to the phenotypic diversity of glioblastoma subclasses. Recent work on genome wide profiling with help of the cancer genome atlas (TCGA) [18] database, using various parameters like copy number analysis, miRNA and mRNA analysis, mutational and methylation analysis, have all led to generation of GBM tumor subtype specific network profiles [19–21]. These sub-types are classical, mesenchymal, neural, and pro-neural. Amongst these four subtypes, the tumors with mesenchymal GBM subtype are aggressive in nature and negatively correlate with patient survival [22]. Several studies have identified microRNAs as potent regulators of subclass-specific gene expression networks in glioblastoma [23]. They serve as important determinants of glioblastoma subclasses through their ability to regulate developmental growth and differentiation programs in several transformed neural precursor cell-types.
In our previous studies, we reported molecular mechanisms for transformation of non-tumorigenic neural stem cell-line HNGC-1 to tumorigenic glioma cancer stem cell line HNGC-2 [24]. Using this cell system we identified differentially expressed miRNAs that were specifically altered during the transformation event. Previously, we demonstrated role of miR-145 as a tumor suppressor in GBM [8]. In this report, we have characterized glioma stem cell-lines – HNGC-2 and NSG-K16 as belonging to the mesenchymal sub-type and shown that miR-34a possesses tumor suppressive function for this glioma sub-type. More importantly, we have identified Rictor, a component of the mTORC2 complex, as a novel target for miR-34a and established that its over-expression contributes to the oncogenic properties of this malignancy. Next, we show that Rictor by inducing AKT phosphorylation inhibits GSK3β activity leading to nuclear activation of β-catenin followed by activation of Wnt signaling pathway. The enhanced tumorigenic potential and invasiveness of glioma stem cells is thereby mainly contributed through activation of Akt and Wnt pathways caused due to loss of miR-34a.
2. Materials and methods
2.1. Tissue samples and clinical data
This study was approved by the Institutional Ethics Committee (IEC) of National Centre for Cell Science (NCCS), Pune, India and KEM Hospital, Mumbai, India. Human glioma tissue samples were collected from KEM Hospital, Mumbai (n = 32) and classified by a neuro-pathologist for respective grades of glioma using WHO criteria (Table S1). The tissue samples comprised of 13 low and 19 high grade glioma tumors as well as normal brain tissues (n = 3) from epileptic resections. Informed consent was sought from the patients for use of tissues in this study. The tumor tissue samples were used for establishment of long term glioma cultures and RNA studies.
2.2. Cell culture
The non-tumorigenic neural stem cell-line HNGC-1 and other glioma stem cell lines HNGC-2 [24] and NSG-K16 [25] were established by us and maintained as described previously. Briefly, HNGC-2 and NSG-K16 cells were cultured in Dulbecco’s modified Eagle’s (DMEM)/Ham’s F12 medium (1:1) (Invitrogen, Carlsbad, CA, USA) with 1× B27 supplement (Invitrogen), epidermal growth factor (EGF) (10 ng/mL) (Invitrogen), basic fibroblast growth factor (bFGF) (20 ng/mL) (Invitrogen), 1× non-essential amino acids (NEAA) (Invitrogen), and 1× Glutamax (Invitrogen) at 37 °C with 5% CO2 in a humidified incubator.
2.3. RNA extraction and quantitative real-time PCR
The tissue samples were homogenized by gentle MACS™ dissociator (MACS Miltenyi Biotec, Auburn, CA, USA) and total RNA was extracted from tissues and cells using Trizol Reagent (Invitrogen) according to manufacturer’s protocol. The RNA concentration was determined with a Biophotometer (Eppendorf, Hamburg, Germany). With help of SuperScript III First strand synthesis system (Invitrogen), 100 ng of total RNA was reverse transcribed into cDNA using miR-34a specific stem-loop primers (Table S2). Expression of mature miR-34a was quantified using SYBR green master mix (Applied Biosystems, Foster City, CA, USA) using 7500 Fast Real Time PCR System (Applied Biosystems). The amplification reaction (10 μL) consisted of 2× SYBR green master mix, gene-specific primers and cDNA. Fold changes in gene expression were calculated using 2−ΔΔCt method with 18S rRNA serving as an internal control.
2.4. Vectors and transfections
Mir-34a stable clones were generated in both HNGC-2 and NSG-K16 cell-lines by stable transfection using Lipofectamine 2000 (Invitrogen) according to manufacturer’s protocol. The level of miR-34a expression was detected in stable miR-34 over-expressing HNGC-2 and NSG-K16 cell-lines. For luciferase assays, 3′UTR of Rictor was amplified using gene specific primers (Table S2). The fragment spanning putative binding site for miR-34a was cloned into luciferase plasmid, pMIR-REPORT miRNA Expression Reporter Vector (Life Technologies, Grand Island, NY, USA). The specificity of the 3′UTR Rictor clone was confirmed by DNA sequencing.
2.5. Proliferation assay
Cell proliferation was measured by MTT assay as described previously [26]. Briefly, cells were plated at density of 1 × 103 cells per well in 96 well plate (BD Biosciences, Bedford, MA, USA). Cell growth was analyzed over a period of 5 days. At every 24 h interval, 20 μL of MTT (5 mg/mL) (Sigma–Aldrich) was added to each well and the plate was incubated for 4 h at 37 °C in a CO2 incubator. The Formazan crystals formed were dissolved in dimethyl sulfoxide (DMSO) (Sigma–Aldrich) and absorbance of each well was measured at 570 nm in a Bio-Rad micro-plate reader 680 (Bio-Rad, Hercules, CA, USA).
2.6. Flow cytometry
The cell cycle kinetics of parental and transfected cells was determined using flow cytometry. For this, cells were harvested and washed with cold 1× PBS (Phosphate Buffered Saline) (HiMedia, Mumbai, MH India) buffer and fixed with 75% ethanol for 30 min on ice. Further, cells were washed 3 times with cold 1× PBS and incubated with 50 μg/mL of propidium iodide (PI) (Invitrogen) and 10 μg/mL of DNase-free RNase A in 500 μL of 1× PBS for 30 min at 37 °C. Later, cells were re-suspended in 500 μL of 1× PBS and acquired on FACSCalibur (BD Bioscience). Cells were acquired and DNA histograms were generated and analyzed by CellQuest™ Pro 5.2.1 software (BD Biosciences). For cell surface marker analyses for CD44 expression, cells were harvested from culture and washed in cold 1× PBS buffer and fixed with 75% ethanol for 30 min on ice. Further, cells were washed with cold 1× PBS and blocked with blocking buffer (5% BSA) for 30 min at 4 °C. Cells were incubated with primary antibody against CD44 (Chemicon International Billerica, MA, USA) for 1 h followed by incubation with secondary antibody Alexa Fluor 488 (Invitrogen). The cells were then washed with cold 1× PBS, re-suspended in 500 μL of cold 1× PBS and acquired on FACSCalibur (BD Bioscience). DNA histograms were generated and analyzed by CellQuest™ Pro 5.2.1 software (BD Bioscience).
2.7. Immuno-staining
The parental and transfected cells were seeded in 24 well plates (BD Bioscience) for 48 h. Later, cells were fixed with 3.7% para-formaldehyde (PFA) (Sigma–Aldrich), for 10 min at room temperature followed by permeabilization using 0.01% Triton X-100 (Sigma–Aldrich). Cells were blocked with 5% BSA (Bovine Serum Albumin) and incubated with primary antibody to Ki67 (1:100; Rabbit) (Chemicon International) for 2 h at room temperature followed by incubation with anti-rabbit Alexa Fluor 594 (1:100) (Invitrogen) for 45 min. The nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole) (Sigma–Aldrich) for 10 min and then mounted on slides with help of mounting medium containing 1% DABCO (1,4-diazabicyclo [2.2.2] octane) (Sigma–Aldrich). The immuno-staining was visualized using confocal microscope Leica TCS SP5II (Leica Microsystems CMS, GmbH, Germany).
2.8. Apoptosis assay
Caspase-3, Caspase-9 and PARP (Poly ADP-ribose polymerases) activities were assayed using apoptotic sampler kit (Cell Signaling Technology, Denvers, MA, USA) as per manufacturer’s instructions. Parental and transfected cells were harvested, washed with 1× PBS buffer, fixed, permeabilized using 0.01% Triton X-100 (Sigma–Aldrich) and blocked with blocking buffer (5% Bovine Serum Albumin) for 30 min. Further, cells were individually incubated with primary antibodies to Caspase 3, Caspase 9 and PARP (1:100) for 1 h followed by incubation with anti-Rabbit Alexa Fluor 594 (1:100) (Invitrogen) for 45 min. Finally, cells were resuspended in 0.5 mL of 1× PBS and acquired on FACSCalibur flow cytometer (BD Bioscience). The data was analyzed with Cell Quest Pro analysis software 5.2.1 (BD Bioscience).
2.9. Xenograft mouse model
The animal experiments were approved by Institutional Animal Ethical Committee (IEC) of NCCS and the experimental procedures followed were according to animal ethical guidelines of NCCS. For in vivo tumorigenicity assay, 6–8 weeks old NOD-SCID mice were used. For subcutaneous injections, 1 × 106 cells of both EV cells and miR-34a expressing cells suspended in 50 μL of 1× PBS were injected into the flanks of mice (n = 9). Animals were monitored for tumor development and end point tumor volume was calculated using formula 4/3 π (√ major axis/2 × minor axis/2).
2.10. Invasion assay
Invasion assay was performed using Matrigel Invasion Chambers (BD Biosciences) with inserts of 8 μm pore size in 24 well plates according to manufacturer’s protocol. Briefly, 5 × 103 cells in 0.5 mL of serum free DMEM medium were seeded on insert comprised of Matrigel. After incubation for 24 h, the non-invading cells from upper chamber were removed mechanically and invasive cells were fixed and stained with 0.1% (weight/volume) crystal violet stain (Sigma–Aldrich). Stained cells were counted from 10 different microscopic fields and the images were acquired using Olympus IX51 microscope (Olympus Imaging PA, USA). The data was quantified with Image J 1.47 software.
2.11. Western blot analysis
To extract proteins, cells were harvested and incubated on ice for 20 min with M-PER reagent (Thermo Scientific Rockford, lL, USA) containing 1× protease inhibitor cocktail (Sigma–Aldrich) The cells were centrifuged at 12,000g for 20 min and the supernatant fractions were collected for western blot analysis. Equal amounts of protein were loaded on SDS–PAGE gel and transferred on to PVDF membranes (Millipore). The blots were probed with various primary antibodies: Rictor, CyclinD1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), β-catenin, GSK-3β, p-AKT, AKT, c-Myc (Cell Signaling Technology), and Actin (MP Biomedicals, OH, USA) at 1:1000 dilution followed by incubation with species specific horseradish peroxidase conjugated secondary antibodies (1:2000). The proteins were detected by Super Signal protein detection kit (Thermo Scientific). Target protein levels were normalized with actin and quantified by Image J Software.
2.12. Luciferase assay
To determine if there was a direct interaction of miR-34a with 3′UTR of Rictor, we co-transfected pMIR-REPORT (Applied Biosystem) luciferase vector and pMIR-REPORT containing Rictor 3′UTR in both EV and miR-34a expressing cells. After 48 h of transfection, luciferase activity was measured by using Dual-Glo Luciferase Assay system (Promega, Madison, WI). Luciferase activity was normalized with Renilla activity.
2.13. TOPflash/FOPflash reporter assay
To determine the effects of miR-34a overexpression on Wnt/β-catenin activity, we performed transfection assays with β-catenin/TCF reporter plasmids according to manufacturer’s protocol (Millipore Billerica, MA, USA). Briefly, 1 × 104 cells were transiently transfected with either 2 μg of pTOPflash (TCF Reporter Plasmid) or pFOPflash (mutant, inactive TCF binding site) plasmids (Millipore) and 0.5 μg of pSV40-Renilla plasmid as an internal control (Promega) using Lipofectamine 2000 (Invitrogen) for 48 h. Both Firefly and Renilla luciferase activities were measured with a Glomax luminometer (Promega) using Dual-Glo Luciferase Assay System (Promega) according to manufacturer’s instructions.
2.14. Bioinformatics analysis
Candidate targets of miR-34a were screened from publically available microRNAs targets using TargetScan (http://www.targetscan.org/), miRanda (http://www.microrna.org/), PicTar (pictar.mdc-berlin.de) and miRDB (http://www.mirdb.org) databases. The miR-34a seed sequence and Rictor 3′UTR alignment was generated by miRanda (http://www.microrna.org/). Kaplan–Meier survival plot for Rictor gene expression data was generated by using REMBRANDT (REpository for Molecular BRAin Neoplasia DaTa) [27].
2.15. Statistical analysis
All statistical analysis was done using GraphPad PRISM version 5.0 (GraphPad Software, Inc., La Jolla, CA, USA). All experiments were performed in triplicates. Data is represented as Means ± SEM (Standard Error of Means). Student’s t test was used to calculate statistical significance between two groups. One-way ANOVA was used for multiple group comparisons (∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.0001).
3. Results
3.1. Glioma stem cell-lines HNGC-2 and NSG-K16 display enriched mesenchymal GBM phenotype
The HNGC-2 and NSG-K16 cell-lines developed by us as glioma stem cell-lines were classified into specific GBM sub-types using molecular markers. This analysis was performed by qRT-PCR wherein the expression of various mesenchymal and pro-neural markers was analyzed and compared to their expression in normal brain tissues. Characteristically, both cell-lines displayed positivity for expression of mesenchymal markers like- RELB, Twist1, ZEB2 and COL5A1 (Fig. 1A); and showed absence of expression for pro-neural related genes i.e., PDGFRA and NKX2-2 (Fig. 1B). The CD44 expression in both HNGC-2 and NSG-K16 cell-lines was high as determined by flow cytometry (Fig. 1C) signifying their aggressive nature. On the basis of marker analyses we confirmed that these cell-lines belonged to the mesenchymal GBM sub-type.
Fig. 1.
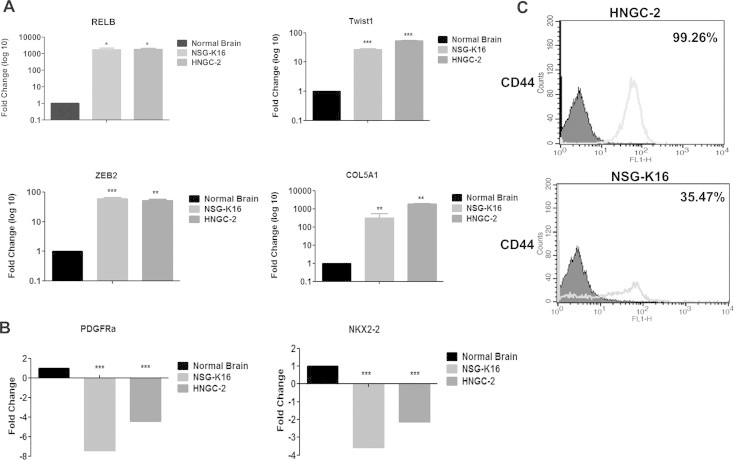
Characterization of glioma stem cell-lines for mesenchymal sub-type. Quantitative Real time PCR (qRT-PCR) analysis for expression of mesenchymal markers RELB, Twist1, ZEB2 and COL5A1 (A) and proneural markers PDGFRa and NKX2-2 (B) in HNGC-2 and NSG-K16 cell-lines. Expression levels were normalized to normal human brain and were set to one. 18S rRNA gene expression served as internal control. Data represents average of three independent experiments. Fold change was calculated by 2−ΔΔCt method. Error bar represents the mean ± SEM. P value was calculated by one way ANOVA and Student’s t test (∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.0001). (C) Flow cytometry analyses for expression of cell surface marker CD44 in HNGC-2 and NSG-K16 cells (representative profile).
3.2. MiR-34a is down-regulated in glioma tumor tissues and glioma cell-lines
We performed stem loop reverse transcription PCR (RT-qPCR) to quantify miR-34a expression in glioma tumor tissues and cell-lines. For this, we compared expression of miR-34a in both low (n = 13) and high grade (n = 19) glial tumors with expression in normal brain tissues (n = 3) (Fig. 2A). A greater than 10-fold decrease in miR-34a levels was demonstrated in both low (P < 0.0001) and high grade glioma tumor tissues (P < 0.0001). A similar pattern of lowered expression of miR-34a was evident in GBM cell lines – HNGC-2 (P = 0.0002) and NSG-K16 (P = 0.0002) (Fig. 2B). To determine, the functional significance of miR-34a in glioma progression, we over-expressed miR-34a in glioma stem cell lines HNGC-2 and NSG-K16 cells and generated miR-34a overexpressing stable cell lines (Fig. 2C). Expectedly, miR-34a was over-expressed in HNGC-2 (P < 0.01) and NSG-K16 (P < 0.01) cells as compared to their respective EV Control cells. Here, EV represents control glioma stem cells transfected with empty vector.
Fig. 2.
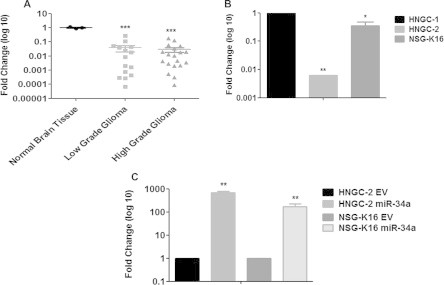
Differential expression of miR-34a in tissues and cell lines. Quantitative Real time PCR (qRT-PCR) analysis of miR-34a expression in (A) low grade (n = 13) and high grade (n = 19) gliomas. Expression levels were normalized to normal human brain and were set to one. (B) Expression in glioma stem cell-lines – HNGC-2 and NSG-K1. Expression levels were normalized to normal human brain and were set to one. (C) HNGC-2 and NSG-K16 cell-lines transfected with miR-34a and empty vector (EV). 18S rRNA served as an internal control. Fold change was calculated by 2−ΔΔCt method. Error bar represents the mean ± SEM of three independent experiments. P value was calculated one way ANOVA and Student’s t test (∗∗P < 0.01, ∗∗∗P < 0.0001).
3.3. MiR-34a inhibits glioma cell proliferation induces cell cycle arrest and apoptosis in vitro
The effects of ectopic over-expression of miR-34a on cell proliferation for glioma stem cell-lines HNGC-2 and NSG-K16 was determined by MTT assay. Overexpression of miR-34 led to inhibition of cell proliferation of both HNGC-2 and NSG-K16 cells as compared to EV control cells (Fig. 3A) analyzed over a 5 day period by MTT assay. Similarly, miR-34a caused decrease in positivity for Ki67 expression in glioma cells as compared to EV cells (Fig. 3B). Cell cycle kinetics using flow cytometry indicated that miR-34a significantly induced cell cycle arrest in both HNGC-2 and NSG-K16 cells (Fig. 3C). The percentages of cells in G0/G1 phase was significantly increased by miR-34a from 55.19 ± 7.4% to 76.48 ± 8.6% in HNGC-2 cells and from 49.03 ± 1.7% to 75.46 ± 5.6% in NSG-K16 cells as compared to their EV counterparts (Fig. 3D). Consequent to G0/G1 arrest, there was an extensive decrease in S phase population by about 2-fold in both HNGC-2 and NSG-K16 cells due to miR-34a (Fig. 3D). The apparent G0/G1 arrest and decrease in S phase population led us to determine possible role of miR-34a in inducing apoptosis. MiR-34a over-expression in both cell-lines caused enhanced apoptosis wherein by flow cytometry we detected presence of activated Caspases 3 and 9 along with cleaved PARP in both HNGC-2 and NSG-K16 (P ⩽ 0.01) cells. The miR-34a overexpression led to distinct peaks representing populations in early and late apoptosis in both cell-lines (Fig. 4A and B), signifying potential of miR-34 to induce apoptosis.
Fig. 3.
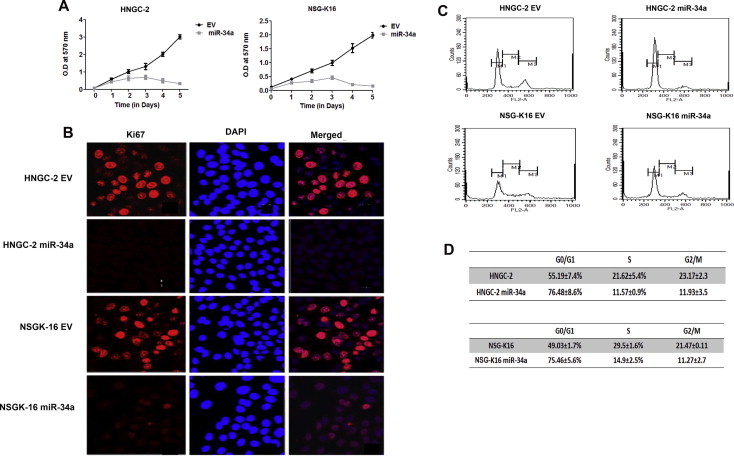
Effects of miR-34a overexpression on cell proliferation and cell cycle. (A) MTT assay of miR-34a over-expressing and EV cells (B) Immuno-staining for proliferation marker Ki67 in miR-34a and EV cells. Nuclei were stained by DAPI (BLUE) stain and Ki67 by AlexFluor 594 (RED). (C) Flow cytometry analysis of EV and miR-34a cells showing distribution of cells in different phases of cell cycle (Upper Panel). (D)The lower panel shows the percentage cells in different phases of cell cycle as represented as means ± SEM.
Fig. 4.
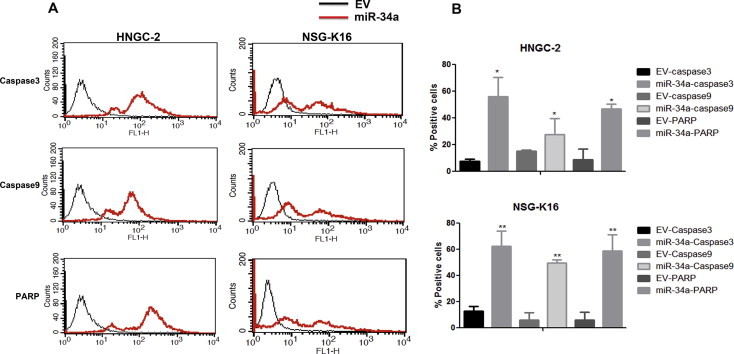
MiR-34a induces caspase dependent apoptosis. (A) HNGC-2 and NSG-K16 cells analyzed for cleaved caspase 3, caspase 9, and PARP by flow cytometry. The peaks for HNGC-2 and NSG-K16 EV cells are depicted by black line graph and for miR-34a glioma cells by red line graph. The multiple peaks in mir-34a expressing cells indicate cells in early and late apoptosis. (B) Quantitative representation of flow cytometry data in (A) showing relative expression of cleaved caspase 3, caspase 9 and PARP in HNGC-2 and NSG-K16 miR-34a transfected and EV control cells. Each bar represents the means ± SEM of 3 independent experiments. ∗P < 0.05, ∗∗∗P < 0.001 vs EV cells generated using t-test.
3.4. MiR-34a suppresses tumor growth and invasion of glioma stem cells
The growth suppression caused by miR-34a over-expression in glioma stem cells led us to evaluate its effect on tumor forming potential in vivo. For this, we injected both HNGC-2-EV cells and miR-34a expressing cells (106) subcutaneously into NOD/SCID mice and monitored tumor growth for 45 days. The HNGC-2 and NSG-K16-EV cells were able to form detectable tumors within 7 days. However, miR-34a over-expressing cells formed significantly reduced tumors as compared to EV cells (Fig. 5A, a and b). The tumors with HNGC-2-miR-34a cells were about 6 folds smaller as compared to EV induced tumors (P < 0.05) (Fig. 5A, c). The notable reduction in tumor volume reaffirmed the tumor suppressive function of miR-34a. MiR-34a also affected invasiveness of glioma cells as was apparent from matrigel invasion chamber assay. The miR-34a overexpressing cells possessed decreased invasive potential in matrigel assay as compared to EV cells (P < 0.0001) (Fig. 5B, a and b). These effects on tumor growth inhibition and invasion potential on glioma cells reiterated role of miR-34a as growth suppressor.
Fig. 5.
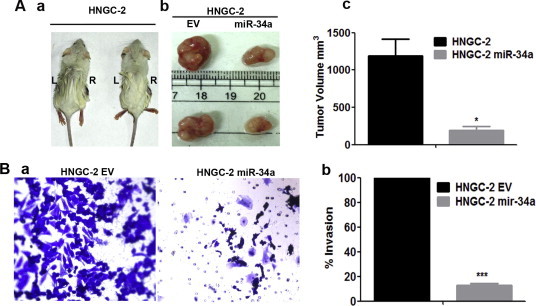
Over-expression of MiR-34a suppresses tumor growth in vivo and inhibits invasion in vitro. (A). Representative images of NOD SCID mice (n = 9) showing tumors formed with HNGC-2-EV (R) and miR-34a cells (L) (a). Tumors excised after 4 weeks are represented in (b) and quantified in (c). (B) Phase contrast micrographs of Invasion assay displaying migratory property of HNGC-2 – EV and miR-34a cells performed using invasion chambers of 8 μ and stained with crystal violet after 24 h (10× magnification) (a) and quantified in (b). The percentages of invading cells were determined by considering migration of HNGC-2 and NSG-K16–EV cells as 100%. Data represents means ± SEM (n = 3) and P value calculated by Student’s t test (∗P < 0.05, ∗∗∗P < 0.0001).
3.5. Rictor is a target for miR-34a
MicroRNAs regulate gene expression at post-transcriptional level either by inhibiting translation of complementary mRNAs and by targeting them for degradation [4]. We used an in silico based approach for identification of novel putative targets for miR-34a that could be responsible for mediating effects on growth suppression of glioma cells. We used three target databases viz: Miranda (http://www.microrna.org/microrna/home.do), TargetScan (http://www.targetscan.org/), and PicTar (http://pictar.mdc-berlin.de/) to identify putative targets for miRNA action. Data from each of these databases led us to identify Rictor as one of the common target for miR-34a. It possessed complementary seed sequence at 3′UTR region for miR-34a and displayed a mirSVR score of −0.6782 (Fig. 6A). Rictor is a core component of the distinct mTOR protein complexes, mTORC2. The mammalian target of rapamycin (mTOR) plays a critical role in the positive regulation of cell growth and survival primarily through direct interaction with raptor (forming mTORC complex 1; mTORC1) or Rictor (forming mTOR complex 2; mTORC2) [28]. The mTORC2 complex with Rictor is a critical regulator of AKT, a critical effector of PI3K signaling pathway and found to be activated in all patients with glioblastoma [18]. Our earlier studies demonstrated higher levels of Rictor in increased glioma grades [29]. In response to miR-34a overexpression in both HNGC-2 and NSG-K16 cells, there was a significant down-regulation of Rictor at protein level indicating it to be a functional target of miR-34a (Fig. 6B). Next, by Luciferase reporter assay we confirmed that Rictor was a direct target of miR-34a in glioma cells. The schematic of luciferase assay is shown in Fig. 6C. Our data indicated that miR-34a directly bound to 3′UTR of Rictor and suppressed the luciferase activity in HNGC-2 and NSG-K16 cells (P < 0.05) (Fig. 6D and E). This data indicated that miR-34a directly targets Rictor in glioma stem cells.
Fig. 6.
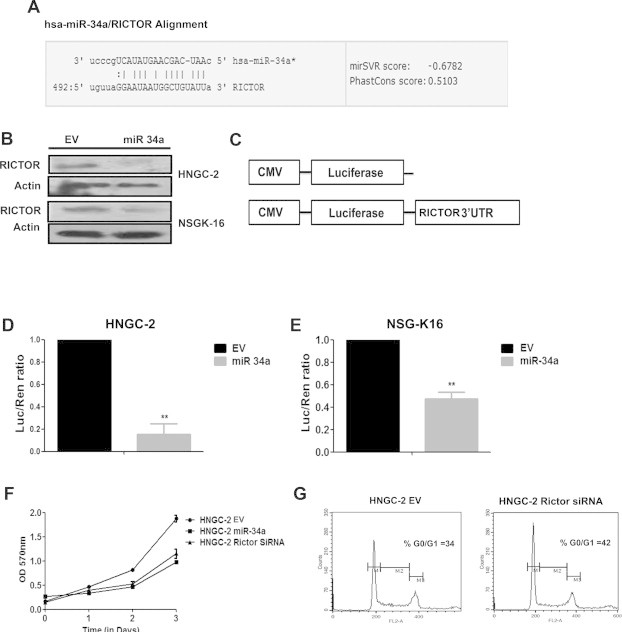
Rictor is a target for miR-34a (A) Alignment of miR-34a seed sequence with 3′UTR of Rictor. (B) Western blot analysis of HNGC-2 and NSG-K16 cells expressing miR-34a and EV and probed for Rictor. The band intensities were calculated with help of Image J software. Actin served as loading control. (C) Schematic representation of Renilla Luciferase Reporter assay with wild-type 3′UTR of Rictor in luciferase experiment. Dual Luciferase assay showing relative luciferase activity in HNGC-2 (D) and NSG-K16 (E) cells expressing EV and miR-34a. Renilla luciferase activity was normalized with firefly luciferase activity as control for transfection. (F) MTT assay showing effect of Rictor siRNA on proliferation of HNGC-2 cells. (G) Flow cytometry profile showing distribution of cells in different phases of cell cycle in HNGC-2 and HNGC-2 Rictor siRNA cells. Error bar represents the means ± SEM and (n = 3). P value is calculated by Students t test (∗∗P < 0.005).
3.6. Inhibition of Rictor mimics effects of miR-34a over-expression
To confirm functional role of Rictor as a miR-34a target involved in mediating effects on cell growth and tumorigenicity in glioma cells, we performed Rictor knock-down experiments. For this we performed a siRNA mediated knock down of Rictor expression in HNGC-2 cells and analyzed whether loss of Rictor activity mimicked the miR-34a effects on glioma stem cells. Here, siRNA against Rictor was transiently transfected into HNGC-2 cells and transfected cells were analyzed for effects on cell proliferation by MTT assay and cell cycle kinetics by flow cytometry. Rictor siRNA inhibited cell proliferation of glioma cells as examined by MTT assay (Fig. 6F). Also, Rictor siRNA caused cell cycle arrest leading to increased G0/G1 population in siRNA treated cells (Fig. 6G) as compared to EV cells. These experiments confirmed that Rictor was a functional target of miR-34a in glioma stem cells.
3.7. MiR-34a affects the PI3K/AKT and WNT/β-Catenin signaling pathway in glioma
Our in vitro and in vivo data provided evidences that miR-34a functioned as a tumor suppressor in high grade gliomas. The ectopic over expression of miR-34a led to decreased aggressiveness in GBM cell-lines by targeting Rictor, an integral component if the mTORC2 complex. Rictor along with mTORC2 leads to the phosphorylation of AKT at serine 473 (ser473) and activates p-AKT [30]. Further, we analyzed whether, over-expression of miR-34a led to activation of AKT signaling pathway. We found a significant down-regulation of p-AKT by miR-34a in HNGC-2 cells as compared to EV cells causing activation of glycogen synthase kinase (GSK)-3β. As GSK-3β functions as a negative regulator of β-catenin stabilization, AKT phosphorylation of GSK-3β leads to β-catenin stabilization and nuclear accumulation. Characteristically, we found a decreased expression of beta-catenin in miR-34a HNGC-2 cells by Western blotting (Fig. 7A). Importantly, complete lack of nuclear beta-catenin expression was obtained by confocal microscopy in miR-34a over-expressing HNGC-2 cells (Fig. 7B). Similarly, the luciferase reporter assay was used to evaluate Wnt activity. There was 3.3-fold decrease in Tcf/Lef activity (P < 0.05) in HNGC-2-miR-34a cells as compared to the control cells (Fig. 7C). The effects of miR-34a leading to inhibition of Wnt signaling caused reduced levels of target proteins of Wnt pathway- Cyclin D1 and c-Myc. We have earlier shown that Wnt signaling is an important player in glioma progression and pathogenesis [25]. Our present data indicates that Rictor is a direct target of miR-34a. Hence, inhibition of miR-34a, through up-regulation of Rictor causes activation of AKT-GSK-3β axis in glioma stem cells leading to activation of WNT/β-catenin signaling pathway. The effects of miR-34a loss on cellular signaling in glioma stem cells leading to enhanced cell proliferation, aggressiveness and tumorigenicity is proposed in Fig. 8.
Fig. 7.
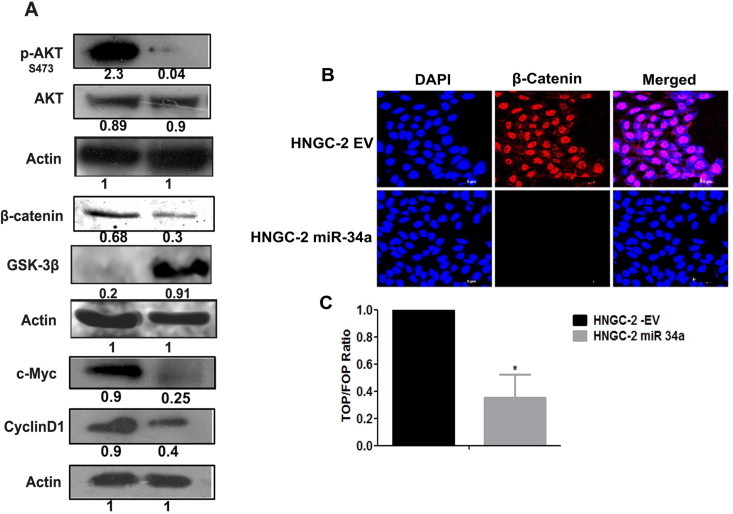
MiR-34a is negative regulator of AKT and WNT signaling pathway. (A) Western blot analyses of HNGC-2-EV and miR-34a cells. Actin was used as loading control; band intensities were calculated by Image J software and normalized to actin. (B) β-Catenin staining in HNGC-2-EV and miR-34a cells, detected with help of goat anti-rabbit Alexa Flour 594 (Red). Nuclei were stained with DAPI (Blue). (C) β-catenin/TCF reporter assay using dual luciferase reporter system. The normalized luciferase activity was used to estimate ratio of TOPflash to FOPflash and represented as a bar graph by considering EV luciferase activity as 1. Data are presented as average values ± SEM (n = 3) and P value is calculated by Student’s t test (∗P < 0.05).
Fig. 8.
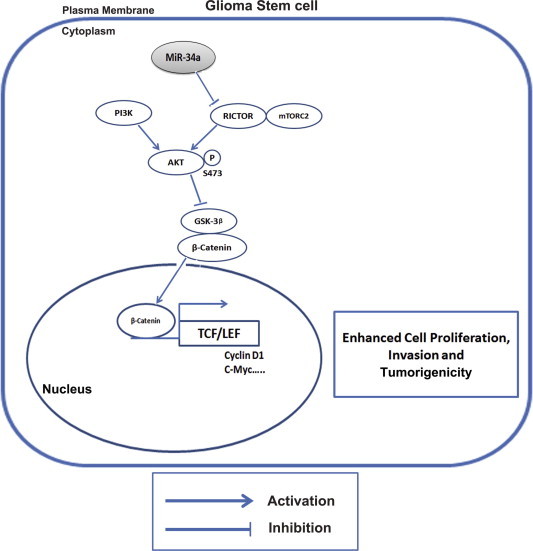
Proposed model depicting mechanism for action of miR-34a in glioma cells.
4. Discussion
Heterogeneity within the tumor, its deep seated location in the brain, aggressive nature and angiogenic behavior makes glioblastoma a difficult CNS tumor to treat. Even though multimodal therapies increase patient survival, GBM prognosis remains challenging [31]. It is believed that the phenotypic and functional heterogeneity within the tumor arises due to presence of a small tumor initiating cell population that propels the tumor.
We have developed in vitro cell culture model systems that mimic glioma tumor progression. These developed cell systems possess a great potential in understanding the signaling events that lead to the advanced disease phenotype in glioma [24,25,32]. The HNGC-2 and NSG-K16 cell-lines used in this study are well characterized by us as glioma stem cell-lines [24,25]. We used these cell-lines to identify a set of miRNA’s that were differentially expressed in glioma stem cell-lines as compared to neural stem cells. We earlier demonstrated function of one of the miRNA’s, miR-145 with tumor suppressive role in GBM with Sox9 and Adducin3 as its novel targets [8]. In the present study, we report functional significance of miR-34a and report Rictor as a novel target for miR-34a using the tumor progression model of HNGC-1 to HNGC-2 cell system developed by us.
It is well established that microRNAs can target multiple genes including oncogenes, and their interactions are cell type specific [33]. Hence, identification of differentially expressed microRNAs and their context dependent targets is of great help in uncovering the network of biological processes governing specific phenotype. MiR-34a is reported to be significantly down-regulated in multiple cancer types and its expression correlates with patient survival. It is localized on chromosome 1p36 and its down-regulation is associated with multiple chromosomal abnormalities like frequent deletion of chromosome 1p36, p53 mutation and CpG methylation of miR-34a promoter itself [34]. Moreover, deregulation of miR-34a affects many biological processes like cell cycle, senescence, apoptosis, differentiation and development [35–37]. These findings emphasize requirement for comprehensive studies to confirm molecular targets for miR-34a and understand its functional contribution to glioma progression. The role of miR-34a as a tumor suppressor in glioma is known [14,38,39] wherein its targets are several oncogenes like c-Met, Notch1, Notch2 and CDK6 [14,40]. Significantly, p53 is an important target for miR-34a and it is shown that expression of miR-34a in mutant p53 is lower as compared to those cell lines bearing wild type p53. It is suggested that role of miR-34a in tumor progression may be closely associated with p53 mutation and is inversely correlated to Bcl-2 expression [6].
In this report, we demonstrate that miR-34a is down-regulated in higher grade gliomas (GBM) and glioma stem cell-lines like – HNGC-2 and NSG-K16. More importantly, we here report identification of Rictor as a novel target for miR-34a. We further demonstrate that over-expression of Rictor in glioma stem cells causes activation of AKT signaling. This via enhancement of GSK-3β axis causes nuclear localization of beta-catenin and over-expression of Wnt target genes- CyclinD1 and c-myc. We show that activation of Wnt cellular signaling pathways causes effects on tumorigenicity and invasiveness in GBM.
Invasion is the hallmark of high grade glioma specifically shown by aggressive mesenchymal GBM phenotype [22,41]. Recently, miR-21 and miR-10b are positively correlated with glioma invasion and miR-34a is listed as one of the anti-invasive microRNAs [5,42,43]. In this study, we characterized the glioma stem cell-lines HNGC-2 and NSG-K16 and showed them to possess the mesenchymal GBM phenotype (Fig. 1). Over-expression of miR-34a in these glioma stem cell-lines reduced their invasive potential by more than 80% signifying an important role for it in glioma invasion.
Our identified miR-34a target, Rictor is significantly involved in glioma tumor progression. It is overexpressed in many cancers and is involved in gliomagenesis via mTOR-dependent and mTOR-independent mechanisms [44–46]. Rictor expression is essentially correlated with aggressiveness of GBM, mainly acting by modulating MMP9 activity. Our previous study has reported that Rictor enhanced MMP9 activity in glioma cell-lines LN-18 and LN-229 via activation of RAF-1-MEK-ERK pathway [29]. With help of REMBRANDT data [27] we generated the Kaplan Meier survival graph for Rictor gene expression. Our analyses showed that 93 patients with higher expression of Rictor had lower survival as compared to patients that showed intermediate Rictor expression (P = 2.47E−08, Supplementary Fig. S1). It is evident from the survival curve that Rictor expression was negatively correlated with survival of patients and played an important role in glioma progression.
Earlier, we showed that Wnt3a mediated Wnt/β catenin signaling was an essential tumorigenic driver in glioma stem cells, indicating that Wnt3a was an oncogene and novel therapeutic target in glioma [25]. Interestingly, here our data shows that miR-34a over-expression down-regulates β-Catenin activity and this was confirmed by us with TOP/FOP flash luciferase assay. The GSK-3β kinase, acts as negative regulator of WNT/β Catenin signaling pathway wherein it induces β-Catenin phosphorylation leading to its proteasome mediated degradation [47]. The interplay between AKT and WNT signaling pathways through AKT-GSK-3β axis was studied previously in skeletal development; twist mediated epithelial-mesenchymal transition (EMT) in cancer stem cells and also in glioma [48–50]. Based on our data, we propose a model of action of miR-34a in glioma stem cells (Fig. 8). Our study demonstrates that miR-34a functionally targets Rictor and acts as tumor suppressor by suppressing AKT followed by WNT signaling pathway. This has crucial implications on cell proliferation, invasion and tumor progression in GBM. Our data provides experimental evidence to suggest that miR-34a is an important therapeutic target for glioma stem cells and thus targeting it has a great potential in inhibiting glioma progression and preventing recurrence thereby improving long term survival in glioblastoma.
Conflict of interest statement
None declared.
Author contributions
Sachin S. Rathod planned and performed experiments, performed REMBRANDT and other bio- informatics experiments and analyzed data.
Sandhya Rani B planned and performed experiments.
Mohsina Khan, data analyses.
Dattatraya Muzumdar provided clinical material for study and analyzed data.
Anjali Shiras planned experiments analyzed data and wrote the paper.
Acknowledgements
We are thankful to Dr. Ramanamurthy for his help in providing us with SCID mice for tumorigenicity assays and Mr. M. L. Shaikh for his technical help in mice tumorigenicity experiments. Grant support for carrying out research was provided by Stem Cell Task Force, Department of Biotechnology (DBT) (Grant no. BT/PR10557/MED/31/29/2008), Government of India, New Delhi, India and National Centre for Cell Science (NCCS), Pune, India. The fellowship of S.R.B was supported by ICMR, New Delhi; India. The fellowship of MK was supported by UGC, New Delhi; India. Fellowship funding for SR was provided by DBT; New Delhi; India.
Appendix A. Supplementary data
Supplementary Fig. S1.
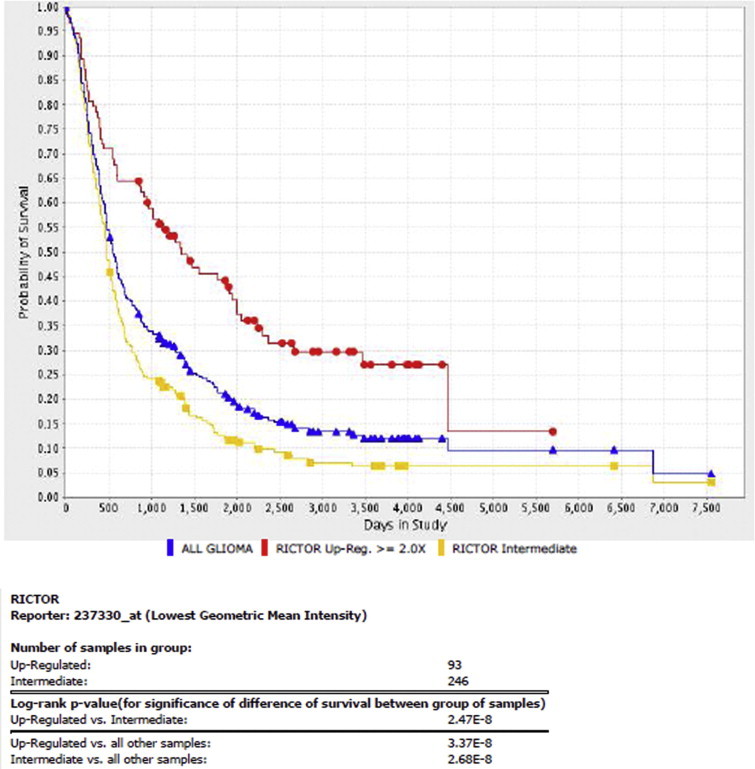
This document contains supplementary figure.
Summary of clinico-pathological features of tumor and normal tissues derived from patients; NBT, normal brain tissue.
Primer sequences.
References
- 1.Wen P.Y., Kesari S. Malignant gliomas in adults. N. Engl. J. Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 2.Meacham C.E., Morrison S.J. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328–337. doi: 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hadjipanayis C.G., Van Meir E.G. Brain cancer propagating cells: biology, genetics and targeted therapies. Trends Mol. Med. 2009;15:519–530. doi: 10.1016/j.molmed.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartel D.P. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moller H.G., Rasmussen A.P., Andersen H.H., Johnsen K.B., Henriksen M., Duroux M. A systematic review of microRNA in glioblastoma multiforme: micro-modulators in the mesenchymal mode of migration and invasion. Mol. Neurobiol. 2013;47:131–144. doi: 10.1007/s12035-012-8349-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao H., Zhao H., Xiang W. Expression level of human miR-34a correlates with glioma grade and prognosis. J. Neurooncol. 2013;113:221–228. doi: 10.1007/s11060-013-1119-1. [DOI] [PubMed] [Google Scholar]
- 7.Gonzalez-Gomez P., Sanchez P., Mira H. MicroRNAs as regulators of neural stem cell-related pathways in glioblastoma multiforme. Mol. Neurobiol. 2011;44:235–249. doi: 10.1007/s12035-011-8196-y. [DOI] [PubMed] [Google Scholar]
- 8.Rani S.B., Rathod S.S., Karthik S., Kaur N., Muzumdar D., Shiras A.S. MiR-145 functions as a tumor-suppressive RNA by targeting Sox9 and adducin 3 in human glioma cells. Neuro-oncology. 2013;15:1302–1316. doi: 10.1093/neuonc/not090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cole K.A., Attiyeh E.F., Mosse Y.P., Laquaglia M.J., Diskin S.J., Brodeur G.M., Maris J.M. A functional screen identifies miR-34a as a candidate neuroblastoma tumor suppressor gene. Mol. Cancer Res. 2008;6:735–742. doi: 10.1158/1541-7786.MCR-07-2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tivnan A., Tracey L., Buckley P.G., Alcock L.C., Davidoff A.M., Stallings R.L. MicroRNA-34a is a potent tumor suppressor molecule in vivo in neuroblastoma. BMC Cancer. 2011;11:33. doi: 10.1186/1471-2407-11-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siemens H., Neumann J., Jackstadt R., Mansmann U., Horst D., Kirchner T., Hermeking H. Detection of miR-34a promoter methylation in combination with elevated expression of c-Met and beta-catenin predicts distant metastasis of colon cancer. Clin. Cancer Res. 2013;19:710–720. doi: 10.1158/1078-0432.CCR-12-1703. [DOI] [PubMed] [Google Scholar]
- 12.Liu C., Kelnar K., Liu B., Chen X., Calhoun-Davis T., Li H., Patrawala L., Yan H., Jeter C., Honorio S., Wiggins J.F., Bader A.G., Fagin R., Brown D., Tang D.G. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat. Med. 2011;17:211–215. doi: 10.1038/nm.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lodygin D., Tarasov V., Epanchintsev A., Berking C., Knyazeva T., Korner H., Knyazev P., Diebold J., Hermeking H. Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle. 2008;7:2591–2600. doi: 10.4161/cc.7.16.6533. [DOI] [PubMed] [Google Scholar]
- 14.Guessous F., Zhang Y., Kofman A., Catania A., Li Y., Schiff D., Purow B., Abounader R. MicroRNA-34a is tumor suppressive in brain tumors and glioma stem cells. Cell Cycle. 2010;9:1031–1036. doi: 10.4161/cc.9.6.10987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ji Q., Hao X., Meng Y., Zhang M., Desano J., Fan D., Xu L. Restoration of tumor suppressor miR-34 inhibits human p53-mutant gastric cancer tumorspheres. BMC Cancer. 2008;8:266. doi: 10.1186/1471-2407-8-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Silber J., Jacobsen A., Ozawa T., Harinath G., Pedraza A., Sander C., Holland E.C., Huse J.T. MiR-34a repression in proneural malignant gliomas upregulates expression of its target PDGFRA and promotes tumorigenesis. PLoS One. 2012;7:e33844. doi: 10.1371/journal.pone.0033844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vo D.T., Qiao M., Smith A.D., Burns S.C., Brenner A.J., Penalva L.O. The oncogenic RNA-binding protein Musashi1 is regulated by tumor suppressor miRNAs. RNA Biol. 2011;8:817–828. doi: 10.4161/rna.8.5.16041. [DOI] [PubMed] [Google Scholar]
- 18.McLendon R., Friedman A., Bigner D. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qiu S., Lin S., Hu D., Feng Y., Tan Y., Peng Y. Interactions of miR-323/miR-326/miR-329 and miR-130a/miR-155/miR-210 as prognostic indicators for clinical outcome of glioblastoma patients. J. Transl. Med. 2013;11:10. doi: 10.1186/1479-5876-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen R., Mo Q., Schultz N., Seshan V.E., Olshen A.B., Huse J., Ladanyi M., Sander C. Integrative subtype discovery in glioblastoma using iCluster. PLoS One. 2012;7:e35236. doi: 10.1371/journal.pone.0035236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang W., Duan J., Zhang J.G., Wang Y.P. Subtyping glioblastoma by combining miRNA and mRNA expression data using compressed sensing-based approach. EURASIP J. Bioinform. Syst. Biol. 2013;2013:2. doi: 10.1186/1687-4153-2013-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verhaak R.G., Hoadley K.A., Purdom E., Wang V., Qi Y., Wilkerson M.D., Miller C.R., Ding L., Golub T., Mesirov J.P., Alexe G., Lawrence M., O’Kelly M., Tamayo P., Weir B.A., Gabriel S., Winckler W., Gupta S., Jakkula L., Feiler H.S., Hodgson J.G., James C.D., Sarkaria J.N., Brennan C., Kahn A., Spellman P.T., Wilson R.K., Speed T.P., Gray J.W., Meyerson M., Getz G., Perou C.M., Hayes D.N. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim T.M., Huang W., Park R., Park P.J., Johnson M.D. A developmental taxonomy of glioblastoma defined and maintained by MicroRNAs. Cancer Res. 2011;71:3387–3399. doi: 10.1158/0008-5472.CAN-10-4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shiras A., Chettiar S.T., Shepal V., Rajendran G., Prasad G.R., Shastry P. Spontaneous transformation of human adult nontumorigenic stem cells to cancer stem cells is driven by genomic instability in a human model of glioblastoma. Stem Cells. 2007;25:1478–1489. doi: 10.1634/stemcells.2006-0585. [DOI] [PubMed] [Google Scholar]
- 25.Kaur N., Chettiar S., Rathod S., Rath P., Muzumdar D., Shaikh M.L., Shiras A. Wnt3a mediated activation of Wnt/beta-catenin signaling promotes tumor progression in glioblastoma. Mol. Cell. Neurosci. 2013;54:44–57. doi: 10.1016/j.mcn.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 26.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 27.Madhavan S., Zenklusen J.C., Kotliarov Y., Sahni H., Fine H.A., Buetow K. Rembrandt: helping personalized medicine become a reality through integrative translational research. Mol. Cancer Res. 2009;7:157–167. doi: 10.1158/1541-7786.MCR-08-0435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oh W.J., Jacinto E. MTOR complex 2 signaling and functions. Cell Cycle. 2011;10:2305–2316. doi: 10.4161/cc.10.14.16586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Das G., Shiras A., Shanmuganandam K., Shastry P. Rictor regulates MMP-9 activity and invasion through Raf-1-MEK-ERK signaling pathway in glioma cells. Mol. Carcinog. 2011;50:412–423. doi: 10.1002/mc.20723. [DOI] [PubMed] [Google Scholar]
- 30.Hresko R.C., Mueckler M. MTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J. Biol. Chem. 2005;280:40406–40416. doi: 10.1074/jbc.M508361200. [DOI] [PubMed] [Google Scholar]
- 31.Huang T.T., Sarkaria S.M., Cloughesy T.F., Mischel P.S. Targeted therapy for malignant glioma patients: lessons learned and the road ahead. Neurotherapeutics. 2009;6:500–512. doi: 10.1016/j.nurt.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shiras A., Bhosale A., Shepal V., Shukla R., Baburao V.S., Prabhakara K., Shastry P. A unique model system for tumor progression in GBM comprising two developed human neuro-epithelial cell lines with differential transforming potential and coexpressing neuronal and glial markers. Neoplasia. 2003;5:520–532. doi: 10.1016/s1476-5586(03)80036-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Di Leva G., Garofalo M., Croce C.M. MicroRNAs in cancer. Annu. Rev. Pathol. 2014;9:287–314. doi: 10.1146/annurev-pathol-012513-104715. (Epub 2013 Sep 25) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei J.S., Song Y.K., Durinck S., Chen Q.R., Cheuk A.T., Tsang P., Zhang Q., Thiele C.J., Slack A., Shohet J., Khan J. The MYCN oncogene is a direct target of miR-34a. Oncogene. 2008;27:5204–5213. doi: 10.1038/onc.2008.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010;17:193–199. doi: 10.1038/cdd.2009.56. [DOI] [PubMed] [Google Scholar]
- 36.Tazawa H., Tsuchiya N., Izumiya M., Nakagama H. Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc. Natl. Acad. Sci. U.S.A. 2007;104:15472–15477. doi: 10.1073/pnas.0707351104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Welch C., Chen Y., Stallings R.L. MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene. 2007;26:5017–5022. doi: 10.1038/sj.onc.1210293. [DOI] [PubMed] [Google Scholar]
- 38.Li Y., Guessous F., Zhang Y., Dipierro C., Kefas B., Johnson E., Marcinkiewicz L., Jiang J., Yang Y., Schmittgen T.D., Lopes B., Schiff D., Purow B., Abounader R. MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res. 2009;69:7569–7576. doi: 10.1158/0008-5472.CAN-09-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luan S., Sun L., Huang F. MicroRNA-34a: a novel tumor suppressor in p53-mutant glioma cell line U251. Arch. Med. Res. 2010;41:67–74. doi: 10.1016/j.arcmed.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 40.Li W.B., Ma M.W., Dong L.J., Wang F., Chen L.X., Li X.R. MicroRNA-34a targets notch1 and inhibits cell proliferation in glioblastoma multiforme. Cancer Biol. Ther. 2011;12:477–483. doi: 10.4161/cbt.12.6.16300. [DOI] [PubMed] [Google Scholar]
- 41.Mikheeva S.A., Mikheev A.M., Petit A., Beyer R., Oxford R.G., Khorasani L., Maxwell J.P., Glackin C.A., Wakimoto H., Gonzalez-Herrero I., Sanchez-Garcia I., Silber J.R., Horner P.J., Rostomily R.C. TWIST1 promotes invasion through mesenchymal change in human glioblastoma. Mol. Cancer. 2010;9:194. doi: 10.1186/1476-4598-9-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin J., Teo S., Lam D.H., Jeyaseelan K., Wang S. MicroRNA-10b pleiotropically regulates invasion, angiogenicity and apoptosis of tumor cells resembling mesenchymal subtype of glioblastoma multiforme. Cell Death. Dis. 2012;3:e398. doi: 10.1038/cddis.2012.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma X., Yoshimoto K., Guan Y., Hata N., Mizoguchi M., Sagata N., Murata H., Kuga D., Amano T., Nakamizo A., Sasaki T. Associations between microRNA expression and mesenchymal marker gene expression in glioblastoma. Neuro-oncology. 2012;14:1153–1162. doi: 10.1093/neuonc/nos145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bashir T., Cloninger C., Artinian N., Anderson L., Bernath A., Holmes B., Benavides-Serrato A., Sabha N., Nishimura R.N., Guha A., Gera J. Conditional astroglial Rictor overexpression induces malignant glioma in mice. PLoS One. 2012;7:e47741. doi: 10.1371/journal.pone.0047741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Masri J., Bernath A., Martin J., Jo O.D., Vartanian R., Funk A., Gera J. MTORC2 activity is elevated in gliomas and promotes growth and cell motility via overexpression of rictor. Cancer Res. 2007;67:11712–11720. doi: 10.1158/0008-5472.CAN-07-2223. [DOI] [PubMed] [Google Scholar]
- 46.Zhang F., Zhang X., Li M., Chen P., Zhang B., Guo H., Cao W., Wei X., Cao X., Hao X., Zhang N. MTOR complex component Rictor interacts with PKCzeta and regulates cancer cell metastasis. Cancer Res. 2010;70:9360–9370. doi: 10.1158/0008-5472.CAN-10-0207. [DOI] [PubMed] [Google Scholar]
- 47.Angers S., Moon R.T. Proximal events in Wnt signal transduction. Nat. Rev. Mol. Cell Biol. 2009;10:468–477. doi: 10.1038/nrm2717. [DOI] [PubMed] [Google Scholar]
- 48.Atkins R.J., Dimou J., Paradiso L., Morokoff A.P., Kaye A.H., Drummond K.J., Hovens C.M. Regulation of glycogen synthase kinase-3 beta (GSK-3beta) by the Akt pathway in gliomas. J. Clin. Neurosci. 2012;19:1558–1563. doi: 10.1016/j.jocn.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 49.Li J., Zhou B.P. Activation of beta-catenin and Akt pathways by Twist are critical for the maintenance of EMT associated cancer stem cell-like characters. BMC Cancer. 2011;11:49. doi: 10.1186/1471-2407-11-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rokutanda S., Fujita T., Kanatani N., Yoshida C.A., Komori H., Liu W., Mizuno A., Komori T. Akt regulates skeletal development through GSK3, mTOR, and FoxOs. Dev. Biol. 2009;328:78–93. doi: 10.1016/j.ydbio.2009.01.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of clinico-pathological features of tumor and normal tissues derived from patients; NBT, normal brain tissue.
Primer sequences.