

1. Disease characteristics
1.1 Name of the disease (synonyms)
Xeroderma pigmentosum (complementation groups XPA, XPB, XPC, XPD, XPE, XPF, XPG, and XP variant (XPV)).
1.2 OMIM# of the disease
XPA #278700, XPB #610651, XPC #278720, XPD #278730, XPE #278740, XPF #278760, XPG #278780, XPV #278750.
1.3 Name of the analyzed genes or DNA/chromosome segments
XPA, ERCC3 (XPB), XPC, ERCC2 (XPD), DDB2 (XPE), ERCC4 (XPF), ERCC5 (XPG), POLH (XPV).
1.4 OMIM# of the gene(s)
XPA *611153, ERCC3 (XPB) *133510, XPC *613208, ERCC2 (XPD) *126340, DDB2 (XPE) *600811, ERCC4 (XPF) *133520, ERCC5 (XPG) *133530, POLH (XPV) *603968.
1.5 Mutational spectrum
XPA: 122 known disease-causing mutations from 101 patients. Mutations: 77 (63%) G>C, 3 (2%) T>A, and 2 (1.5%) G>T transversions, 30 (25%) transitions (20 of these are c.682C>T stop codon mutations; GenBank accession number: NC_000009.11), 8 (7%) deletions, and 2 (1.5%) insertions. Consequences: frameshifts, protein truncations, and functional relevant amino-acid substitutions.1, 2, 3
XPB/ERCC3: eight known disease-causing mutations from five patients. Mutations: 1 (12.5%) C>A transversion, 1 (12.5%) A>C transversion, 1 (12.5%) G>A transition, 3 (37,5%) T>C transitions, 1 (12.5%) deletion (c.807-808delT_T), and 1 (12.5%) insertion (c.1421-1422insA). Consequences: two functional relevant amino-acid substitutions (c.296T>C (p.(Phe99Ser)) and c.355A>C (p.(Thr199Pro))), and six frameshift/protein truncations (GenBank accession number: NC_000002.11).4
XPC: 51 known disease-causing mutations from 114 patients. Mutations: transitions, transversions, deletions, insertions, complex deletion/insertion mutations, and splice-site mutations. Consequences: frameshifts, protein truncations, and functional relevant amino-acid substitutions. Here, a phenotype correlation emerges indicating that XPC mutations only result in a classical XP phenotype.5, 6, 7, 8
XPD/ERCC2: 48 known disease-causing mutations from 32 XP patients (36 trichothiodystrophy patients are excluded). Mutations: 43 (90%) base exchanges and 5 (10%) deletions. Consequences: frameshifts/protein truncations and functional relevant amino-acid substitutions. A genotype–phenotype correlation could already be established indicating that XPD mutations can result in six different clinical entities: classical XP, XP with neurological symptoms, trichothiodystrophy (TTD), XP/TTD complex, XP/Cockayne syndrome complex, or the cerebro-oculo-facio-skeletal syndrome (COFS).9, 10, 11
XPE/DDB2: nine known disease-causing mutations from 12 patients. Mutations: four (44%) transitions, two (23%) transversions, and three deletions (33%). Consequences: functional relevant amino-acid substitutions, frameshifts/protein truncations.12
XPF/ERCC4: 15 known disease-causing mutations from nine patients. Mutations: nine (60%) transitions and one (7%) transversion, four (26%) deletions, and one (7%) insertion. Consequences: functional relevant amino-acid substitutions, frameshifts/protein truncations.1, 2, 3
XPG/ERCC5: 25 known disease-causing mutations from 19 patients. Mutations: 12 (48%) transitions, 5 (20%) transversions, and 8 (32%) deletions. Consequences: Functional relevant amino-acid substitutions, frameshifts/protein truncations.13, 14
POLH: 32 known disease-causing mutations from 22 patients (three patients are completely uncharacterized). Mutations: 5 (16%) transitions, 4 (13%) transversions, 18 (55%) deletions, and 5 (16%) insertions. Consequences: functional relevant amino-acid substitutions, frameshifts/protein truncations.15
1.6 Analytical methods
Xeroderma pigmentosum is clinically diagnosed. Hallmarks of the disease include severe sun-sensitivity since birth, freckling in sun-exposed skin in the first years of life, and skin cancer development at a mean age of 8 years.16, 17
As the clinical entity xeroderma pigmentosum (XP) can be caused by eight different defective genes (XPA-XPG and POLH), there are functional DNA-repair assays available to define the defective gene (complementation group assignment—see section 3.1.1.).
Finally, base sequence analysis of the identified gene identifies the exact disease-causing mutation and ultimately proves the diagnosis. DNA material for sequencing is derived from cultured primary patient fibroblasts established from a skin punch biopsy or from peripheral venous blood. cDNA sequencing can confirm the mutation and detect the expression of variant alleles. Exons and flanking intron regions are targets for sequencing for validating the genotype of the XP patient. To date, knowledge of the exact disease-causing mutation(s) has no therapeutic consequence. If available, the mutations should be validated in the healthy parents or other siblings of the affected patient.
Knowledge about the mutated mRNA and/or protein levels may be helpful, too. To this end primary fibroblasts can be used for mRNA level measurements via qPCR techniques. Western blotting can give an idea about the amount of (mutated) protein expression and its size. In case of frequently occurring and disease-causing base exchanges (eg founder mutations in XPA in Japan) mutation detection can be facilitated by applying restriction fragment length polymorphism (RFLP) techniques. Methods may differ from lab to lab and new ‘high throughput' techniques like exome sequencing or whole-genome sequencing may soon be available more broadly.
1.7 Analytical validation
The mutation, once identified, is confirmed by sequencing another independently prepared sample. The genetic investigation of the direct relatives of the patients assigns the origin to the parents and manifests the XP genotype(s).
1.8 Estimated frequency of the disease
Incidence at birth (‘birth prevalence') or population prevalence (if known to be variable between ethnic groups, please report):
XP occurs worldwide an in all ethnic groups. The worldwide frequency of XP is very low and differs in different regions in the world (North America 1:1 000 000, North Africa/Middle East 1:50 000, Northern Europe 1:450 000). XP-A, XP-C, XP-D, and XP-V patients prevail compared with the other complementation groups. Patients are reported from Japan, USA, Europe, and the Middle East. In the Japanese population, XPA gene mutations are the most common cause of XP (confirmed founder mutations). XP-B is very rare and appears equally distributed throughout all populations. XP-C is the most frequent complementation group worldwide and especially in USA, Europe, and the Middle East (confirmed founder mutation as detected by microsatellite analysis). A very high incidence (∼1/5000) of black XP-C patients was reported in the Mayotte population in the Indian Ocean, whereas XP-C has rarely been reported in Japan. XP-D occurs at a frequency of 20% worldwide. A confirmed XPD founder mutation is common among Iraqi Jews causing a very mild XP phenotype. XP-F is very rare, patients have mainly reported from Japan or Europe. XP-G patients have also been infrequently described, mostly in Europe, Japan, and USA. The largest group (one-third) of all patients with XP are those of the variant type with defective polymerase eta. Carriers of the mutated polymerase eta gene have been mostly identified in Europe and USA.1, 3, 16, 17, 18, 19
1.9 Diagnostic setting

Comment:Genetic testing for XP is also applied in the context of population screening.
2. Test characteristics
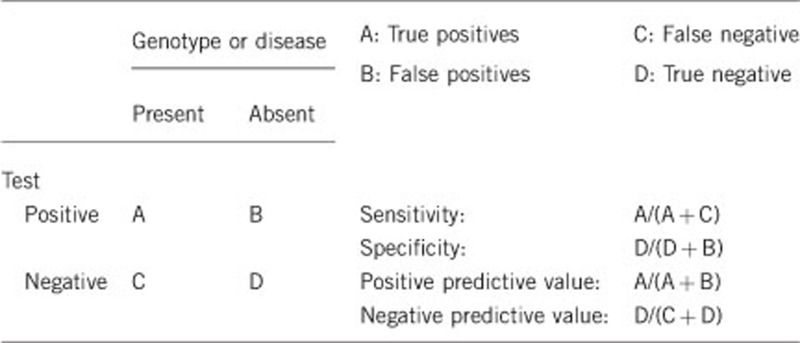
2.1 Analytical sensitivity
(proportion of positive tests if the genotype is present)Parallel genomic DNA and mRNA sequencing exhibit an estimated positive predictive value above 95%.
2.2 Analytical specificity
(proportion of negative tests if the genotype is not present)Parallel genomic DNA and mRNA sequencing exhibit an estimated negative predictive value above 95%.
2.3 Clinical sensitivity
(proportion of positive tests if the disease is present)The clinical sensitivity can be dependent on variable factors such as age or family history. In such cases, a general statement should be given, even if quantification can only be made case by case.
The clinical sensitivity of parallel genomic DNA and mRNA sequencing can be estimated at 80–90%.
2.4 Clinical specificity
(proportion of negative tests if the disease is not present)The clinical specificity can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
The clinical specificity of parallel genomic DNA and mRNA sequencing can be estimated at >95%.
2.5 Positive clinical predictive value
(life-time risk to develop the disease if the test is positive)The life-time risk to develop the disease if the genetic testing was positive is 100% however, there can be considerable individual variation in the severity of the disease expression.
2.6 Negative clinical predictive value
(probability not to develop the disease if the test is negative)Assume an increased risk based on family history for a non-affected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested:
If an index case in the family has been positively tested (XP-causing gene and mutation known) a negative test (not homozygous, compound heterozygous or hemizygous for disease-causing mutation) in another family member can be attributed with a nearly 100% negative clinical predictive value. To date, heterozygous mutation carriers are regarded as healthy individuals.3, 16
Index case in that family had not been tested:
Genetic testing of other family members without clinical symptoms and not testing the affected index patient is unusual and not recommended. XP is an autosomal-recessive disease. If there is no index case in the family, the probability of not suffering from XP, if there are suggestive clinical symptoms and the genetic testing was negative, may be estimated at 80–90%. This includes disease-causing alterations that cannot be detected with exonic and cDNA sequencing (eg promoter mutations, epigenetic alterations) and yet unidentified XP-causing genes. Eight XP-causing genes have been identified, but more than 20 proteins are involved in the nucleotide excision repair pathway.
3. Clinical utility
3.1 (Differential) diagnostics: the tested person is clinically affected
(To be answered if in 1.9 ‘A' was marked)
3.1.1 Can a diagnosis be made other than through a genetic test?

XP patients (except XP-V patients) show a defect in the nucleotide-excision repair (NER) pathway. This is a repair mechanism that enables the removal of bulky DNA lesions. NER almost exclusively removes the bulky DNA lesions that result from UV light exposure (DNA photoproduct removal).20
Unscheduled DNA synthesis (UDS) is a method to functionally measure the cellular NER capacity. The readout is the amount of incorporated labeled nucleoside analogs into the DNA after irradiation of (patient) cells with UV light (corresponds to the last gap-filling step of NER after excision of the DNA damage-carrying oligonucleotide). These analogs are either radioactively or fluorescently labeled and can be quantified per nucleus. UDS is reduced in XP patient cells, indicating reduced gap filling (DNA synthesis) after reduced excision repair. Like UDS, host cell reactivation (HCR) can also be utilized for functional cellular NER assessment and for complementation group assignment (XP-A to XP-G). Here, patient cells are transfected with a UV-irradiated reporter gene plasmid (coding for firefly luciferase). XP host cells show a markedly decreased luciferase expression compared with wild-type cells due to their inability to repair transcription-blocking UV-photoproducts from the reporter gene plasmid. Transfection of an expression plasmid containing wild-type cDNA of the respective XP gene is able to restore, at least in part, the NER capacity of XP patient cells. This in turn results in increased UDS or luciferase expression (HCR) and can therefore be used to assign XP patients complementation groups. For XP-V patients (no repair defect; defect in translesional synthesis) a post-UV cell-survival assay can be performed. This proliferation assay determines the general ability of cells to cope with cellular stress like UV-light exposure. In case of XP-V patients, the polymerase eta is unable to perform accurate translesion synthesis. Translesion synthesis occurs only during S phase. Caffeine reverses the cell cycle checkpoint and allows cells to enter S phase again. Only if caffeine is added to the cell cultural medium XP-V cells exhibit a reduced post-UV cell survival. Under these conditions, the defect of polymerase eta becomes detectable.3, 16
In addition, it is possible to diagnose XP-C patients using quantitative Real-Time PCR. The amount of XPC mRNA expression corresponds to the diseased alleles (reduction of one third in XPC heterozygotes and of more than two thirds in XPC homozygotes or compound heterozygotes). This holds true only for XPC but not the other XP genes.5, 6
3.1.2 Describe the burden of alternative diagnostic methods to the patient
The burden of skin punch biopsies is very low for the patients (bleeding, possibly infection, and pain) and will result in a tiny scar. The burden of drawing venous blood is negligible.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
Complementation group assignment applying functional tests (followed by gene sequencing) is the preferred process to minimize cost and time. However, the diagnosis of XP is primarily based on clinical examinations. To confirm the clinical diagnosis, genetic testing can be performed and allows to pinpoint the disease-causing gene as well as mutation. Due to the fact that XP is a highly diverse disease with at least eight disease-causing genes, abandonment of alternative diagnostics (complementation group assignment) is not advisable until new technologies such as next-generation sequencing may lower costs and turn-around time.
3.1.4 Will disease management be influenced by the result of a genetic test?

3.2 Predictive setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.9 ‘B' was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
If the test result is positive (please describe)
If the test result is negative (please describe)
3.2.2 Which options in view of lifestyle and prevention does a person at risk have if no genetic test has been done (please describe)?
Not applicable.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.9 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Yes.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
Yes. If the disease-causing mutation is known it is sufficient to test only for this mutation, for example, by rapid RFLP, to identify mutation carriers.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
No. As XP patients exhibit clinical symptoms starting with birth a positive genetic test result in an index patient enables prenatal diagnostics (s. 3.4).
3.4 Prenatal diagnosis
(To be answered if in 1.9 ‘D' was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Yes.
4. If applicable, further consequences of testing
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe).
Genetic XP testing is quite useful. First, the patients and his family know exactly the disease-causing gene and its mutation, and they better understand the reason for their symptoms. Second, siblings of the XP patients can be assured of being disease carriers or non-carriers. Third, as XP research advances genotype–phenotype correlations are beginning to emerge. In the future, doctors may be able to advice patients towards certain behaviors (eg strictness of sun avoidance) based on the type of mutation in a certain XP-causing gene. Knowing the causative gene and mutation enables accurate genetic counseling to the patient and his family, and further provides an option for prenatal diagnosis when desired. Several XP support groups worldwide (USA, UK, Germany, France, South Africa, Tunisia, Spain) with the overall goal to improve the quality of life of those affected with XP through education and support do exist. Concerning XP mutational databases the following web-sites may be useful: Genetics Home Reference (http://ghr.nlm.nih.gov/condition/xeroderma-pigmentosum), the Xeroderma Pigmentosum and Cockayne Syndrome Human Mutation Database (http://www.xpmutations.org), the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/hgmd0.html), and other databases like PubMed or those mentioned on the DNA Repair Interest Group web site (http://sigs.nih.gov/DNA-repair/Pages/default.aspx).
Acknowledgments
This work was supported by EuroGentest2 (Unit 2: ‘Genetic testing as part of health care'), a Coordination Action under FP7 (Grant Agreement Number 261469) and the European Society of Human Genetics. This work was supported in part by the Deutsche Forschungsgemeinschaft DFG, the Deutsche Krebshilfe e.V., the Niedersächsische Krebsgesellschaft e.V., and the Forschungsförderungsprogramm der Universitätsmedizin Göttingen (to SE).
The authors declare no conflict of interest.
References
- DiGiovanna JJ, Kraemer KH. Shining a light on xeroderma pigmentosum. J Invest Dermatol. 2012;132:785–796. doi: 10.1038/jid.2011.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford PT, Goldstein AM, Tamura D, et al. Cancer and neurologic degeneration in xeroderma pigmentosum: Long term follow-up characterizes the role of DNA repair. J Med Genet. 2011;48:168–176. doi: 10.1136/jmg.2010.083022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmert S.Xeroderma pigmentosum, Cockaynés Syndrome, and TrichothiodystrophyIn: Irvine AD, Hoeger P, Yan A (eds)Oxford (UK): Harper's Textbook of Pediatric Dermatology; 2011135.1–135.24. [Google Scholar]
- Oh K-S, Khan SG, Jaspers NGJ, et al. Phenotypic heterogeneity in the XPB DNA helicase gene (ERCC3): Xeroderma pigmentosum without and with Cockayne syndrome. Hum Mutat. 2006;27:1092–1103. doi: 10.1002/humu.20392. [DOI] [PubMed] [Google Scholar]
- Schäfer A, Hofmann L, Gratchev A, et al. Functional molecular-genetic analysis of 16 XP-C patients from Germany: environmental factors predominately contribute to phenotype variations. Exp Dermatol. 2013;22:24–29. doi: 10.1111/exd.12052. [DOI] [PubMed] [Google Scholar]
- Khan SG, Oh K-S, Shahlavi T, et al. Reduced XPC DNA repair gene mRNA levels in clinically normal parents of xeroderma pigmentosum patients. Carcinogenesis. 2006;27:84–94. doi: 10.1093/carcin/bgi204. [DOI] [PubMed] [Google Scholar]
- Khan SG, Levy HL, Legerski R, et al. Xeroderma pigmentosum group c splice mutation associated with autism and hypoglycinemia. J Invest Dermatol. 1998;111:791–796. doi: 10.1046/j.1523-1747.1998.00391.x. [DOI] [PubMed] [Google Scholar]
- Slor H, Batko S, Khan SG, et al. Clinical, cellular, and molecular features of an Israeli xeroderma pigmentosum family with a frameshift mutation in the XPC gene: sun protection prolongs life. J Invest Dermatol. 2000;115:974–980. doi: 10.1046/j.1523-1747.2000.00190.x. [DOI] [PubMed] [Google Scholar]
- Lehmann AR. The xeroderma pigmentosum group D (XPD) gene: one gene, two functions, three diseases. Genes Dev. 2001;15:15–23. doi: 10.1101/gad.859501. [DOI] [PubMed] [Google Scholar]
- Schäfer A, Hofmann L, Gratchev A, et al. Functional molecular-genetic analysis of 9 newly identified XPD-deficient patients reveals a novel mutation resulting in TTD as well as in XP/CS complex phenotypes. Exp Dermatol. 2013;22:486–489. doi: 10.1111/exd.12166. [DOI] [PubMed] [Google Scholar]
- Falik-Zaccai TC, Erel-Segal R, Horev L, et al. A novel XPD mutation in a compound heterozygote; the mutation in the second allele is present in three homozygous patients with mild sun sensitivity. Environ Mol Mutagen. 2012;53:505–514. doi: 10.1002/em.21716. [DOI] [PubMed] [Google Scholar]
- Oh K-S, Emmert S, Tamura D, DiGiovanna JJ, Kraemer KH. Multiple skin cancers in adults with mutations in the XP-E (DDB2) DNA repair gene. J Invest Dermatol. 2011;131:785–788. doi: 10.1038/jid.2010.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer A, Schubert S, Gratchev A, et al. Characterization of 3 XPG-defective patients identifies 3 missense mutations that impair repair and transcription. J Invest Dermatol. 2013;133:1841–1849. doi: 10.1038/jid.2013.54. [DOI] [PubMed] [Google Scholar]
- Thorel F, Constantinou A, Dunand-Sauthier Isabelle, et al. Definition of a short region of xpg necessary for TFIIH interaction and stable recruitment to sites of UV damage. Mol Cell Biol. 2004;24:10670–10680. doi: 10.1128/MCB.24.24.10670-10680.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inui H, Oh K-S, Nadem C, et al. Xeroderma variant patients from America, Europe and Asia. J Invest Dermatol. 2008;128:2055–2068. doi: 10.1038/jid.2008.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bootsma D, Kraemer KH, Cleaver JE, Hoeijmakers JH.Nucleotide excision repair syndromes: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophyIn: Vogelstein B, Kinzler KW (eds)New York, USA: The Genetic Basis of Human Cancer; 2002211–237. [Google Scholar]
- Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol. 1987;123:241–250. doi: 10.1001/archderm.123.2.241. [DOI] [PubMed] [Google Scholar]
- De Boer J, Hoeijmakers JH. Nucleotide excision repair and human syndromes. Carcinogenesis. 2000;21:453–460. doi: 10.1093/carcin/21.3.453. [DOI] [PubMed] [Google Scholar]
- Thoms KM, Kuschal C, Emmert S. Lessons learned from DNA repair defective syndromes. Exp Dermatol. 2007;16:532–544. doi: 10.1111/j.1600-0625.2007.00559.x. [DOI] [PubMed] [Google Scholar]
- Falik-Zaccai TC, Keren Z, Slor H. The versatile DNA nucleotide excision repair (NER) and its medical significance. Pediatr Endocrinol Rev. 2009;7:37–42. [PubMed] [Google Scholar]
- Tamura D, DiGiovanna JJ, Kraemer KH. Founder mutations in xeroderma pigmentosum. J Invest Dermatol. 2010;130:1491–1493. doi: 10.1038/jid.2010.76. [DOI] [PMC free article] [PubMed] [Google Scholar]