

1. Disease characteristics
1.1 Name of the disease (synonyms)
Hyperlipoproteinaemia, Type II
Hypercholesterolaemia, Familial
Hyperlipoproteinemia, Type IIA
Hyper-Low-Density-Lipoproteinaemia
1.2 OMIM# of the disease
143890.
1.3 Name of the analysed genes or DNA/chromosome segments:
LDLR (LRG_274), APOB (NM_000384.2) and PCSK9 (LRG_275).
1.4 OMIM# of the gene(s):
LDLR (606945), APOB (107730), PCSK9 (607786).
1.5 Mutational spectrum
About 1.300 variants in the most commonly affected gene in the autosomal dominant form of Familial Hypercholesterolemia (FH), LDLR, are listed in the UCL LDLR variant database (http://www.ucl.ac.uk/ldlr/Current/index.php?select_db=LDLR) and the LOVDv2.0 platform (https://grenada.lumc.nl/LOVD2/UCL-Heart/home.php?select_db=LDLR). These variants are equally distributed over the gene and include exonic substitutions, small exonic rearrangements, large rearrangements, promoter variants, intronic variants and a variant in the 3' untranslated sequence, point mutations, splice site mutations, large deletions, with approximately 80% being likely to be disease causing.1 Another reference database with known LDLR variants is maintained by Inserm (http://www.umd.be/LDLR/).
One major disease causing mutation in the APOB gene, c.10580 G>A (p.Arg3527Gln).2, 3, 4
Only a few PCSK9 mutations, all of the missense type and resulting in a ‘gain of function', have been found to be associated with the Hypercholesterolaemia phenotype, with some of them being restricted to certain ethnic groups.5
1.6 Analytical methods
Genetic testing: DNA extraction from peripheral blood (leucocytes), amplification of all 18 exons (LDLR) or 12 exons (PCSK9) and the promoter sequences or the part of APOB containing the major mutation, c.10580 G>A (p.Arg3527Gln), by PCR using flanking oligonucleotides, followed by direct DNA sequencing. Other methods such as Multiplex Ligation-dependent Probe Amplification can be used to detect deletions and duplications of one or more exons in the LDLR gene. A stepwise procedure according to the proportion of mutations found in the three genes involved in the disease has been recommended (LDLR>APOB>PCSK9), however, with the development of next generation sequencing methods in diagnostic laboratories, the approach of library capture of all exons promoters etc. of the three genes and simultaneous sequencing is becoming feasible and should be considered.6 Where homozygous FH or compound heterozygous FH is suspected and neither of the three genes have been found mutated, consider sequence analysis of the LDLRAP1 gene, responsible for the autosomal-recessive form of FH (ARH).
1.7 Analytical validation
The analysis of an independent biological sample is recommended in order to confirm an identified mutation. Analysis of samples from family members for segregation of the identified variant with the disease might be helpful, especially when the identified variation was not described before.
1.8 Estimated frequency of the disease
(Incidence at birth (‘birth prevalence') or population prevalence. If known to be variable between ethnic groups, please report):
The general prevalence is reported to be about 1:500, with a higher frequency because of founder effects in the following populations: Lebanese, Afrikaners in South Africa and French Canadians (reviewed in Liyanage KE et al.7), Finnish/North Karelia8 and Danish.9 Based on this frequency FH is dramatically underdiagnosed in most countries.3
1.9 Diagnostic setting

Comment: Prenatal testing is possible, but not carried out frequently because testing can be done soon after birth. Prenatal testing for homozygosity might be considered in families presenting a relevant constellation, however, there are differences in laws on prenatal testing between countries.
2. Test characteristics
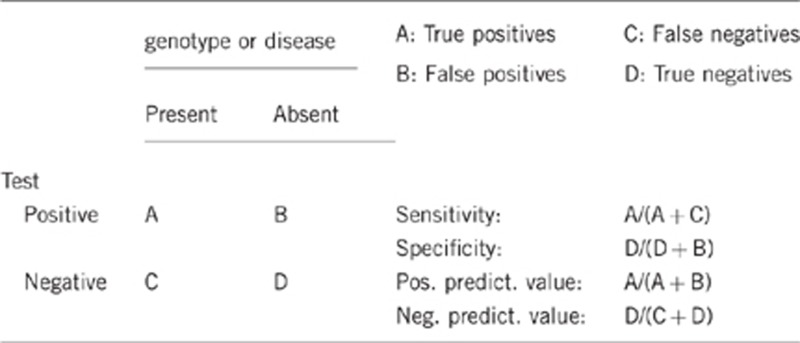
2.1 Analytical sensitivity
(proportion of positive tests if the genotype is present) Close to 100%.
2.2 Analytical specificity
(proportion of negative tests if the genotype is not present) Close to 100%.
2.3 Clinical sensitivity
(proportion of positive tests if the disease is present) The clinical sensitivity can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
Based on clinical criteria patients can be classified as definite FH or possible FH. In a UK study the mutation detection rate was roughly 60% for patients with definite FH and 30% for patients with possible FH.10 When patients are classified on the basis of the Dutch Lipid Clinic Network Criteria (DLCNC) score as ‘possible'(>3 and <5), ‘probable' (>5 and<8), or ‘definite' FH (>8), 70% of ‘definite' FH patients were found to carry a mutation, only 29% of 'probable' and 11% of ‘possible' FH patients were mutation-positive.11 Considering a similar mutation screening strategy these rates can be expected to be similar for most European populations.
2.4 Clinical specificity
(proportion of negative tests if the disease is not present) The clinical specificity can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
Approximately 100%. A negative FH diagnosis based on the LDL-C level made before the age of 2 (reviewed in Haney EM et al.12) and the rare paradox phenotype of relatively low LDL-C levels in people with a positive genetic test are reasons for not reaching the 100%.
2.5 Positive clinical predictive value
(life time risk to develop the disease if the test is positive)
Close to 100%. The paradox phenotype of relatively low LDL-C levels in genetically diagnosed FH patients was observed in different studies with a very low frequency, e.g. with a proportion of 1.6% in a Dutch cohort.13
2.6 Negative clinical predictive value
(Probability not to develop the disease if the test is negative) Assume an increased risk based on family history for a non-affected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested:
Close to 100%.
Index case in that family had not been tested:
Close to 100%.
3. Clinical Utility
3.1 (Differential) diagnostics: The tested person is clinically affected
(To be answered if in 1.9 ‘A' was marked)
3.1.1 Can a diagnosis be made other than through a genetic test?

3.1.2 Describe the burden of alternative diagnostic methods to the patient
The clinical diagnosis of definite FH or possible FH can be made based on the National Institute for Clinical Health and Excellence (NICE)-endorsed Simon Broome Criteria or the Dutch Lipid Clinic Network Criteria. The criteria consider LDL-Cholesterol levels, clinical findings (Xanthoma, premature atherosclerosis) and family history of hypercholesterolaemia or premature coronary heart disease.14, 15
There is an overlap with the autosomal-recessive form of hypercholesterolemia (OMIM #603813) and polygenic hypercholesterolaemia which can only be ruled out by a genetic diagnosis.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
Not applicable.
3.1.4 Will disease management be influenced by the result of a genetic test?

3.2 Predictive Setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.9 ‘B' was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
If the test result is positive (please describe): Yes, the patient would be aware of an increased risk for atherosclerotic events which might be prevented by changes in lifestyle, including fat modified diet with a high content of polyunsaturated fatty acids, Mediterranean diet, physical activity and lipid-lowering medication treatment.23
If the test result is negative (please describe): Persons with affected family members who suffered from early myocardial infarction or other consequences of FH will be relieved by the knowledge of being unaffected.
3.2.2 Which options in view of lifestyle and prevention does a person at-risk have if no genetic test has been done (please describe)?
There is no difference in the options with respect to lifestyle and prevention between genetically tested and non-tested individuals.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.9 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
A genetic test would show which family members are affected and need treatment.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
A positive genetic test will lead to additional genetic testing when cascade screening is initiated. However, knowledge of the mutation segregating in the family of the index patient will simplify genetic testing in further family members.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Yes.
3.4 Prenatal diagnosis
(To be answered if in 1.9 ‘D' was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Not applicable.
4. If applicable, further consequences of testing
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe)
Even if LDL-C is currently low such that statin treatment is not recommended, the person is aware that he is a carrier and therefore at risk for developing elevated LDL-C in later life, and this may encourage maintaining a healthy life style, and lead to regular monitoring of lipid levels by their doctor. Their first degree relatives (ie their children and brothers and sisters) who are at 50% risk of also having inherited the FH-causing mutation, may not also have inherited the genetic factors protecting the index case from having elevated LDL-C and should certainly be advised to have their genotype and lipid levels tested.
Acknowledgments
This work was supported by EuroGentest2 (Unit 2: ‘Genetic testing as part of health care'), a Coordination Action under FP7 (Grant Agreement Number 261469) and the European Society of Human Genetics. ID and EST were supported by the German Federal Ministry of Education and Research (#16SV5536K) and SEH is funded by the British Heart Foundation (PG08/008).
Elisabeth Steinhagen-Thiessen received honoraria for lectures, research grants and consultancy fees from the following companies (within the past two years): Aegerion, Fresenius, Medac, MSD Sharp&Dohme, Novartis, Pfizer and Sanofi.
References
- Usifo E, Leigh SE, Whittall RA, et al. Low-density lipoprotein receptor gene familial hypercholesterolemia variant database: update and pathological assessment. Annals of human genetics. 2012;76:387–401. doi: 10.1111/j.1469-1809.2012.00724.x. [DOI] [PubMed] [Google Scholar]
- Soria LF, Ludwig EH, Clarke HR, Vega GL, Grundy SM, McCarthy BJ. Association between a specific apolipoprotein B mutation and familial defective apolipoprotein B-100. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:587–591. doi: 10.1073/pnas.86.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: Consensus Statement of the European Atherosclerosis Society European heart journal 2013. doi: 10.1093/eurheartj/eht273 [DOI] [PMC free article] [PubMed]
- Varret M, Abifadel M, Rabes JP, Boileau C. Genetic heterogeneity of autosomal dominant hypercholesterolemia. Clinical genetics. 2008;73:1–13. doi: 10.1111/j.1399-0004.2007.00915.x. [DOI] [PubMed] [Google Scholar]
- Abifadel M, Rabes JP, Devillers M, et al. Mutations and polymorphisms in the proprotein convertase subtilisin kexin 9 (PCSK9) gene in cholesterol metabolism and disease. Human mutation. 2009;30:520–529. doi: 10.1002/humu.20882. [DOI] [PubMed] [Google Scholar]
- Futema M, Plagnol V, Whittall RA, et al. Use of targeted exome sequencing as a diagnostic tool for Familial Hypercholesterolaemia. Journal of medical genetics. 2012;49:644–649. doi: 10.1136/jmedgenet-2012-101189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liyanage KE, Burnett JR, Hooper AJ, van Bockxmeer FM. Familial hypercholesterolemia: epidemiology, Neolithic origins and modern geographic distribution. Critical reviews in clinical laboratory sciences. 2011;48:1–18. doi: 10.3109/10408363.2011.565585. [DOI] [PubMed] [Google Scholar]
- Vuorio AF, Turtola H, Piilahti KM, Repo P, Kanninen T, Kontula K. Familial hypercholesterolemia in the Finnish north Karelia. A molecular, clinical, and genealogical study. Arteriosclerosis, thrombosis, and vascular biology. 1997;17:3127–3138. doi: 10.1161/01.atv.17.11.3127. [DOI] [PubMed] [Google Scholar]
- Benn M, Watts GF, Tybjaerg-Hansen A, Nordestgaard BG. Familial hypercholesterolemia in the danish general population: prevalence, coronary artery disease, and cholesterol-lowering medication. The Journal of clinical endocrinology and metabolism. 2012;97:3956–3964. doi: 10.1210/jc.2012-1563. [DOI] [PubMed] [Google Scholar]
- Taylor A, Wang D, Patel K, et al. Mutation detection rate and spectrum in familial hypercholesterolaemia patients in the UK pilot cascade project. Clinical genetics. 2010;77:572–580. doi: 10.1111/j.1399-0004.2009.01356.x. [DOI] [PubMed] [Google Scholar]
- Hooper AJ, Nguyen LT, Burnett JR, et al. Genetic analysis of familial hypercholesterolaemia in Western Australia. Atherosclerosis. 2012;224:430–434. doi: 10.1016/j.atherosclerosis.2012.07.030. [DOI] [PubMed] [Google Scholar]
- Haney EM, Huffman LH, Bougatsos C, Freeman M, Steiner RD, Nelson HD. Screening and treatment for lipid disorders in children and adolescents: systematic evidence review for the US Preventive Services Task Force. Pediatrics. 2007;120:e189–e214. doi: 10.1542/peds.2006-1801. [DOI] [PubMed] [Google Scholar]
- Huijgen R, Kindt I, Fouchier SW, et al. Functionality of sequence variants in the genes coding for the low-density lipoprotein receptor and apolipoprotein B in individuals with inherited hypercholesterolemia. Human mutation. 2010;31:752–760. doi: 10.1002/humu.21258. [DOI] [PubMed] [Google Scholar]
- Wierzbicki AS, Humphries SE, Minhas R. Guideline Development G: Familial hypercholesterolaemia: summary of NICE guidance. Bmj. 2008;337:a1095. doi: 10.1136/bmj.a1095. [DOI] [PubMed] [Google Scholar]
- van Aalst-Cohen ES, Jansen AC, Tanck MW, et al. Diagnosing familial hypercholesterolaemia: the relevance of genetic testing. European heart journal. 2006;27:2240–2246. doi: 10.1093/eurheartj/ehl113. [DOI] [PubMed] [Google Scholar]
- European Association for Cardiovascular P, Rehabilitation. Reiner Z, Catapano AL, De Backer G, et al. ESC/EAS Guidelines for the management of dyslipidaemias: the Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS) European heart journal. 2011;32:1769–1818. doi: 10.1093/eurheartj/ehr158. [DOI] [PubMed] [Google Scholar]
- Shafiq N, Singh M, Kaur S, Khosla P, Malhotra S. Dietary treatment for familial hypercholesterolaemia. Cochrane database of systematic reviews. 2010;1:CD001918. doi: 10.1002/14651858.CD001918.pub2. [DOI] [PubMed] [Google Scholar]
- Lahera V, Goicoechea M, de Vinuesa SG, et al. Endothelial dysfunction, oxidative stress and inflammation in atherosclerosis: beneficial effects of statins. Current medicinal chemistry. 2007;14:243–248. doi: 10.2174/092986707779313381. [DOI] [PubMed] [Google Scholar]
- Cholesterol Treatment Trialists C. Baigent C, Blackwell L, Emberson J, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170 000 participants in 26 randomised trials. Lancet. 2010;376:1670–1681. doi: 10.1016/S0140-6736(10)61350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompsen J, Thompson PD. A systematic review of LDL apheresis in the treatment of cardiovascular disease. Atherosclerosis. 2006;189:31–38. doi: 10.1016/j.atherosclerosis.2006.02.030. [DOI] [PubMed] [Google Scholar]
- Starr B, Hadfield SG, Hutten BA, et al. Development of sensitive and specific age- and gender-specific low-density lipoprotein cholesterol cutoffs for diagnosis of first-degree relatives with familial hypercholesterolaemia in cascade testing. Clinical chemistry and laboratory medicine: CCLM/FESCC. 2008;46:791–803. doi: 10.1515/CCLM.2008.135. [DOI] [PubMed] [Google Scholar]
- Lee P, Hegele RA. Current Phase II proprotein convertase subtilisin/kexin 9 inhibitor therapies for dyslipidemia. Expert opinion on investigational drugs. 2013;22:1411–1423. doi: 10.1517/13543784.2013.822485. [DOI] [PubMed] [Google Scholar]
- Versmissen J, Oosterveer DM, Yazdanpanah M, et al. Efficacy of statins in familial hypercholesterolaemia: a long term cohort study. Bmj. 2008;337:a2423. doi: 10.1136/bmj.a2423. [DOI] [PMC free article] [PubMed] [Google Scholar]