

Abstract
Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired bone marrow disorder caused by expansion of a clone of hematopoietic cells lacking glycosylphosphatidylinositol (GPI)-anchored membrane proteins. Multiple lines of evidence suggest immune attack on normal hematopoietic stem cells provides a selective growth advantage to PNH clones. Recently, frequent loss of HLA alleles associated with copy number-neutral loss of heterozygosity in chromosome 6p (CN-6pLOH) in aplastic anemia (AA) patients was reported, suggesting that AA hematopoiesis “escaped” from immune attack by loss of HLA alleles. We report here the first case of CN-6pLOH in a Japanese PNH patient only in GPI-anchored protein positive (59%) granulocytes but not in GPI-anchored protein negative (41%) granulocytes. CN-6pLOH resulted in loss of the alleles A*02:06-DRB1*15:01-DQB1*06:02, which have been reported to be dominant in Japanese PNH patients. Our patient had maintained nearly normal blood count for several years. Our case supports the hypothesis that a hostile immune environment drives selection of resistant hematopoietic cell clones, and indicates that clonal evolution may occur also in normal phenotype (non-PNH) cells in some cases.
Keywords: paroxysmal nocturnal hemoglobinuria, array comparative genomic hybridization, loss of heterozygosity, clonal evolution, bone marrow failure syndromes
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare, life-threatening bone marrow failure syndrome which is characterized by three major features: intravascular hemolytic anemia, bone marrow failure, and thrombosis (1). PNH is an acquired clonal disorder of the hematopoietic stem cell (HSC) caused by a somatic mutation of the X-linked phosphatidylinositol glycan class A (PIGA) gene in one or a few hematopoietic stem cells (2). Even healthy individuals were reported to have very small number of PNH cells (3). The mechanism of clonal expansion of PNH cells is not understood, but the close association between PNH and aplastic anemia (AA) suggests that immune-mediated attack to hematopoietic stem cells underlies the pathogenesis of the association. Multiple evidences have accumulated to support the model of PNH clone expansion based on autoimmunity. PNH clones were less sensitive to NK and T cell killing due to a lack of expression of stress-inducible GPI-anchored proteins ULBP1 and ULBP2 in vitro and with patients granulocytes (4, 5), and inefficient T lymphocyte response was observed to GPI (−) cells in vitro and in mouse models (6). Recently, frequent loss of HLA alleles associated with copy number-neutral loss of heterozygosity of the 6p arms (CN-6pLOH) in AA patients was reported (7). Here we report the first case of a PNH patient with CN-6pLOH in GPI (+) granulocytes but not in GPI (−) granulocytes.
Patient and Method
A 33-year-old male presented to hospital for mild thrombocytopenia (130 × 109/l), and PNH was diagnosed by flow cytometry (1). The patient has not been treated for 2 years and 6 months due to lack of symptoms of anemia or thrombosis, though he had experienced hematuria several times a year since the diagnosis. PNH clone sizes were 49.0% and 22.0% in granulocytes and red blood cells at diagnosis, and were 45.2% and 28.5% respectively twelve months after the diagnosis. The LDH remained elevated since diagnosis (500 – 600 U/l). At the time of Array Comparative Genomic Hybridization (aCGH) analysis, twenty-four months after the diagnosis, the proportions of GPI-negative cells were 40.9%, 25.7%, and 4.7% in granulocytes, red blood cells, and T cells. Blood count included leukocyte 3.7 × 109/l, (38.8% neutrophils, 48.0% lymphocytes, 8.7% monocytes, 0.8% eosinophils, 0.5% basophils), hemoglobin 14.4 g/dl, MCV 101.9 fl, platelets 113 × 109/l, and reticulocyte count 112 × 109/l. The LDH was elevated at 620 U/l (normal range up to 229 U/l). Informed consent was obtained from the patient in accordance with the protocols approved by the Institutional Review Boards of Osaka University Hospital. Red blood cells were analyzed for GPI negativity with anti CD55 and anti CD59 antibodies in a CD235 positive population. Peripheral blood granulocytes (CD11b+7AAD-) and T cells (CD3+7AAD-) were separated into GPI (+) and GPI (−) cells by Flaer (Pinewood Scientific Services, BC, Canada) staining positivity or negativity. After sorting, each cell population of granulocytes was subjected to DNA extraction with the QIAamp DNA Blood Mini kit or the QIAamp DNA Micro kit (QIAGEN, Hilden, Germany). High-resolution genome-wide DNA copy number analysis was performed with both GPI (+) and GPI (−) granulocytes using the CytoScan®HD Array (Affymetrix, CA, USA). Sample processing was performed at Coriell Genotyping and Microarray Center, Coriell Institute for Medical Research (NJ, USA). The data was analyzed with Affymetrix Chromosome Analysis Suite (CHAS). For the analysis of clonal lesions, loss of heterozygosity (LOH) was called when the size was more than 25Mb and involved telomere (8). Alleles at HLA-A, -B, -DRB1, -DQB1, and -DPB1 loci were identified by PCR and sequence-specific oligonucleotide probes (PCR-SSOP) method using the WAKFlow HLA Typing kit (Wakunaga Phamaceutical Co., Ltd., Hiroshima, Japan) at the HLA Foundation Laboratory (Kyoto, Japan), as described previously (9). Briefly, target DNA was PCR-amplified with 5′-biotin-labeled primers that are highly specific to sequences of HLA genes. Amplified DNA was denatured and hybridized to locus-specific probes conjugated to microbeads labeled with streptavidin-phycoerythrin. The fluorescent intensity of phycoerhythrin on each coded oligobead was measured by the Luminex® 100 system (Luminex, TX, USA). The data analysis was performed using the WAKFlow® Typing Software (Wakunaga Phamaceutical Co., Ltd.). The haplotype of 6 loci was inferred based on the data of haplotype frequencies of a Japanese population (701 families; n=2,972) estimated by direct counting method. The data is available at the web site of the HLA Foundation Laboratory (http://hla.or.jp/haplo/haplonavi.php?type=haplo&lang=en).
Results and Discussion
The Affymetrix CytoScan® HD Array contains more than 2.4 million markers for copy numbers and 750 thousand SNPs, enabling detection of high resolution copy number, LOH detection, and breakpoint estimation across the genome. We employed the CytoScan®HD Array for aCGH analysis of submicroscopic aberrations of genomes in three Japanese PNH patients who had both GPI (+) and GPI (−) cells in granulocytes. Remarkably, CN-6pLOH was detected in a GPI (+) granulocyte population, but not in a GPI (−) granulocyte population, in a single patient (Fig. 1). Mosaicism was observed near the centromere of 6p, suggesting there were at least two clones with CN-6pLOH in GPI (+) cells. Both of the CN-LOH covered HLA class I and II genes (6p21.2 – 6p25.3, and 6p11.2 – 6p25.3). HLA typing of the patient by PCR-SSOP method was A*02:06, A*26:02, B*35:01, B*40:06, C*03:03, C*08:01, DRB1*09:01, DRB1*15:01, DQB1*03:03, DQB1*06:02, DPB1*02:01, DPB1*05:01. The haplotype of A*02:06-B*35:01-C*03:03-DRB1*15:01-DQB1*06:02- DPB1*05:01 was lost in 80% – 90% of GPI (+) granulocytes, due to 6pLOH. High frequency of CN-6pLOH in AA patients was reported by a Japanese group (7), and particular alleles including HLA-A*02:06 were dominantly missing, suggesting 6pLOH hematopoietic stem cells escape from the autoimmunity mediated by cytotoxic T cells (CTLs) which recognize unknown autoantigens on class I HLA molecules. HLA alleles A*02:06, DRB1*15:01, and DQB1*06:02 were reported to be frequent among Japanese PNH patients (10, 11), suggesting an immunological mechanism underlies the expansion of PNH clones. Autoimmune mechanisms have long been hypothesized for the expansion of PNH clones, and accumulated evidences support the hypothesis in literatures. PNH clones were reported to be less sensitive to NK and T cell killing (4, 5) or CD4+ T cells (6). GPI itself was suggested to be an autoantigen recognized by GPI-specific T cells (12, 13). GPI (+) granulocytes reflect purportedly “normal” hematopoiesis, but our previous studies showed frequent chromosomal abnormalities and apoptotic gene expression in this “normal” population. Based on these observations, in this patient, 80% to 90% of GPI (+) granulocytes were derived from clones that might have escaped immune attack by loss of the haplotype and therefore able to maintain nearly normal hematopoiesis for several years. The PNH clone in this patient propagated without loss of the alleles, supporting the hypothesis that PNH clones are less susceptible to immune attack even when they express autoantigens. HLA-restricted CTLs are supposed to play an important role in the clonal selection of GPI (+) cells in our patient, but how the PNH cells in the same patient escaped the attack is an open question. 6pLOH could also happen to the GPI (−) clone as a result of mitotic recombination, but it is still to be clarified if additional CN-6pLOH endows GPI (−) cells with comparative growth advantage under HLA-restricted immune attack. Clonal evolution in both the GPI (+) and GPI (−) cells may have caused the balanced cohabitation with nearly normal blood count. Further analysis would be necessary to examine if this phenomenon is common in other PNH patients, and assessment of both clones in the patient would be needed. In conclusion, our case supports the hypothesis that a hostile immune environment drives selection of resistant hematopoietic cell clones, and indicates that clonal evolution may occur also in normal phenotype (non-PNH) cells in some cases.
Fig. 1.
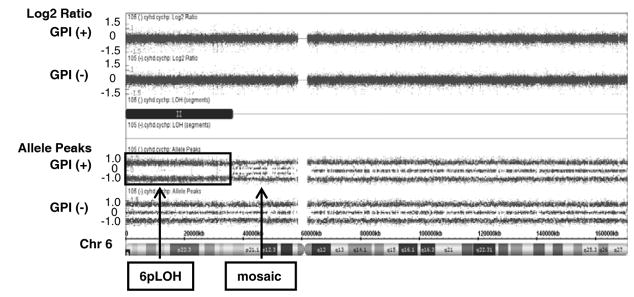
Acquired CN-6pLOH in GPI (+) granulocytes but not in GPI (−) granulocytes. Upper panel shows copy number status by log2 ratio: theoretically, the log 2 ratio of normal (copy number-neutral) clones is log2 (2/2) = 0, and of single copy losses is log2 (1/2) = −1. Lower panel shows allele frequency calculated as the difference between the signals of the A allele minus B allele. A homozygous AA maps to approximately +1, and a homozygous BB allele maps to approximately −1, with the heterozygote mapping to approximately 0. Single A and B allele maps to 0.5 and −0.5 respectively. Copy number was neutral in 6p arm, but loss of heterozygosity (disappearance of heterozygote signal) was observed with mosaicism near the centromere.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, the NHLBI, and in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology in Japan. The authors thank Satoru Hayashi for excellent technical assistance.
Footnotes
Authorship contributions
NSY was the principal investigator and takes primary responsibility for the paper; YU, SK, and NSY designed the research; YU and SK performed the laboratory work for this study; JN, YM and YK recruited the patients in Japan and provided vital patients samples and clinical information; TK, YK, and NSY coordinated and supervised the study; YU and NSY wrote the manuscript. All authors approved the final version of the manuscript.
Conflict of interest disclosures
The authors report no potential conflicts of interest.
References
- 1.Parker C, Omine M, Richards S, Nishimura J, Bessler M, Ware R, Hillmen P, Luzzatto L, Young N, Kinoshita T, Rosse W, Socie G. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106(12):3699–709. doi: 10.1182/blood-2005-04-1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Takeda J, Miyata T, Kawagoe K, Iida Y, Endo Y, Fujita T, Takahashi M, Kitani T, Kinoshita T. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993;73:703–11. doi: 10.1016/0092-8674(93)90250-t. [DOI] [PubMed] [Google Scholar]
- 3.Araten DJ, Nafa K, Pakdeesuwan K, Luzzatto L. Clonal populations of hematopoietic cells with paroxysmal nocturnal hemoglobinuria genotype and phenotype are present in normal individuals. Proc Natl Acad Sci USA. 1999;96:5209–14. doi: 10.1073/pnas.96.9.5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagakura S, Ishihara S, Dunn DE, Nishimura J, Kawaguchi T, Horikawa K, Hidaka M, Kagimoto T, Eto N, Mitsuya H, Kinoshita T, Young NS, Nakakuma H. Decreased susceptibility of leukemic cells with PIG-A mutation to natural killer cells in vitro. Blood. 2002;100(3):1031–7. doi: 10.1182/blood.v100.3.1031. [DOI] [PubMed] [Google Scholar]
- 5.Hanaoka N, Kawaguchi T, Horikawa K, Nagakura S, Mitsuya H, Nakakuma H. Immunoselection by natural killer cells of PIGA mutant cells missing stress-inducible ULBP. Blood. 2006;107(3):1184–91. doi: 10.1182/blood-2005-03-1337. [DOI] [PubMed] [Google Scholar]
- 6.Murakami Y, Kosaka H, Maeda Y, Nishimura J, Inoue N, Ohishi K, Okabe M, Takeda J, Kinoshita T. Inefficient response of T lymphocytes to GPI-anchor-negative cells: implications for paroxysmal nocturnal hemoglobinuria. Blood. 2002;100:4116–22. doi: 10.1182/blood-2002-06-1669. [DOI] [PubMed] [Google Scholar]
- 7.Katagiri T, Sato-Otsubo A, Kashiwase K, Morishima S, Sato Y, Mori Y, Kato M, Sanada M, Morishima Y, Hosokawa K, Sasaki Y, Ohtake S, Ogawa S, Nakao S. Frequent loss of HLA alleles associated with copy number-neutral 6pLOH in acquired aplastic anemia. Blood. 2011;118(25):6601–9. doi: 10.1182/blood-2011-07-365189. [DOI] [PubMed] [Google Scholar]
- 8.Maciejewski JP, Tiu RV, O’Keefe C. Application of array-based whole genome scanning technologies as a cytogenetic tool in haematological malignancies. Br J Haematol. 2009;146(5):479–88. doi: 10.1111/j.1365-2141.2009.07757.x. [DOI] [PubMed] [Google Scholar]
- 9.Itoh Y, Mizuki N, Shimada T, Azuma F, Itakura M, Kashiwase K, Kikkawa E, Kulski JK, Satake M, Inoko H. High-throughput DNA typing of HLA-A, -B, -C, and -DRB1 loci by a PCR-SSOPLuminex method in the Japanese population. Immunogenetics. 2005;57(10):717–29. doi: 10.1007/s00251-005-0048-3. [DOI] [PubMed] [Google Scholar]
- 10.Shichishima T, Okamoto M, Ikeda K, Kaneshige T, Sugiyama H, Terasawa T, Osumi K, Maruyama Y. HLA class II haplotype and quantitation of WT1 RNA in Japanese patients with paroxysmal nocturnal hemoglobinuria. Blood. 2002;100(1):22–8. doi: 10.1182/blood.v100.1.22. [DOI] [PubMed] [Google Scholar]
- 11.Shichishima T, Noji H, Ikeda K, Akutsu K, Maruyama Y. The frequency of HLA class I alleles in Japanese patients with bone marrow failure. Haematologica. 2006;91(6):856–7. [PubMed] [Google Scholar]
- 12.Karadimitris A, Luzzatto L. The cellular pathogenesis of paroxysmal nocturnal haemoglobinuria. Leukemia. 2001;15:1148–52. doi: 10.1038/sj.leu.2402180. [DOI] [PubMed] [Google Scholar]
- 13.Gargiulo L, Papaioannou M, Sica M, Talini G, Chaidos A, Richichi B, Nikolaev AV, Nativi C, Layton M, de la Fuente J, Roberts I, Luzzatto L, Notaro R, Karadimitris A. Glycosylphosphatidylinositol-specific, CD1d-restricted T cells in paroxysmal nocturnal hemoglobinuria. Blood. 2013;121(14):2753–61. doi: 10.1182/blood-2012-11-469353. [DOI] [PubMed] [Google Scholar]