

Abstract
It is increasingly clear that the interaction between host and microbiome profoundly affects health. There are 10 times more bacteria in and on our bodies than the total of our own cells, and the human intestine contains approximately 100 trillion bacteria. Interrogation of microbial communities by using classic microbiology techniques offers a very restricted view of these communities, allowing us to see only what we can grow in isolation. However, recent advances in sequencing technologies have greatly facilitated systematic and comprehensive studies of the role of the microbiome in human health and disease. Comprehensive understanding of our microbiome will enhance understanding of disease pathogenesis, which in turn may lead to rationally targeted therapy for a number of conditions, including autoimmunity.
Ankylosing spondylitis
Ankylosing spondylitis (AS) belongs to a common group of arthritides called spondyloarthopathies (SpA). AS targets primarily the spine and pelvis and is characterized histopathologically by entheseal inflammation. Disease progression in AS is characterized by excessive bone formation (ankylosis) that gradually bridges the gap between joints, eventually fusing joints and causing stiff ness, pain, significant morbidity, and increased mortality [1].
The coexistence of AS and intestinal inflammation has been known for some time [2]. Between 5% and 10% of patients with AS develop clinically diagnosed inflammatory bowel disease (IBD), and a further 70% of patients with AS develop subclinical gut inflammation [1,2]. In reactive arthritis, a member of the SpA family, inflammatory arthritis develops following urogenital infection with Chlamydia trachomatis or gastrointestinal infection with Campylobacter, Salmonella, Shigella, or Yersinia [3]. Such cause-and-effect relationships are not established for other SpAs.
Genetic overlap between ankylosing spondylitis and gut disease
Strong genetic overlap exists between AS and IBD, and the two conditions commonly occur together in families [4]. Danoy and colleagues [5] (2010) studied genes known to be associated with IBD in a large AS cohort. New loci and genes were identified, and of particular note were genes involved in the interleukin-23 (IL-23) pathway, such as STAT3, IL-23 receptor (IL23R), and IL12B (which encodes IL-12p40, the share subunit of IL-23 and IL-12) [5-7]. How these genetic lesions influence gut homeostasis and function remains unclear. Major differences also exist between the genetics of IBD and AS, and AS has shown no association to date with NOD2/CARD15 or the autophagy gene ATG16L1, which are major susceptibility factors in IBD, whereas IBD shows no association with HLA-B or ERAP1, which are the strongest AS susceptibility genes [8]. Although no association has been shown with NOD2/CARD15 and AS specifically, polymorphisms in NOD2/CARD15 have been associated with an increased risk of gut disease in patients with SpA [9].
The gut, barrier regulation, and intestinal epithelial cells
Homeostasis of the normal microbial flora in the gut is essential for intestinal health. The gastrointestinal tract is heavily populated with microbes and is the primary site for interaction between these microorganisms and the immune system [10,11]. Furthermore, microbes found in the gut help to shape host immune systems from an early age. The incomplete development of the immune system in neonates and under germ-free (GF) conditions tells us that microbiota sculpt the host immune system [12,13].
Maintenance of intestinal and microbial homeostasis is increasingly recognized as playing a pivotal role in overall health [14], and dysregulation of either gut or microbial homeostasis may play a role in autoimmunity. Physiological processes in the host that maintain gut homeostasis and respond to perturbance in the gut microenvironment involve both the adaptive and innate immune system and the barrier function of the intestines themselves.
The physical barrier
The human gastrointestinal tract is not a complete barrier. It is composed of a single layer of intestinal epithelial cells (IECs), which form a physical barrier separating the intestinal lumen from the lamina propria (Figure 1). IECs secrete soluble factors that are crucial to intestinal homeostasis, such as mucins and anti-microbial peptides, including lysozymes, defensins, cathelicidins, lipocalins, and C-type lectins such as RegIIIγ [15-17]. Release of these molecules into luminal crypts is thought to prevent microbial invasion into the crypt micro-environment as well as limit bacteria-epithelial cell contact [16,18]. Compared with healthy controls, Crohn's disease patients with active disease have pronounced decreases in the human α-defensins DEFA5 and DEFA6 in the ileum, resulting in altered mucosal function and overgrowth or dysregulation of commensal microbial flora [18,19]. Conversely, overexpression of anti-microbials, including α-defensins, are reported in sub-clinically inflamed ileum of AS patients compared with Crohn's disease patients and healthy controls [20]. However, it remains unclear whether changes in innate mucosal defenses lead to alterations in gut-resident microbial flora or whether early changes in the microbiome sculpt intestinal host responses. Furthermore, depletion of the mucin layer leads to an IBD-like phenotype and endoplasmic reticulum stress, potentially driving IL-23 production [21]. IL-23 excess alone is sufficient to induce spondyloarthritis in mice [22], and genetic evidence, in particular, indicates that the cytokine plays a key role in the development of spondyloarthritis in humans.
Figure 1.
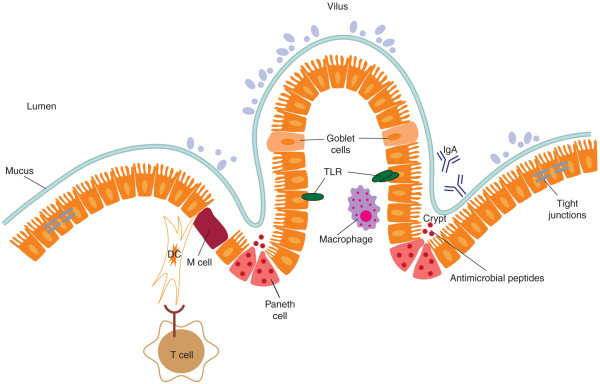
The physical barrier. Separating the intestinal lumen and its inhabiting commensal bacteria from the underlying lamina propria is a single layer of intestinal epithelial cells (IECs). These IECs are stitched together, creating a tight junction and regulating the paracellular flux. IECs also secrete soluble factors that are crucial to intestinal homeostasis, such as mucins and anti-microbial peptides (AMPs), including lysozymes, IgA, defensins, and C-type lectins such as RegIIIγ. Release of these molecules into luminal crypts is thought to prevent microbial invasion into the crypt microenvironment as well as limit bacteria-epithelial cell contact. Toll-like receptors (TLRs) are also expressed on IECs to sense a breach of barrier or bacterial invasion. Underneath the IECs, the lamina propria contains T cells, bacteria-sampling dendritic cells (DCs), and macrophages.
Permeability and disease
The dynamic crosstalk between IECs, microbes, and the local immune cells is fundamental to intestinal homeostasis but is also suggested to play a part in disease pathogenesis [23]. In IBD and celiac disease, tight junctions are dysregulated, causing increased permeability between IECs, resulting in a 'leaky gut' [24,25]. Studies looking at first-degree relatives of patients with AS showed that they too have increased gut permeability, so it is likely that there is an underlying genetic process operating in the gut [26,27].
The immune barrier
The complexity of the intestinal immune system is the subject of many reviews, but here we will focus on some intestinal immune populations that are believed to be important in rheumatic disorders.
Innate immunity
Dendritic cells (DCs) densely populate the intestinal lamina propria and form a widespread microbe-sensing network. DCs recognize a broad repertoire of bacteria, sensing with receptors such as Toll-like receptors (TLRs) and monitoring the bacteria on the mucosal surface [28]. Intestinal DCs orchestrate production of intestine-specific IgA to limit bacterial contact with the intestinal epithelial cell surface [29]. Activated DCs can secrete a number of cytokines and chemokines, including IL-23 and IL-6, that are involved in inflammation and migration of DCs [30].
Macrophages patrol the gastrointestinal systems in high numbers. They frequently come in contact with 'stray' bacteria, including commensals that have breached the epithelial cell barrier. Macrophages phagocytose and rapidly kill such bacteria by using mechanisms that include the production of antimicrobial proteins and reactive oxygen species [31]. Intestinal macrophages have several unique characteristics, including the expression of the anti-inflammatory cytokine IL-10, both constitutively and after bacterial stimulation [32,33]. This makes intestinal macrophages non-inflammatory cells that still have the capacity to phagocytose. The importance of this pathway is highlighted by the fact that loss-of-function mutations in IL10R lead to early-onset IBD [34]. However, not all intestinal macrophages are non-inflammatory. In patients with Crohn's disease, a population of intestinal macrophages secrete inflammatory cytokines such as IL-23, tumor necrosis factor-alpha (TNF-α), and IL-6. These macrophages contribute to an inflammatory microenvironment in patients with Crohn's disease [35]. Intestinal macrophages also play a role in the restoration of the physical integrity of the epithelial cell barrier following injury or bacterial insult [24]. Re-establishing this barrier after injury is imperative to avoid bacterial penetration and sepsis in such a microbe-laden environment [36].
Adaptive immunity
IL-23-responsive cells
IL-23 is a key cytokine in the development of IL-17- and IL-22-secreting cells. IL-23 signals through a receptor consisting of the specific IL-23R subunit and IL-12Rβ1, also shared with IL-12R [37]. Loss-of-function polymorphisms in IL23R are associated with protection from AS[7], psoriasis [38], and IBD [39], and many other genes in the IL-23 pathway are associated with these diseases. Under physiological conditions, IL-23-, IL-17-, and IL22-producing cells are enriched in gut mucosa, and IL-17 and IL-22 are known to be important regulators of intestinal 'health'. IL-17 plays important roles in intestinal homoeostasis in several ways, including maintenance of epithelial barrier tight junctions [40] and induction of anti-microbial proteins such as β-defensins, S100 proteins, and REG proteins. IL-22 induces secretion of anti-microbial peptides [41]. In the gut, innate-like immune cells act as sentinels, responding very rapidly to alterations to the microbial composition of the gut with rapid secretion of IL-17. Key among these sentinels are γδ T cells, natural killer T (NKT) cells, mucosaassociated invariant T (MAIT) cells, and lymphoid tissue inducer (LTi)-like cells (Figure 2).
Figure 2.
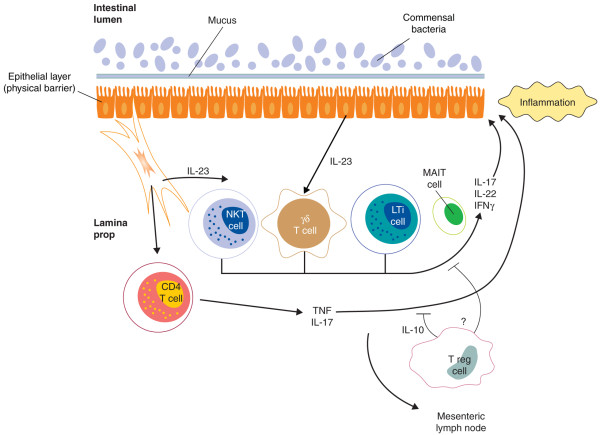
The immune barrier. Dendritic cells (DCs) and macrophages patrol the gastrointestinal tract in high numbers. They densely populate the intestinal lamina propria and form a widespread microbe-sensing network. Activated DCs can secrete a number of cytokines and chemokines, including interleukin-23 (IL-23), IL-6, and IL-1, activating IL-23-responsive cells. IL-23-, IL-17-, and IL-22-producing cells are enriched in gut mucosa, and IL-17 and IL-22 are known to be important regulators of intestinal 'health'. IL-17 plays important roles in intestinal homoeostasis in several ways, including maintenance of epithelial barrier tight junctions. LTi, lymphoid tissue inducer; MAIT, mucosa-associated invariant T; NKT, natural killer T; T reg, regulatory T; TNF, tumor necrosis factor.
γδ T cells
γδ T cells are found in large numbers at epithelial surfaces such as the gut and skin, where they can account for up to 50% of T cells. γδ T cells not only bear an antigen-specific T-cell receptor (TCR) but also have many properties of cells of the innate immune system, including expression of the major innate immunity receptors and TLRs [42] and dectin-1 [43], which recognizes microbial β-glucans. Expression of these receptors supports a role for γδ T cells in early responses to microbes. Of further relevance, we and others have recently confirmed that CARD9, part of the dectin-1 response pathway, is a susceptibility gene for AS and IBD [44]. γδ T cells are potent producers of inflammatory cytokines such as interferon-gamma (IFN-γ), TNF-α, and IL-17 [45,46].
They are pathogenic in the collagen-induced arthritis model [47] and mouse models of colitis [48], and IL-17secreting γδ T cells are expanded in patients with AS[49]. Intraepithelial γδ T cells also play an important role in modulating intestinal epithelial growth through secretion of fibroblast growth factor [50]. Alterations to γδ T-cell numbers or functions, therefore, may have profound effects on intestinal health.
Natural killer T cells
NKT cells are characterized by expression of an invariant TCR, Vα24Jα18 in humans and the orthologous Vα14Jα18 in mice. NKT cells recognize glycolipid structures presented to them by the non-classic antigen-presenting molecule CD1d. Like γδ T cells, NKT cells are rapid responders to antigenic stimuli and are capable of producing a range of immunoregulatory cytokines [51-54]. Within the gut, NKT cells are protective in T helper 1 (TH1)-mediated models of IBD but are pathogenic in TH2 models [55,56]. Recently, it has been shown that microbial stimulation of NKT cells in the gut of mice affects NKT cell phenotypic and functional maturation [57,58]. Given that NKT cells have protective roles in models of arthritis [59] and SpA [60], their functional maturation in the gut provides evidence for a role for mucosal T-cell priming in inflammatory joint disease.
Mucosa-associated invariant T cells
MAIT cells are a population of innate-like T cells that are abundant in human gut, liver, and blood and secrete a range of inflammatory cytokines such as IL-17 and IFN-γ in response to antigenic stimulation [61-63]. Like NKT cells, MAIT cells bear an invariant TCR (Vα7.2 in humans) that recognizes antigen presented by the non-classic MHC-like molecule, MR1 [64]. In blood, MAIT cells display a memory phenotype [63] and express the transcription factor ZBTB16 [65], allowing them to rapidly secrete cytokine in response to antigenic stimuli. Furthermore, they express high levels of IL-23R [66].
MAIT cells react to a wide range of microbial stimuli, including Gram-positive and Gram-negative bacteria as well as yeasts [61,62]. Although the precise role of MAIT cells in maintenance of the mucosal barrier remains unclear, the rapid postnatal expansion of MAIT cells and their acquisition of phenotypic markers of memory likely represent an interaction with developing commensal microflora [67].
Lymphoid tissue inducer-like cells and innate lymphoid cells
LTi-like cells are found in spleen, lymph node, and gut lamina propria. LTi-like cells constitutively express hallmarks of IL-17-secreting cells, including IL-23R, RORγt, AHR, and CCR6 [68]. Stimulation of LTi-like cells withIL-23 induces IL-17 secretion [68], whereas PMA and ionomycin stimulation invokes secretion of IL-22 [69]. LTi cells appear to be related to an increasingly interesting and heterogeneous population of innate lymphoid cells (ILCs). ILCs have been linked to gut inflammation through colitis models in which IL-23-responsive ILCs secrete IL-17 and IFN-γ and promote intestinal inflammation [70]. NKp46+ ILCs are involved in host defense against Citrobacter rodentium infection through secretion of IL-22 [71].
The human microbiome revolution
Together, the multi-factorial components of the immune system shape the microbial population that inhabits the gut and (to an extent) vice versa, with each side pushing to establish a stable state. Understanding the yin and yang of this relationship is at the heart of current micro-biome research.
Microbiome refers to the totality of microbes, their genetic elements, and environmental interaction in a defined environment [72]. Projects such as the Human Microbiome Project, run by the National Institutes of Health, and the Metagenomics of the Human Intestinal Tract (MetaHit) consortium aim to determine what constitutes a 'normal' or healthy microbiome. Research in this field has recently been greatly accelerated by the development of high throughput sequencing methods for microbial profiling, which can profile microbial populations whether or not the microbes present can be cultured.
16S rRNA sequencing
16S rRNA is a section of prokaryotic DNA found in all bacteria and archaea. 16S rRNA sequences are used to differentiate between organisms across all major phyla of bacteria and to classify strains down to the species level [73]. 16S rRNA sequencing has dramatically changed our understanding of phylogeny and microbial diversity because it provides an unbiased assessment of the microbiome and is not restricted by the ability to culture the bacteria present.
Further improvements in sequencing technologies have reduced the need for targeted studies such as 16S sequencing and enabled high-throughput shotgun sequencing. This latter type of sequencing randomly samples all genes present in a habitat rather than just 16S rRNA. Shotgun sequencing provides more information about the micro-biome than just the 16S characterization, which is of particular benefit in determining the functional capacity of the microbial community rather than just its phylogeny. However, shotgun sequencing is more complex to analyze, owing in part to the challenge of distinguishing between host and bacterial genomic material, and requires far more sequencing to be performed. Therefore, most studies to date have relied on 16S rRNA sequencing approaches.
Advances in tools for metagenomic studies
Several sequencing technologies have been developed for human genetic studies and have since been adapted to metagenomics. The Roche 454 sequencing platform(Roche, Basel, Switzerland) uses large-scale parallel pyrosequencing to produce reads of between 450 and 1000 base pairs (bp) in length. Read lengths produced by the 454 platform are well suited to 16S rRNA amplicon metagenomics studies as well as shotgun sequencing as they are easily aligned to reference bacterial genetic data sets. Sequencing platforms from Illumina (San Diego, CA, USA), the HiSeq2000 and MiSeq, produce shorter reads than the Roche 454. The HiSeq was designed primarily for human genomic sequencing, and current chemistry produces 100-bp paired-end reads. Illumina sequencing is best suited for shotgun sequencing or indexed amplicon sequencing of multiple samples.
This is a rapidly developing field. Advances in chemistry are predicted to increase both read lengths and output for both of these platforms over the next 12 to 24 months, particularly for the Illumina platforms that are less mature than the Roche 454. New platforms coming to the market are likely to have a major impact on metagenomics study design. For example, the Pacific Biosciences sequencing technology (Pacific Biosciences, Menlo Park, CA, USA) provides reads of approximately 3,000 bases, which will make it particularly suited to this field, despite its lower overall output (approximately 100 Mb of sequence per run). Life Technologies Ion Torrent technology (Life Technologies, Grand Island, NY, USA) is reported to produce up to 400 base reads and up to 1 Gb of sequence per run. The relative positions of these competing technologies in metagenomics have yet to be established.
The normal human gut microbiome
To date, only a handful of studies have examined in any depth the function as well as the composition and diversity of the human gut microbiome. Two large studies interrogating and cataloguing microbiomes from various regions of the body in health and disease have been undertaken by the National Institutes of Health Human Microbiome Project in the US and the European MetaHit project [74-76]. The European MetaHit consortium combined published data sets from around the world and added 22 newly sequenced fecal metagenomes from four different European countries. They identified three robust clusters, termed 'enterotypes', that were not nation- or continent-specific. These enterotypes characterized the microbial phylogenetic variation as well as the function variation of the clusters at gene and functional class levels [74]. Each enterotype had a dominant bacterial genus: enterotype 1 was dominated by the genus Bacteriodes, enterotype 2 by Prevotella, and enterotype 3 by Ruminococcus. The three enterotypes were also shown to be functionally different. For example, enterotype 2, which is Prevotella-dominant, also contains Desfulfovibrio, which may act in synergy with Prevotella to degrade mucin glycoproteins present in the mucosal layer of the gut. It may be that different enterotypes may be associated with diseases such as obesity and IBD rather than necessarily specific bacterial species, given their differing functional capacities. Further studies will be required to determine whether enterotypes are consistently found in expanded data sets and in studies of intestinal biopsies as well as the fecal samples used in the MetaHit study.
The microbiome in immune-mediated arthritis
The major findings of microbial profiling studies in major immune-mediated diseases associated with arthritis are summarised in Table 1. To date, no study investigating the gut microbiome by using sequencing-based methods in AS has been reported. Many studies largely using antibody tests have suggested an increased carriage of Klebsiella species in patients with AS, but this has not been universally supported [77]. One study using denaturing gradient gel electrophoresis to profile the microbiome by using fecal samples found no differences between AS cases and healthy controls [78].
Table 1.
Alterations in gut microbiota associated with immune-mediated diseases
| Associated microbes | Microbiota changes | References |
|---|---|---|
| IBD - Crohn's disease | Reduction in microbial diversity when compared with controls | [95] |
| Gut microbiome | ||
| Bacteroides ovatus ↑ | ||
| Bacteroides vulgatus ↑ | ||
| Bacteroides uniformis ↓ | ||
| IBD | Associated with overall community shift and dysbiosis | [96] |
| Gut microbiome | ||
| Bacteroidetes ↓ | ||
| Lachnospiraceae ↓ | ||
| Actinobacteria ↑ | ||
| Proteobacteria ↑ | ||
| Clostridium ↓ | ||
| Firmicutes/Bacteroidetes ratio ↓ | ||
| Bifi dobacteria ↓ | ||
| Celiac disease | Overall higher diversity of microbes in patients with celiac disease compared with controls | [97] |
| Gut microbiome | ||
| Bacteroides vulgatus ↑ | ||
| Escherichia coli ↓ | ||
| Clostridium coccoides ↓ | ||
| Psoriasis | Overrepresentation of Firmicutes and an underrepresentation of Actinobacteria and Proteobacteria when compared with controls | [98,99] |
| Skin microbiome | ||
| Firmicutes ↑ | ||
| Bacteroidetes ↑ | ||
| Actinobacteria ↓ | ||
| Proteobacteria ↓ | ||
| Rheumatoid arthritis | Dysbiosis and increased diversity | [100,101] |
| Oral microbiome | ||
| Porphyromonas gingivalis ↑ |
IBD, inflammatory bowel disease.
Rheumatoid arthritis (RA) is the only inflammatory arthritis for which modern metagenomic studies have been reported. Community profiling studies of gut flora of patients with RA reveal differences in the composition of gut microbiota of patients with RA compared with those of healthy controls, and a lower abundance of Bifidobacterium and Bacteroides bacteria was observed in RA cases [79,80]. However, these studies used fecal samples and not intestinal biopsies, possibly influencing the populations observed [81]. Also, microbial profiling data suggest that gingival infection may be important, particularly in anti-citrullinated peptide antibody-positive RA, although the studies suggesting this have generally used antibody tests rather than sequencing-based approaches (reviewed in [82]).
Several lines of evidence indicate that the gut micro-biome plays an important role in IBD, including the association of genes involved in mucosal immunity with IBD (such as CARD15, CARD9, IL-23R, and ATG16L1), the therapeutic effect of antibiotics on the condition, and the beneficial effect of fecal stream diversion in Crohn's disease. Previous studies of human IBD have been undertaken by using standard culture techniques (for example, [83]) or molecular analysis (for example, [84,85]). These studies noted alterations in intestinal microbiota when compared with non-IBD patients, a finding recently confirmed by using 16S rRNA sequencing of intestinal biopsies [86]. This sequencing study, however, did not identify an IBD-specific microbial profile, perhaps because of insufficient resolution of the sequencing performed or small sample size (12 patients and 5 controls). Much larger studies will be required to dissect the relationship between the host genetic factors determining risk of IBD and the gut microbiome.
The microbiome is thought to play a significant role in psoriasis, another AS-related condition. It has long been suggested that streptococcal infection, especially throat infections, may trigger psoriasis in a genetically susceptible individual [87]. Recent studies using 16S rRNA sequencing have found significant differences between the cutaneous microbiota of psoriasis cases and controls and in involved and control skin in psoriasis cases, and less staphylococci and propionic bacteria have been observed in cases and in affected skin [88]. Again, further studies will be required to determine whether there is a particular microbial profile or specific bacterial species involved in psoriasis.
These studies are thus consistent with the hypothesis that the microbiome contributes to the etiopathogenesis of immune-mediated arthritis or seronegative diseases like IBD and psoriasis. However, at this point, there is no definitive evidence of specific bacterial infections or changes in microbial profile that play a causative role in these conditions (with the exception of reactive arthritis).
Chicken and the egg
Whether the changes noted in these early metagenomic studies of immune-mediated disease are a consequence of disease or are involved in its development or persistence is unclear. This distinction may prove impossible to dissect in human studies, but considerable evidence in studies in mice supports a role for the microbiome in driving immune-mediated diseases.
In the B27 rat model of AS, rats housed under GF conditions did not develop disease [89], demonstrating that microbes in this model are important for disease penetrance. In contrast, in the New Zealand black model of systemic lupus erythematosus, mice maintained under GF conditions produced higher levels of antinuclear antibodies and developed worse disease [90], demonstrating a protective role for commensal microbes.
Given the IL-17/IL-22 cytokines, which are of relevance to AS, IBD, and psoriasis in particular, strong evidence from murine studies indicates that interaction between the gut microbiome and the host determines the overall level of activation of the immune cells producing these cytokines. Segmented filamentous bacteria (SFB) are com mensal bacteria that induce IL-17 secretion. Mice lacking SFB have low levels of intestinal IL-17 and are susceptible to infection with pathogenic Citrobacter spp. Restoration of SFB in these mice increased the number of gut-resident IL-17-producing cells and enhanced resistance to infection [12]. Salzman and colleagues [91] illustrated that α-defensins modulate mucosal T-cell response by regulating the composition, but not the total numbers, of bacteria in the intestinal microbiome.
Examining the intestinal microbiota of mice expressing the human α-defensin gene, they demonstrated significant α-defensin-dependent alterations in commensal composition, leading to a loss of SFB and fewer IL-17producing lamina propria T cells [91]. Furthermore, using recolonization studies, investigators recently demonstrated that, in neonatal mice, commensal microbes influence invariant NKT (iNKT) cell intestinal infiltration and activation, establishing mucosal iNKT cell tolerance to later environmental exposures [92].
The mechanism by which the microbiome influencesIL-17-producing cell activation is still being determined. Ivanov and colleagues [12] demonstrated that serum amyloid A, produced in the terminal ileum, can induce TH17 differentiation of CD4+ T lymphocytes. It has also been shown that development of TH17 lymphocytes in the intestine is stimulated by microbiota-induced IL-1β (but not IL-6) production [93]. Colonization with Clostridial species has been shown to stimulate intestinal transforming growth factor-beta (TGF-β) production, in turn increasing IL-10+ CTLA4high Treg (regulatory T)activation [94]. Clostridial colonization of neonatal mice reduced severity of induced colitis by using dextran sulphate sodium or oxazolone and reduced serum IgE levels in adulthood. So it is likely that alterations to the gut microflora or invasion of the gut by pathogenic bacteria influences the balance of IL-17- and IL-22producing cells and other immune cells, influencing susceptibility to local and systemic immune-mediated disease.
The above studies highlight that changes in the micro-biome can lead to inflammation which may have far-reaching effects and demonstrate experimental approaches by which findings of metagenomic studies in mice and humans can be explored to successfully dissect the role of the microbiome in human immune-mediated diseases.
Conclusions
The human gut microbiome is a dynamic and complex ecosystem that is only now beginning to be understood. It is increasingly clear that the interaction between host and microbiome profoundly affects health. It is still unclear how interactions between host genes, microbes,and environmental factors can predispose patients to the development autoimmune diseases such as AS. We are only beginning to grasp the influence the microbiome has on health. Improved knowledge of the composition and function of the gut microbiome in patients with AS and how the microbiome shapes the immune response and influences inflammation, both local and systemic, will likely provide important insights into early events in the pathogenesis of AS.
Abbreviations
AS: ankylosing spondylitis; bp: base pair(s); DC: dendritic cell; GF: germ-free; IBD: inflammatory bowel disease; IEC: intestinal epithelial cell; IFNγ: interferon-gamma; IL: interleukin; IL-23R: interleukin-23 receptor; ILC: innate lymphoid cell; iNKT: invariant natural killer T; LTi: lymphoid tissue inducer; MAIT: mucosaassociated invariant T; MetaHit: Metagenomics of the Human Intestinal Tract; NKT: natural killer T; RA: rheumatoid arthritis; SFB: segmented filamentous bacteria; SpA: spondyloarthopathies; TCR: T-cell receptor; TH: T helper; TLR: Toll-like receptor; TNF-α: tumor necrosis factor-alpha.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Mary-Ellen Costello, Email: m.costello5@uq.edu.au.
Dirk Elewaut, Email: Dirk.Elewaut@Ugent.be.
Tony J Kenna, Email: t.kenna@uq.edu.au.
Matthew A Brown, Email: matt.brown@uq.edu.au.
Acknowledgements
The authors thank Murray Hargrave and Philip Robinson for their assistance in the preparation of this review. TJK is supported by an Arthritis Australia Heald Fellowship. MAB is supported by Australian National Health and Medical Research Council Senior Principal Research Fellowship APP1024879. DE is supported by grants of the Fund for Scientific Research Flanders and the Research Council of Ghent University.
References
- Thomas GP, Brown MA. Genetics and genomics of ankylosing spondylitis. Immunol Rev. 2010;15:162–180. doi: 10.1111/j.0105-2896.2009.00852.x. [DOI] [PubMed] [Google Scholar]
- Mielants H, Veys EM, Goemaere S, Goethals K, Cuvelier C, De Vos M. Gut inflammation in the spondyloarthropathies: clinical, radiologic, biologic and genetic features in relation to the type of histology. A prospective study. J Rheumatol. 1991;15:1542–1551. [PubMed] [Google Scholar]
- Uotila T, Antonen J, Laine J, Kujansuu E, Haapala AM, Lumio J, Vuento R, Oksa H, Herrala J, Kuusi M, Mustonen J, Korpela M. Pirkanmaa Waterborne Outbreak Study Group. Reactive arthritis in a population exposed to an extensive waterborne gastroenteritis outbreak after sewage contamination in Pirkanmaa, Finland. Scand J Rheumatol. 2011;15:358–362. doi: 10.3109/03009742.2011.562533. [DOI] [PubMed] [Google Scholar]
- Thjodleifsson B, Geirsson AJ, Bjornsson S, Bjarnason I. A common genetic background for inflammatory bowel disease and ankylosing spondylitis: a genealogic study in Iceland. Arthritis Rheum. 2007;15:2633–2639. doi: 10.1002/art.22812. [DOI] [PubMed] [Google Scholar]
- Danoy P, Pryce K, Hadler J, Bradbury LA, Farrar C, Pointon J. Australo-Anglo-American Spondyloarthritis Consortium; Ward M, Weisman M, Reveille JD, Wordsworth BP, Stone MA. Spondyloarthritis Research Consortium of Canada; Maksymowych WP, Rahman P, Gladman D, Inman RD, Brown MA. Association of variants at 1q32 and STAT3 with ankylosing spondylitis suggests genetic overlap with Crohn's disease. PLoS Genet. 2010;15:e1001195. doi: 10.1371/journal.pgen.1001195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austral o-Anglo-American Spondyloarthritis Consortium (TASC); Reveille JD, Sims AM, Danoy P, Evans DM, Leo P, Pointon JJ, Jin R, Zhou X, Bradbury LA, Appleton LH, Davis JC, Diekman L, Doan T, Dowling A, Duan R, Duncan EL, Farrar C, Hadler J, Harvey D, Karaderi T, Mogg R, Pomeroy E, Pryce K, Taylor J, Savage L, Deloukas P, Kumanduri V, Peltonen L, Ring SM, Whittaker P, Glazov E, Thomas GP, Maksymowych WP, Inman RD, Ward MM, Stone MA, Weisman MH, Wordsworth BP, Brown MA. Genome-wide association study of ankylosing spondylitis identifies non-MHC susceptibility loci. Nat Genet. 2010;15:123–127. doi: 10.1038/ng.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium; Australo-Anglo-American Spondylitis Consortium (TASC); Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, Kwiatkowski DP, McCarthy MI, Ouwehand WH, Samani NJ, Todd JA, Donnelly P, Barrett JC, Davison D, Easton D, Evans DM, Leung HT, Marchini JL, Morris AP, Spencer CC, Tobin MD, Attwood AP, Boorman JP, Cant B, Everson U, Hussey JM, Jolley JD, Knight AS, Koch K, Meech E. et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet. 2007;15:1329–1337. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson PC, Brown MA. The genetics of ankylosing spondylitis and axial spondyloarthritis. Rheum Dis Clin North Am. 2012;15:539–553. doi: 10.1016/j.rdc.2012.08.018. [DOI] [PubMed] [Google Scholar]
- Laukens D, Georges M, Libioulle C, Sandor C, Mni M, Vander Cruyssen B, Peeters H, Elewaut D, De Vos M. Evidence for significant overlap between common risk variants for Crohn's disease and ankylosing spondylitis. PLoS ONE. 2010;15:e13795. doi: 10.1371/journal.pone.0013795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;15:313–323. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;15:1258–1270. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, Tanoue T, Imaoka A, Itoh K, Takeda K, Umesaki Y, Honda K, Littman DR. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;15:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan J, Chung H, Troy E, Kasper DL. Microbial colonization drives expansion of IL-1 receptor 1-expressing and IL-17-producing gamma/delta T cells. Cell Host Microbe. 2010;15:140–150. doi: 10.1016/j.chom.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nat Rev Genet. 2012;15:10. doi: 10.1038/nrg3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;15:298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- Koslowski MJ, Beisner J, Stange EF, Wehkamp J. Innate antimicrobial host defense in small intestinal Crohn's disease. Int J Med Microbiol. 2010;15:34–40. doi: 10.1016/j.ijmm.2009.08.011. [DOI] [PubMed] [Google Scholar]
- Wehkamp J, Koslowski M, Wang G, Stange EF. Barrier dysfunction due to distinct defensin deficiencies in small intestinal and colonic Crohn's disease. Mucosal Immunol. 2008;15:S67–S74. doi: 10.1038/mi.2008.48. [DOI] [PubMed] [Google Scholar]
- Simms LA, Doecke JD, Walsh MD, Huang N, Fowler EV, Radford-Smith GL. Reduced α-defensin expression is associated with inflammation and not NOD2 mutation status in ileal Crohn's disease. Gut. 2008;15:903–910. doi: 10.1136/gut.2007.142588. [DOI] [PubMed] [Google Scholar]
- Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, Shen B, Schaeffeler E, Schwab M, Linzmeier R, Feathers RW, Chu H, Lima H Jr, Fellermann K, Ganz T, Stange EF, Bevins CL. Reduced Paneth cell α-defensins in ileal Crohn's disease. Proc Natl Acad Sci USA. 2005;15:18129–18134. doi: 10.1073/pnas.0505256102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia F, Bombardieri M, Rizzo A, Principato A, Giardina AR, Raiata F, Peralta S, Ferrante A, Drago S, Cottone M, Pitzalis C, Triolo G. Over-expression of paneth cell-derived anti-microbial peptides in the gut of patients with ankylosing spondylitis and subclinical intestinal inflammation. Rheumatology. 2010;15:2076–2083. doi: 10.1093/rheumatology/keq239. [DOI] [PubMed] [Google Scholar]
- Heazlewood CK, Cook MC, Eri R, Price GR, Tauro SB, Taupin D, Thornton DJ, Png CW, Crockford TL, Cornall RJ, Adams R, Kato M, Nelms KA, Hong NA, Florin TH, Goodnow CC, McGuckin MA. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 2008;15:e54. doi: 10.1371/journal.pmed.0050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherlock JP, Joyce-Shaikh B, Turner SP, Chao CC, Sathe M, Grein J, Gorman DM, Bowman EP, McClanahan TK, Yearley JH, Eberl G, Buckley CD, Kastelein RA, Pierce RH, Laface DM, Cua DJ. IL-23 induces spondyloarthropathy by acting on ROR-gammat+ CD3+CD4-CD8- entheseal resident T cells. Nat Med. 2012;15:1069–1076. doi: 10.1038/nm.2817. [DOI] [PubMed] [Google Scholar]
- Artis D. E pithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat Rev Immunol. 2008;15:411–420. doi: 10.1038/nri2316. [DOI] [PubMed] [Google Scholar]
- Reid G, Younes JA, Van der Mei HC, Gloor GB, Knight R, Busscher HJ. Microbiota restoration: natural and supplemented recovery of human microbial communities. Nat Rev Micro. 2011;15:27–38. doi: 10.1038/nrmicro2473. [DOI] [PubMed] [Google Scholar]
- Lubrano E, Ciacci C, Ames PR, Mazzacca G, Oriente P, Scarpa R. The arthritis of coeliac disease: prevalence and pattern in 200 adult patients. Rheumatology. 1996;15:1314–1318. doi: 10.1093/rheumatology/35.12.1314. [DOI] [PubMed] [Google Scholar]
- Vaile JH, Meddings JB, Yacyshyn BR, Russell AS, Maksymowych WP. Bowel permeability and CD45RO expression on circulating CD20+ B cells in patients with ankylosing spondylitis and their relatives. J Rheumatol. 1999;15:128–135. [PubMed] [Google Scholar]
- Martínez-G onzález O, Cantero-Hinojosa J, Paule-Sastre P, Gómez-Magán JC, Salvatierra-Ríos D. Intestinal permeability in patients with ankylosing spondylitis and their healthy relatives. Rheumatology. 1994;15:644–647. doi: 10.1093/rheumatology/33.7.644. [DOI] [PubMed] [Google Scholar]
- Chabot S, Wagner JS, Farrant S, Neutra MR. TLRs regulate the gatekeeping functions of the intestinal follicle-associated epithelium. J Immunol. 2006;15:4275–4283. doi: 10.4049/jimmunol.176.7.4275. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Ha SA, Tsuji M, Fagarasan S. Intestinal IgA synthes is: a primitive form of adaptive immunity that regulates microbial communities in the gut. Semin Immunol. 2007;15:127–135. doi: 10.1016/j.smim.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Wesa A, Galy A. Increased production of pro-inflammatory cytokines and enhanced T cell responses after activation of human dendritic cells with IL-1 and CD40 ligand. BMC Immunology. 2002;15:14. doi: 10.1186/1471-2172-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem. 2007;15:2871–2879. doi: 10.1074/jbc.M608083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirotani T, Lee PY, Kuwata H, Yamamoto M, Matsumoto M, Kawase I, Akira S, Takeda K. The nuclear IkappaB protein IkappaBNS selectively inhibits lipopolysaccharide-induced IL-6 production in macrophages of the colonic lamina propria. J Immunol. 2005;15:3650–3657. doi: 10.4049/jimmunol.174.6.3650. [DOI] [PubMed] [Google Scholar]
- Denning TL, Wang Y-c, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol. 2007;15:1086–1094. doi: 10.1038/ni1511. [DOI] [PubMed] [Google Scholar]
- Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schäffer AA, Noyan F, Perro M, Diestelhorst J, Allroth A, Murugan D, Hatscher N, Pfeifer D, Sykora KW, Sauer M, Kreipe H, Lacher M, Nustede R, Woellner C, Baumann U, Salzer U, Koletzko S, Shah N, Segal AW, Sauerbrey A, Buderus S, Snapper SB, Grimbacher B, Klein C. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;15:2033–2045. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada N, Hisamatsu T, Okamoto S, Chinen H, Kobayashi T, Sato T, Sakuraba A, Kitazume MT, Sugita A, Koganei K, Akagawa KS, Hibi T. Unique CD14+ intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-γ axis. J Clin Invest. 2008;15:2269–2280. doi: 10.1172/JCI34610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pull SL, Doherty JM, Mills JC, Gordon JI, Stappenbeck TS. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc Natl Acad Sci USA. 2005;15:99–104. doi: 10.1073/pnas.0405979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, Zonin F, Vaisberg E, Churakova T, Liu M, Gorman D, Wagner J, Zurawski S, Liu Y, Abrams JS, Moore KW, Rennick D, de Waal-Malefyt R, Hannum C, Bazan JF, Kastelein RA. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;15:715–725. doi: 10.1016/S1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- Cargill M, Schrodi SJ, Chang M, Garcia VE, Brandon R, Callis KP, Matsunami N, Ardlie KG, Civello D, Catanese JJ, Leong DU, Panko JM, McAllister LB, Hansen CB, Papenfuss J, Prescott SM, White TJ, Leppert MF, Krueger GG, Begovich AB. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Gen. 2007;15:273–290. doi: 10.1086/511051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M, Griffiths A, Dassopoulos T, Bitton A, Yang H, Targan S, Datta LW, Kistner EO, Schumm LP, Lee AT, Gregersen PK, Barmada MM, Rotter JI, Nicolae DL, Cho JH. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;15:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa A, Andoh A, Araki Y, Bamba T, Fujiyama Y. Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced colitis in mice. Clin Immunol. 2004;15:55–62. doi: 10.1016/j.clim.2003.09.013. [DOI] [PubMed] [Google Scholar]
- Zheng SG, Wang J, Horwitz DA. Cutting edge: Foxp3+CD4+CD25+ regulatory T cells induced by IL-2 and TGF-beta are resistant to Th17 conversion by IL-6. J Immunol. 2008;15:7112–7116. doi: 10.4049/jimmunol.180.11.7112. [DOI] [PubMed] [Google Scholar]
- Mokuno Y, Matsuguchi T, Takano M, Nish imura H, Washizu J, Ogawa T, Takeuchi O, Akira S, Nimura Y, Yoshikai Y. Expression of Toll-like receptor 2 on γδ T cells bearing invariant Vγ6/Vδ1 induced by Escherichia coli infection in mice. J Immunol. 2000;15:931–940. doi: 10.4049/jimmunol.165.2.931. [DOI] [PubMed] [Google Scholar]
- Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. Interleukin-17- producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity. 2009;15:321–330. doi: 10.1016/j.immuni.2009.06.020. [DOI] [PubMed] [Google Scholar]
- Evans DM, Spencer CC, Pointon JJ, Su Z, Harvey D, Kochan G, Oppermann U, Dilthey A, Pirinen M, Stone MA, Appleton L, Moutsianas L, Leslie S, Wordsworth T, Kenna TJ, Karaderi T, Thomas GP, Ward MM, Weisman MH, Farrar C, Bradbury LA, Danoy P, Inman RD, Maksymowych W, Gladman D, Rahman P. Spondyloarthritis Research Consortium of Canada (SPARCC); Morgan A, Marzo-Ortega H, Bowness P. et al. Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibility. Nat Genet. 2011;15:6. doi: 10.1038/ng.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhart E, Green AM, Flynn JL. IL-17 produ ction is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol. 2006;15:4662–4669. doi: 10.4049/jimmunol.177.7.4662. [DOI] [PubMed] [Google Scholar]
- Shibata K, Yamada H, Hara H, Kishihara K, Yoshikai Y. Resident Vdelta1+ gammadelta T cells control early infiltration of neutrophils after Escherichia coli infection via IL-17 production. J Immunol. 2007;15:4466–4472. doi: 10.4049/jimmunol.178.7.4466. [DOI] [PubMed] [Google Scholar]
- Roark CL, French JD, Taylor MA, Bendele AM, Born WK, O'Brien RL. Exacerbation of collagen-induced arthritis by oligoclonal, IL-17-producing gamma delta T cells. J Immunol. 2007;15:5576–5583. doi: 10.4049/jimmunol.179.8.5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanno M, Kanari Y, Naito T, Inoue N, Hisamatsu T, Chinen H, Sugimoto K, Shimomura Y, Yamagishi H, Shiohara T, Ueha S, Matsushima K, Suematsu M, Mizoguchi A, Hibi T, Bhan AK, Ishikawa H. Exacerbating role of gammadelta T cells in chronic colitis of T-cell receptor alpha mutant mice. Gastroenterology. 2008;15:481–490. doi: 10.1053/j.gastro.2007.11.056. [DOI] [PubMed] [Google Scholar]
- Kenna TJ, Davidson SI, Duan R, Bradbury LA, McFarlane J, Smith M, Weedon H, Street S, Thomas R, Thomas GP, Brown MA. Enrichment of circulating interleukin-17-secreting interleukin-23 receptor-positive gamma/delta T cells in patients with active ankylosing spondylitis. Arthritis Rheum. 2012;15:1420–1429. doi: 10.1002/art.33507. [DOI] [PubMed] [Google Scholar]
- Boismenu R, Havran WL. Modulation of epithelial cell growth by intraepithelial gamma delta T cells. Science. 1994;15:1253–1255. doi: 10.1126/science.7973709. [DOI] [PubMed] [Google Scholar]
- Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Ann Rev Immunol. 2007;15:297–336. doi: 10.1146/annurev.immunol.25.022106.141711. [DOI] [PubMed] [Google Scholar]
- Rachitskaya AV, Hansen AM, Horai R, Li Z, Villasmil R, Luger D, Nussenblatt RB, Caspi RR. Cutting edge: NKT cells constitutively express IL-23 receptor and RORgammat and rapidly produce IL-17 upon receptor ligation in an IL-6- independent fashion. J Immunol. 2008;15:5167–5171. doi: 10.4049/jimmunol.180.8.5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbari O, Stock P, Meyer E, Kronenberg M, Sidobre S, Nakayama T, Taniguchi M, Grusby MJ, DeKruyff RH, Umetsu DT. Essential role of NKT cells producing IL-4 and IL-13 in the development of allergen-induced airway hyperreactivity. Nat Med. 2003;15:582–588. doi: 10.1038/nm851. [DOI] [PubMed] [Google Scholar]
- Baxter AG, Kinder SJ, Hammond KJ, Scollay R, Godfrey DI. Association between alphabetaTCR+CD4-CD8- T-cell deficiency and IDDM in NOD/Lt mice. Diabetes. 1997;15:572–582. doi: 10.2337/diabetes.46.4.572. [DOI] [PubMed] [Google Scholar]
- Heller F, Fuss IJ, Nieuwenhuis EE, Blumberg RS, Strober W. Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13- producing NK-T cells. Immunity. 2002;15:629–638. doi: 10.1016/S1074-7613(02)00453-3. [DOI] [PubMed] [Google Scholar]
- Ueno Y, Tanaka S, Sumii M, Miyake S, Tazuma S, Taniguchi M, Yamamura T, Chayama K. Single dose of OCH improves mucosal T helper type 1/T helper type 2 cytokine balance and prevents experimental colitis in the presence of Vα14 natural killer T cells in mice. Inflamm Bowel Dis. 2005;15:35–41. doi: 10.1097/00054725-200501000-00005. [DOI] [PubMed] [Google Scholar]
- Coppieters KT, Elewaut D. Natural killer T cells: born in the thymus, raised in the gut. Gastroenterology. 2012;15:293–296. doi: 10.1053/j.gastro.2012.06.024. [DOI] [PubMed] [Google Scholar]
- Wingender G, Stepniak D, Krebs P, Lin L, McBride S, Wei B, Braun J, Mazmanian SK, Kronenberg M. Intestinal microbes affect phenotypes and functions of invariant natural killer T cells in mice. Gastroenterology. 2012;15:418–428. doi: 10.1053/j.gastro.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppieters K, Van Beneden K, Jacques P, Dewint P, Vervloet A, Vander Cruyssen B, Van Calenbergh S, Chen G, Franck RW, Verbruggen G, Deforce D, Matthys P, Tsuji M, Rottiers P, Elewaut D. A single early activation of invariant NK T cells confers long-term protection against collagen-induced arthritis in a ligand-specific manner. J Immunol. 2007;15:2300–2309. doi: 10.4049/jimmunol.179.4.2300. [DOI] [PubMed] [Google Scholar]
- Jacques P, Venken K, Van Beneden K, Hammad H, Seeuws S, Drennan MB, Deforce D, Verbruggen G, Apostolaki M, Kollias G, Lambrecht BN, De Vos M, Elewaut D. Invariant natural killer T cells are natural regulators of murine spondylarthritis. Arthritis Rheum. 2010;15:988–999. doi: 10.1002/art.27324. [DOI] [PubMed] [Google Scholar]
- Gold MC, Cerri S, Smyk-Pearson S, Cansler ME, Vogt TM, Delepine J, Winata E, Swarbrick GM, Chua WJ, Yu YY, Lantz O, Cook MS, Null MD, Jacoby DB, Harriff MJ, Lewinsohn DA, Hansen TH, Lewinsohn DM. Human mucosal associated invariant T cells detect bacterially infected cells. PLoS Biol. 2010;15:e1000407. doi: 10.1371/journal.pbio.1000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bourhis L, Martin E, Péguillet I, Guihot A, Froux N, Coré M, Lévy E, Dusseaux M, Meyssonnier V, Premel V, Ngo C, Riteau B, Duban L, Robert D, Huang S, Rottman M, Soudais C, Lantz O. Antimicrobial activity of mucosal associated invariant T cells. Nat Immunol. 2010;15:701–708. doi: 10.1038/ni.1890. [DOI] [PubMed] [Google Scholar]
- Dusseaux M, Martin E, Serriari N, Péguillet I, Premel V, Louis D, Milder M, Le Bourhis L, Soudais C, Treiner E, Lantz O. Human MAIT cells are xenobiotic resistant, tissue-targeted, CD161hi IL-17-secreting T cells. Blood. 2011;15:1250–1259. doi: 10.1182/blood-2010-08-303339. [DOI] [PubMed] [Google Scholar]
- Treiner E, Duban L, Bahram S, Radosavljevic M, Wanner V, Tilloy F, Affaticati P, Gilfillan S, Lantz O. Selection of evolutionarily conserved mucosalassociated invariant T cells by MR1. Nature. 2003;15:164–169. doi: 10.1038/nature01433. [DOI] [PubMed] [Google Scholar]
- Savage AK, Constantinides MG, Han J, Picard D, Martin E, Li B, Lantz O, Bendelac A. The transcription factor PLZF directs the effector program of the NKT cell lineage. Immunity. 2008;15:391–403. doi: 10.1016/j.immuni.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billerbeck E, Kang YH, Walker L, Lockstone H, Grafmueller S, Fleming V, Flint J, Willberg CB, Bengsch B, Seigel B, Ramamurthy N, Zitzmann N, Barnes EJ, Thevanayagam J, Bhagwanani A, Leslie A, Oo YH, Kollnberger S, Bowness P, Drognitz O, Adams DH, Blum HE, Thimme R, Klenerman P. Analysis of CD161 expression on human CD8+ T cells defines a distinct functional subset with tissue-homing properties. Proc Natl Acad Sci USA. 2010;15:3006–3011. doi: 10.1073/pnas.0914839107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bourhis L, Guerri L, Dusseaux M, Martin E, Soudais C, Lantz O. Mucosal associated invariant T cells: unconventional development and function. Trends Immunol. 2011;15:212–218. doi: 10.1016/j.it.2011.02.005. [DOI] [PubMed] [Google Scholar]
- Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Littman DR, O'Shea JJ. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J Exp Med. 2009;15:35–41. doi: 10.1084/jem.20072713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupedo T, Crellin NK, Papazian N, Rombouts EJ, Weijer K, Grogan JL, Fibbe WE, Cornelissen JJ, Spits H. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat Immunol. 2009;15:66–74. doi: 10.1038/ni.1668. [DOI] [PubMed] [Google Scholar]
- Buonocore S, Ahern PP, Uhlig HH, Littman DR, Maloy KJ, Powrie F. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. 2010;15:1371–1375. doi: 10.1038/nature08949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK, Doherty JM, Mills JC, Colonna M. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature. 2009;15:722–725. doi: 10.1038/nature07537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederberg J, McCray AT. 'Ome sweet 'omics - A genealogical treasury of words. Scientist. 2001;15:8. [Google Scholar]
- Woese CR. Bacterial evolution. Microbiol Rev. 1987;15:221–271. doi: 10.1128/mr.51.2.221-271.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, Bertalan M, Borruel N, Casellas F, Fernandez L, Gautier L, Hansen T, Hattori M, Hayashi T, Kleerebezem M, Kurokawa K, Leclerc M, Levenez F, Manichanh C, Nielsen HB, Nielsen T, Pons N, Poulain J, Qin J, Sicheritz-Ponten T, Tims S. et al. Enterotypes of the human gut microbiome. Nature. 2011;15:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIH HMP Working Group; Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, Bonazzi V, McEwen JE, Wetterstrand KA, Deal C, Baker CC, Di Francesco V, Howcroft TK, Karp RW, Lunsford RD, Wellington CR, Belachew T, Wright M, Giblin C, David H, Mills M, Salomon R, Mullins C, Akolkar B, Begg L, Davis C, Grandison L, Humble M, Khalsa J, Little AR. et al. The NIH Human Microbiome Project. Genome Res. 2009;15:2317–2323. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P. et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;15:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone MA, Payne U, Schentag C, Rahman P, Pacheco-Tena C, Inman RD. Comparative immune responses to candidate arthritogenic bacteria do not confirm a dominant role for Klebsiella pneumonia in the pathogenesis of familial ankylosing spondylitis. Rheumatology. 2004;15:148–155. doi: 10.1093/rheumatology/keg482. [DOI] [PubMed] [Google Scholar]
- Stebbings S, Munro K, Simon MA, Tannock G, Highton J, Harmsen H, Welling G, Seksik P, Dore J, Grame G, Tilsala-Timisjarvi A. Comparison of the faecal microflora of patients with ankylosing spondylitis and controls using molecular methods of analysis. Rheumatology. 2002;15:1395–1401. doi: 10.1093/rheumatology/41.12.1395. [DOI] [PubMed] [Google Scholar]
- Eerola E, Mottonen T, Hannonen P, Luukkainen R, Kantola I, Vuori K, Tuominen J, Toivanen P. Intestinal flora in early rheumatoid arthritis. Rheumatology. 1994;15:1030–1038. doi: 10.1093/rheumatology/33.11.1030. [DOI] [PubMed] [Google Scholar]
- Vaahtovuo J, Munukka E, Korkeamaki M, Luukainen R, Toivanen P. Fecal microbiot a in early rheumatoid arthritis. J Rheumatol. 2008;15:1500–1505. [PubMed] [Google Scholar]
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;15:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikuls TR. Help stop tooth decay...and prevent RA? J Rheumatol. 2010;15:1083–1085. doi: 10.3899/jrheum.100173. [DOI] [PubMed] [Google Scholar]
- Giaffer MH, Holdsworth CD, Duerden BI. The assessment of faecal flora in patients with inflammatory bowel disease by a simplified bacteriological technique. J Med Microbiol. 1991;15:238–243. doi: 10.1099/00222615-35-4-238. [DOI] [PubMed] [Google Scholar]
- Seksik P, Rigottier-Gois L, Gramet G, Sutren M, Pochart P, Marteau P, Jian R, Dore J. Alterations of the dominant faecal bacterial groups in patients with Crohn's disease of the colon. Gut. 2003;15:237–242. doi: 10.1136/gut.52.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott SJ, Musfeldt M, Wenderoth DF, Hampe J, Brant O, Folsch UR, Timmis KN, Schreiber S. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut. 2004;15:685–693. doi: 10.1136/gut.2003.025403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AW, Sanderson JD, Churcher C, Parkes GC, Hudspith BN, Rayment N, Brostoff J, Parkhill J, Dougan G, Petrovska L. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011;15:7. doi: 10.1186/1471-2180-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger JG. The immunologic basis for the treatment of psoriasis with new biologic agents. J Am Acad Dermatol. 2002;15:1–26. doi: 10.1067/mjd.2002.120568. [DOI] [PubMed] [Google Scholar]
- Fahlen A, Engstrand L, Baker BS, Powles A, Fry L. Comparison of bacterial microbiota in skin biopsies from normal and psoriatic skin. Arch Dermatol Res. 2012;15:15–22. doi: 10.1007/s00403-011-1189-x. [DOI] [PubMed] [Google Scholar]
- Rath HC, Herfarth HH, Ikeda JS, Grenther WB, Hamm TE, Balish E, Taurog JD, Hammer RE, Wilson KH, Sartor RB. Normal luminal bacteria, especially Bacteroides species, mediate chronic colitis, gastritis, and arthritis in HLA-B27/human beta2 microglobulin transgenic rats. J Clin Invest. 1996;15:945–953. doi: 10.1172/JCI118878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unni K, Holley K, McDuffie F, Titus J. Comparative study of NZB mice under germf ree and conventional conditions. J Rheumatol. 1975;15:8. [PubMed] [Google Scholar]
- Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjöberg J, Amir E, Teggatz P, Barman M, Hayward M, Eastwood D, Stoel M, Zhou Y, Sodergren E, Weinstock GM, Bevins CL, Williams CB, Bos NA. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol. 2010;15:76–82. doi: 10.1038/ni.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, Glickman JN, Siebert R, Baron RM, Kasper DL, Blumberg RS. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012;15:489–493. doi: 10.1126/science.1219328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw MH, Kamada N, Kim YG, Nunez G. Microbiota-induced IL-1beta, but not IL-6, is critical for the development of steady-state TH17 cells in the intestine. J Exp Med. 2012;15:251–258. doi: 10.1084/jem.20111703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, Taniguchi T, Takeda K, Hori S, Ivanov II, Umesaki Y, Itoh K, Honda K. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;15:337–341. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dicksved J, Halfvarson J, Rosenquist M, Jarnerot G, Tysk C, Apajalahti J, Engstrand L, Jansson JK. Molecular analysis of the gut microbiota of identical twins with Crohn's disease. ISME J. 2008;15:716–727. doi: 10.1038/ismej.2008.37. [DOI] [PubMed] [Google Scholar]
- Spor A, Koren O, Ley R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat Rev Micro. 2011;15:279–290. doi: 10.1038/nrmicro2540. [DOI] [PubMed] [Google Scholar]
- Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, Flavell RA. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;15:745–757. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Tseng C-h, Strober BE, Pei Z, Blaser MJ. Substantial alterations of the cutaneous bacterial biota in psoriatic lesions. PLoS ONE. 2008;15:e2719. doi: 10.1371/journal.pone.0002719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice EA, Segre JA. The skin microbiome. Nat Rev Micro. 2011;15:244–253. doi: 10.1038/nrmicro2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchon CA, Chandad F, Ferucci ED, Willemze A, Ioan-Facsinay A, van der Woude D, Markland J, Robinson D, Elias B, Newkirk M, Toes RM, Huizinga TW, El-Gabalawy HS. Antibodies to porphyromonas gingivalis are associated with anticitrullinated protein antibodies in patients with rheumatoid arthritis and their relatives. J Rheumatol. 2010;15:1105–1112. doi: 10.3899/jrheum.091323. [DOI] [PubMed] [Google Scholar]
- Scher JU, Ubeda C, Equinda M, Khanin R, Buischi Y, Viale A, Lipuma L, Attur M, Pillinger MH, Weissmann G, Littman DR, Pamer EG, Bretz WA, Abramson SB. Periodontal disease and the oral microbiota in new-onset rheumatoid arthritis. Arthritis Rheum. 2012;15:3083–3094. doi: 10.1002/art.34539. [DOI] [PMC free article] [PubMed] [Google Scholar]