

Abstract
Mutations in the PCSK1 gene encoding prohormone convertase 1/3 (PC1/3) are strongly associated with obesity in humans. The PC1/3N222D mutant mouse thus far represents the only mouse model that mimics the PC1/3 obesity phenotype in humans. The present investigation addresses the cell biology of the N222D mutation. Metabolic labeling experiments reveal a clear defect in the kinetics of insulin biosynthesis in islets from PC1/3N222D mutant mice, resulting in an increase in both proinsulin and its processing intermediates, predominantly lacking cleavage at the Arg-Arg site. Although the mutant PC1/3 zymogen is correctly processed to the 87-kDa form, pulse-chase immunoprecipitation experiments, labeling, and immunohistochemical experiments using uncleavable variants all demonstrate that the PC1/3-N222D protein is largely mislocalized compared with similar wild-type (WT) constructs, being predominantly retained in the endoplasmic reticulum. The PC1/3-N222D mutant also undergoes more efficient degradation via the ubiquitin-proteasome system than the WT enzyme. Lastly, the mutant PC1/3-N222D protein coimmunoprecipitates with WT PC1/3 and exerts a modest effect on intracellular retention of the WT enzyme. These profound alterations in the cell biology of PC1/3-N222D are likely to contribute to the defective insulin biosynthetic events observed in the mutant mice and may be relevant to the dramatic contributions of polymorphisms in this gene to human obesity.
Prohormone convertase 1/3 (PC1/3) (also known as sPC3 and PC1) is one of 2 major enzymes that accomplish the processing and maturation of prohormones and neuropeptide precursors within the regulated secretory pathway (reviewed in Ref. 1). PC1/3, encoded by the gene PCSK1, is synthesized in the endoplasmic reticulum (ER) as an inactive 94-kDa zymogen, where it is rapidly cleaved into a partially active 87-kDa precursor (2–4). When this protein reaches the secretory granules, the maturation process continues to generate a 74-kDa species and ends with further cleavage to a fully active, although enzymatically unstable, 66-kDa protein (2, 5, 6). Proinsulin, proglucagon, and proopiomelanocortin have all been shown to be substrates of PC1/3, which, often in combination with prohormone convertase 2, are efficiently cleaved into glucagon, insulin, and ACTH (7), peptides involved in a wide variety of key metabolic functions. In agreement, the lack of human PC1/3 observed in patients with inactivating mutations (8–10) results in both distinct metabolic alterations, particularly related to obesity, such as reactive hypoglycemia, and many other endocrine dysfunctions. More recently, the relationship between various single nucleotide polymorphisms (SNPs) of human PC1/3 has been analyzed in the context of obesity (11). To that end, a total of 16 SNPs were evaluated for obesity prevalence using a metaanalysis on more than 6000 Europeans; this study concluded that mutations causing partial PCSK1 deficiency increase the risk of obesity by 8.7-fold (11). Therefore, it appears that PCSK1 constitutes one of the most prevalent monogenic contributors to extreme obesity in European (11) as well as several other populations (12, 13).
Unexpectedly, PCSK1 null mice do not display an obesity phenotype, although they show growth retardation and multiple neuroendocrine disorders (14). However, an obese mouse model with a mutation in the PCSK1 gene was discovered in a random chemical mutagenesis experiment (15). This mutation consists of a single asparagine to aspartate change at residue 222 (N222D). Mice bearing this mutation show multiple endocrine defects and increased deposition of fat in white adipose tissue. Steady-state radiolabeling analysis of pancreatic islets obtained from these animals indicated a general defect in insulin maturation but not in proinsulin production; the proinsulin cleavage sites affected by loss of PC1/3 activity were not examined (15).
It has been proposed that the altered phenotype of the PC1/3N222D mutant mouse is primarily a consequence of decreased autocatalytic maturation of the mutant enzyme, as observed by the reduction in the level of fully mature 66-kDa PC1/3 (15). In the present study, we have investigated the biosynthesis and cellular trafficking of the mouse PC1/3-N222D mutant and wild-type (WT) enzymes in various secretory cell lines. Using a version of the N222D mutant that cannot be cleaved, we evaluated the transit of the form of 87-kDa PC1/3 through the secretory pathway irrespective of its maturation to the 66-kDa form. The characterization presented here demonstrates that the cell biology of PC1/3-N222D differs greatly from that of its WT counterpart in the early secretory pathway.
Materials and Methods
Labeling of pancreatic islets
The islets were isolated from mice of mixed sex and metabolically labeled as previously described (16). Briefly, the handpicked islets were recovered in RPMI 1640 medium containing 10% fetal bovine serum (FBS) overnight. One hundred islets were preincubated in DMEM containing 25.5mM glucose for 30 minutes. Islets were washed twice with prewarmed Met/Cys-deficient DMEM. The islets were then metabolically labeled with 35S-labeled Met/Cys in the same medium for 20 minutes. The labeled islets were split in half: one was lysed immediately in lysis buffer containing proteinase inhibitors, and another half was chased in RPMI 1640 medium for the times indicated. Newly synthesized proinsulin, conversion intermediates, and insulin were immunoprecipitated with antiinsulin and analyzed by Tris-tricine-urea-SDS-PAGE under reducing and nonreducing conditions, as indicated.
Recombinant DNA procedures
For expression in cells, mouse PC1/3 cDNA (GeneCopoeia) was subcloned in frame into the XbaI and BamH1 restriction sites in pcDNA3.1/Hygro(−) (Invitrogen). A single Flag (DYKDDDDK) tag was inserted at the C terminus before the stop codon using site-directed mutagenesis (Stratagene). Two substitution mutations were generated in the above construct (to replace amino acid residues 617–618 RR to KH and amino acid residues 654–655 RR to KA, respectively) by site-directed mutagenesis to generate the “uncleavable” form of the protein. This construct is termed PC1/3-WT-Flag. A single influenza hemagglutinin antigen (HA) (YPYDVPDYA) tag, replacing the Flag tag, was generated in this construct using oligo-directed mutagenesis (Stratagene). This construct is considered as WT (PC1/3-WT-HA) in the present study. The N222D amino acid substitution was generated in PC1/3-WT-Flag and PC1/3-WT-HA to obtain PC1/3-N222D-Flag and PC1/3-N222D-HA, respectively, using site-directed mutagenesis, as described earlier. The α-1 antitrypsin (AAT1) expression plasmid, Ubiquitin-Flag (Ub-Flag), and β-secretase 1 (BACE1)-HA (HA-BACE1-WT) were described earlier (17–19).
Cell culture and transfections
Neuro2a cells were purchased from the American Type Culture Collection and were maintained in DMEM high glucose: Opti-MEM (1:1) medium supplemented with 10% FBS (Atlanta Biologicals) at 37°C in a humidified atmosphere containing 5% CO2. Rin5f cells were maintained in DMEM low-glucose medium (Invitrogen) containing 10% FBS. Either Lipofectamine 2000 (Invitrogen) or FuGENE HD (Promega) transfection reagents were used for transfections, as per the manufacturer's instructions. HEK cells were maintained in DMEM high-glucose medium containing 10% FBS.
Immunofluorescence
Rin5f cells or Neuro2a cells were grown on coverslips overnight in 24-well plates with a density of 105 cells per well. The next day, cells were transfected with 0.20 μg per well of each PC1/3 expression vector using FuGENE (Promega). Forty-eight hours after transfection, cells were fixed with 4% paraformaldehyde for 20 minutes at room temperature, permeabilized (0.2% Triton X-100 in PBS) for 10 minutes, and blocked (0.2% porcine skin gelatin in PBS) for 30 minutes. Rin5f cells were then incubated with a 1:100 dilution of anti-HA mouse monoclonal (Covance) to immunolabel PC1/3-WT-HA and PC1/3-N222D-HA and colabeled either with a 1:50 dilution of goat anti-calreticulin (PA1–84481; Thermo Fisher/Pierce) or a 1:100 dilution of sheep anti-trans-Golgi network (TGN)38 (AHP499G; Serotec) for 1 hour at 37°C. Neuro2a cells were incubated with a 1:1000 dilution of 2B5 PC1/3 rabbit polyclonal antiserum to immunolabel the untagged WT PC1/3 and PC1/3-N222D proteins and colabeled either with a 1:50 dilution of goat anti-calreticulin (PA1–84481; Thermo Fisher/Pierce) or a 1:100 dilution of sheep anti-TGN38 (AHP499G; Serotec) for 1 hour at 37°C. After washing, cells were incubated with either Cy3- or Cy2-conjugated donkey antimouse IgG, Cy3- or Cy2-conjugated donkey antirabbit IgG, Cy2-conjugated donkey antigoat IgG, or Cy3-conjugated donkey antisheep IgG (1:250; Jackson ImmunoResearch) in blocking solution for 1 hour at 37°C. Cells were incubated with DAPI Nucleic Acid Stain (Invitrogen). Cells were then rinsed in PBS and embedded with Fluoromount G (Electron Microscopy Sciences).
Confocal microscopy and colocalization analysis
Immunofluorescent images were obtained with a Fluoview500 confocal microscope (Olympus) and Fluoview version 6.0 software. Images were obtained with a ×60 objective (N.A 1.4 oil) using a sequential mode of laser scanning with 21 slices per cell (Z-step of 200 nm). Under these conditions, each pixel corresponds to 103 nm. Images were processed in the ImageJ software. Deconvolution and colocalization analyses were made using the Iterative Deconvolve 3D and JaCoP plugins (20). In order to analyze the subcellular colocalizations of the different PC1/3 proteins, Pearson's correlation coefficients were determined; these coefficients were then analyzed using an unpaired t test with GraphPad Prism 5 software.
Cycloheximide (CHX) and MG132 experiments
Two × 105 Neuro2a cells per well were plated into a 12-well plate and transiently transfected with PC1/3-WT-HA and PC1/3-N222D-HA in triplicate using Lipofectamine 2000 (Invitrogen). For the CHX experiment, cells were cultured in plating media overnight and the next day exposed to 0.1-mg/mL CHX (in plating media) for 0, 1, 2, 3, or 4 hours, respectively. For the MG132 experiment, cells were cultured in the presence or absence of 5μM MG132 for 12 hours. Cells were briefly washed with chilled PBS, harvested in Laemmli sample buffer, and electrophoresed on a 10% Tris-glycine gel (Bio-Rad). After transfer to nitrocellulose, Western blots were probed with 1:1000 dilution of anti-HA antibody (101MP; Covance) followed by antimouse horseradish peroxidase (HRP) secondary at 1:20 000 (A5278; Sigma), or HRP conjugated anti-Flag antibody (A8592; Sigma). Bands were visualized using enhanced chemiluminescence “Dura” substrate (Thermo Fisher). A two-tailed unpaired Student's t test (GraphPad Prism) was used to show differences for the different chase times (mean ± SD shown; n = 3; P < .05). Linear regression analysis of these data (GraphPad Prism) was used to show that the decay over time of the band intensities of the 2 proteins (WT and N222D) was also statistically different (P < .05). Confidence limits (95%) for this determination were 587–428 AU/h (wild type) and 469–338 AU/h (N222D).
Pulse-chase and immunoprecipitation
Neuro2a cells were plated in 12-well plates and transfected with PC1/3-encoding constructs (as indicated). Twenty-four hours after transfection, cells were washed with PBS and incubated for 20 minutes at 37°C and 5% CO2 in starvation medium (DMEM lacking methionine or cysteine). For the pulsed samples, the cells were incubated with 500 μL of labeling media (starvation media supplemented with 0.25 mCi per well 35S Met/Cys translabel) for 20 minutes. After washing with 1 mL of methionine-containing DMEM, cells were either extracted immediately in immunoprecipitation buffer or chased in cold methionine-containing medium for 2 hours. Immunoprecipitation was performed as described previously (21, 22). After overnight incubation at 4°C with 4 μL of 2B5, an N-terminally directed PC1/3 antiserum (22), immunoprecipitates were separated on a 7.5% SDS-PAGE gel, and radiolabeled PC1/3 was detected using a Storm phosphoimager.
Ubiquitination assay
Neuro2a or HEK cells grown on 6-well plates at the density of 5 × 105 cells were transiently transfected using Lipofectamine 2000 reagent (Invitrogen) with PC1/3-WT-HA and PC1/3-N222D-HA in the presence or absence of cDNA encoding Flag-Ub (17). Cells were incubated with 5μM lactacystin in plating media overnight. The next day, the cells were washed with chilled PBS and lysed in cell lysis buffer (50mM Tris-HCl [pH 7.4], 150mM NaCl, 2mM EDTA, and 1% Triton X-100) in the presence of protease inhibitor cocktail (Roche “Complete”). Cells were shaken for 30 minutes on ice, cleared by centrifugation for 20 minutes at 14 000 rpm at 4°C to eliminate insoluble material, and the supernatant was used for immunoprecipitation. Cell lysates were precleared over 50 μL of prewashed Protein A-Sepharose beads (Sigma-Aldrich) for 1 hour at 4°C. The precleared cell lysates were passed over another 50 μL of prewashed Protein A beads for 1 hour at 4°C followed by incubation with anti-HA monoclonal antibody overnight. Bound beads were washed once with cell lysis buffer, twice with PBS, and resuspended in 2× Laemmli sample buffer containing 5% β-mercaptoethanol. Samples were electrophoresed on a 10% Tris-glycine gel (Bio-Rad), transferred to nitrocellulose, and probed with anti-HA monoclonal and anti-Flag antibodies at 1:1000, followed by antimouse HRP secondary antibodies at 1:20 000. Bands were visualized using enhanced chemiluminescence “Dura” substrate (Pierce).
Coimmunoprecipitation
Neuro2a cells grown in 6-well plates at a density of 5 × 105 cells were transiently transfected using FuGENE HD reagent (Promega) with PC1/3-WT-HA in the presence and absence of PC1/3-N222D-Flag. An unrelated HA-tagged bait, HA-BACE1-WT (19), was used as a negative control. Cells were washed twice with chilled PBS and lysed using ice-cold cell lysis buffer (25mM Tris-HCl [pH 7.4], 100mM NaCl, 1mM EDTA, and 1% Triton X-100) in the presence of a protease inhibitor cocktail (Roche “Complete”). Coimmunoprecipitations and analyses were carried out as described in the ubiquitination assay, except that 15 μL of anti-HA monoclonal antibody was used per immunoprecipitation, and protein G-Sepharose beads were used.
Statistical analysis
All statistical analyses were conducted using GraphPad Prism software. Data are presented as mean ± SD or SE, as indicated.
Results
The PC1/3N222D mutation affects insulin biosynthesis in vivo
PC1/3N222D mutant mice represent a valuable tool to delineate the obesity phenotype seen in humans with PC1/3 deficiency. An earlier study demonstrated defective insulin levels and increased proinsulin levels in PC1/3N222D mutant mice. Moreover, some studies have suggested that PC1/3 WT ordinarily cleaves the B-chain/C-peptide junction of proinsulin first, whereas PC2 tends to cleave the C-peptide/A-chain junction thereafter (23, 24). Thus, fully active PC2 with less active PC1/3N222D might potentially produce abnormally increased amounts of proinsulin processing intermediate cleaved properly at the C-peptide/A-chain junction but with the B-chain uncleaved (so-called “intermediate 1”) (Figure 1A); if PC2-mediated proinsulin cleavage is stimulated by previous PC1/3-mediated cleavage, then both cleavages might be affected in PC1/3N222D mutant mice.
Figure 1.

The PC1/3N222D mutation affects insulin biosynthesis in vivo. A, Schematic showing proinsulin processing. Proinsulin intermediate 1 is cleaved at the C-A junction, and proinsulin intermediate 2 is cleaved at the B-C junction. B, One hundred pancreatic islets were preincubated in DMEM containing 25.5mM glucose for 30 minutes. After being briefly washed twice with Cys/Met-deficient DMEM medium, the islets were pulse labeled with 35S-Cys/Met in the same medium for 20 minutes. The labeled islets were split in half: 1 was lysed immediately, and the other was chased in RPMI 1640 for 4 hours. Newly synthesized proinsulin, conversion intermediates, and insulin were recovered by immunoprecipitation with antiinsulin and analyzed by Tris-tricine-urea-SDS-PAGE under nonreducing conditions. C, Islets were labeled for 20 minutes and chased in RPMI 1640 medium for times indicated. The islets were lysed. The lysates and chase media were combined and immunoprecipitated with antiinsulin followed by Tris-tricine-urea-SDS-PAGE under reducing conditions.
We analyzed the dynamic biosynthetic profile of insulin in pancreatic islets from PC1/3N222D mutant mice using WT mouse islets as a control. A pulse-chase assay carried out in the presence of glucose was analyzed by immunoprecipitation, Tris-tricine-urea-SDS-PAGE under reducing and nonreducing conditions, and phosphorimaging. As shown in Figure 1B, at 4 hours of chase after pulse labeling, there was a considerable amount of unprocessed intact proinsulin and also proinsulin intermediates, especially intermediate 1, remaining in PC1/3N222D mutant mouse islets. By contrast, most proinsulin and its intermediates were cleaved to insulin in WT mouse islets (as seen under both reducing and nonreducing conditions) (Figure 1, B and C). Although some insulin was produced by the mutant PC1/3 at 4 hours of chase, these data clearly reveal a defect in the kinetics of insulin biosynthesis in the islets of PC1/3N222D mutant mice, resulting in an increase of both proinsulin and its processing intermediates.
The PC1/3 N222D mutant protein is localized to the ER, whereas WT PC1/3 is present in the Golgi and in the tips of endocrine cells
We pursued a cell biological approach to address the defective biosynthesis of insulin in N222D mutant mice. We first asked whether there was a difference in the intracellular distribution of the N222D variant as compared with WT protein in cells. We generated PC1/3-WT-HA and PC1/3-N222D-HA expression constructs that carried a single HA-tag at their C termini. Both constructs were mutated (amino acids 617–618, RR to KH; and amino acids 654–655, RR to KA) to obtain an 87-kDa protein that cannot be further cleaved to the 74- to 66-kDa forms (described in Materials and Methods) (Figure 2A). This uncleavable version of PC1/3 was generated to ensure visualization of the 87-kDa active form of PC1/3 while eliminating trafficking differences that arise from differential cleavage to mature protein. Plasmids encoding PC1/3-WT-HA and PC1/3-N222D-HA were transiently transfected into rat pancreatic islet tumor cells (Rin5f) and analyzed using immunostaining and confocal microscopy (Figure 2B). In Rin5f cells, PC1/3-N222D-HA was predominantly localized to the ER, as confirmed by its colocalization with the ER marker calreticulin, whereas PC1/3-WT-HA was localized mainly to the Golgi apparatus (as assessed by its colocalization with TGN38) (Figure 2B), as expected (25).
Figure 2.
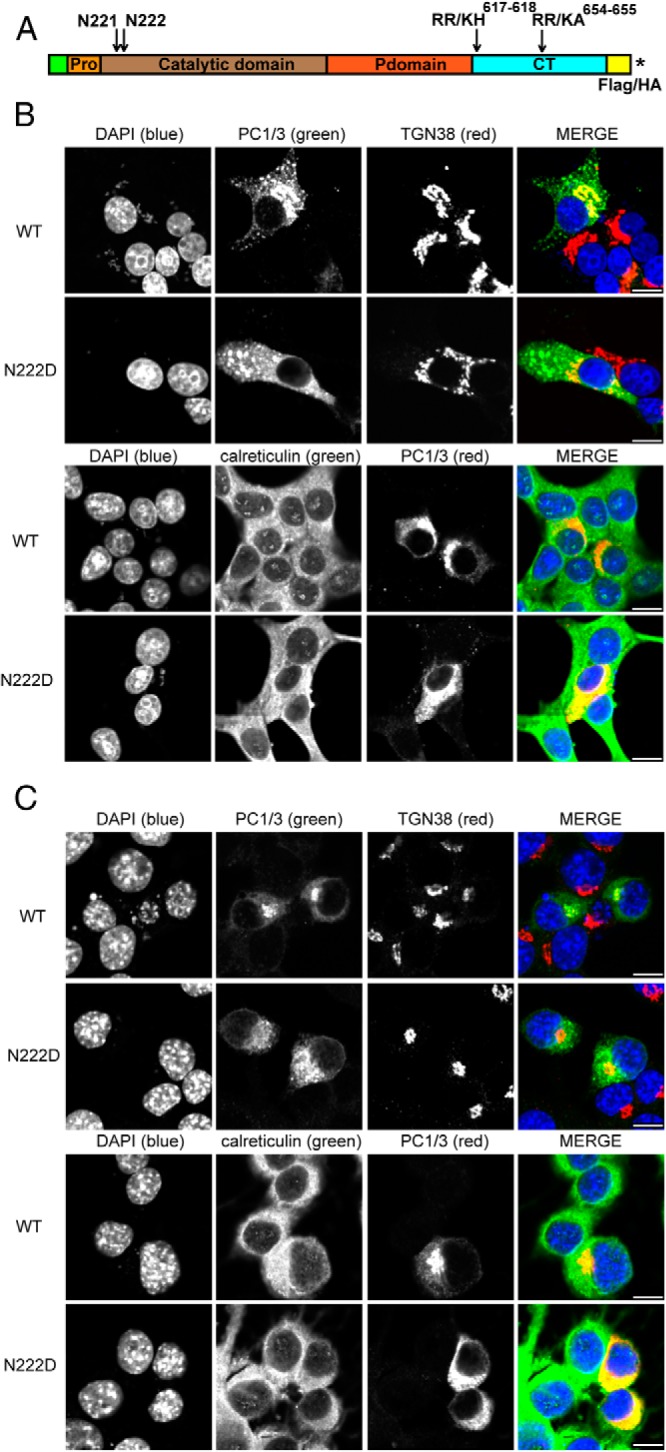
The N222D variant of PC1 is localized to the ER, whereas the WT protein is present in the Golgi and in the tips of endocrine cells. A, Schematic representation of PC1/3 showing the prodomain, catalytic domain, P domain, C-terminal domain, mutations incorporated, and the insertion positions of tags. An asterisk represents the stop codon. B, Rin5f cells were transiently transfected with PC1/3-WT-HA and PC1/3-N222D-HA constructs. Cells were fixed with 4% paraformaldehyde and processed as described (19). Colocalization was determined by immunostaining using a 1:100 dilution of monoclonal antibody to HA, a 1:100 dilution of sheep polyclonal antibody anti-TGN38, or a 1:50 dilution of goat polyclonal anti-calreticulin; these were followed by incubation with a 1:250 dilution of Cy2 (green)- or Cy3 (red)-conjugated antimouse immunoglobulin G, Cy2-conjugated antigoat, and Cy3-conjugated antisheep, respectively. C, In order to confirm that differences are due to the N222D mutation, Neuro2a cells were transiently transfected with PC1/3 constructs lacking the mutations that block carboxy-terminal processing. PC1/3-WT-Flag and PC1/3-N222D (untagged) were transfected and were treated as in B, except that PC1/3 was imaged using a rabbit polyclonal anti-PC1/3 antibody (2B5) together with either Cy2- or Cy3-conjugated antirabbit IgG. Scale bar, 10 μm.
To rule out the possibility that the ER accumulation phenotype is due merely to the epitope tag and the mutations that render it uncleavable, we carried out a similar experiment, in which we transiently transfected Neuro2a cells with cDNAs encoding cleavable (ie, lacking blockade mutations) FLAG-tagged PC1/3-WT and cleavable untagged PC1/3-N222D constructs (Figure 2C). We found a similar ER localization phenotype, indicating that the ER accumulation of the N222D variant is indeed due solely to the N222D mutation. Quantitation of colocalization confirmed these subcellular patterns; the Pearson's coefficients of colocalization indicated that PC1/3-N222D had a higher level of colocalization with calreticulin than PC1/3-WT in Rin5PC1/3-WT in Rin5f cells (WT: 0.328 ± 0.041; N222D: 0.548 ± 0.036; t test; *, P < .01, mean ± SE, n = 8 cells) and in Neuro2a cells (WT: 0.082 ± 0.029; N222D: 0.359 ± 0.044; t test; *, P < .001, mean ± SE, n = 5 cells). The inverse phenomenon was observed when we analyzed the colocalization of the different PC1/3 constructs and TGN38. In this case, the Pearson's coefficients showed that PC1/3-N222D had a lower level of colocalization with TGN38 than PC1/3 WT in Rin5F cells (WT: 0.350 ± 0.021; N222D: 0.262 ± 0.021; t test; *, P < .05, mean ± SE, n = 8 cells) and in Neuro2a cells (WT: 0.515 ± 0.027; N222D: 0.304 ± 0.027; t test; *, P < .001, mean ± SE, n = 8 cells).
WT PC1/3 and the N222D variant physically interact, but their association does not inhibit intracellular transport of WT PC1/3
To test whether accumulation of N222D variant in the ER can confer ER retention to the WT protein, we first analyzed whether these proteins can interact with each other. We expressed PC1/3-WT-HA and an unrelated HA-tagged protein, β-secretase 1 (HA-BACE1) and coexpressed either Flag-tagged WT PC1/3 or the PC1/3 N222D variant in Neuro2a cells (Figure 3A). Anti-HA monoclonal antibody was used to immunoprecipitate either PC1/3-WT-HA or HA-BACE1, and coprecipitated proteins were detected using anti-Flag antibody. Both PC1/3-WT-Flag and PC1/3-N222D-Flag specifically coprecipitated with PC1/3-WT-HA but not with the unrelated control protein HA-BACE1 (even though substantially more of this protein was present in the control). Our data demonstrate that WT PC1/3 molecules can physically interact with each other, consistent with oligomerization in the secretory pathway (26).
Figure 3.

PC1/3 WT and the PC1/3 N222D variant interact: coimmunoprecipitation. A, Neuro2a cells expressing PC1/3-WT-HA alone or coexpressing either PC1/3-WT-Flag or PC1/3-N222D-Flag were cultured overnight; an unrelated HA-BACE1-WT protein was expressed similarly. Cell lysates were subjected to immunoprecipitation with anti-HA monoclonal antibody and analyzed by tag immunoblotting as indicated. B and C, Neuro2a cells were subjected to transfections in triplicate with plasmids as indicated. Secretion was analyzed by culturing cells overnight in the presence of 0.1 mg/mL aprotinin. Conditioned media and cells were harvested and analyzed by immunoblotting with AAT1 and 2B5 antisera to detect AAT1 and total PC1/3, respectively; or anti-HA antibodies to detect only WT PC1/3. D, Quantification of expression and secretion of WT PC1/3 (HA-tag only) in the presence of N222D PC1/3; WT PC1/3 was normalized to 100%. Data are expressed as the mean ± SD, n = 3. P < .125 for cells (top panel) and P < .01 for media (bottom panel) using an unpaired Student's t test. CO-IP, co-immunoprecipitation; IP, immunoprecipitation.
Notably, the PC1/3-N222D variant was also able to associate with a WT PC1/3 partner (Figure 3A). Because of its trafficking defect and its association with WT PC1/3, we considered whether the N222D mutant might exert a dominant-negative effect on the WT enzyme. Once again, epitope-tagged partners were coexpressed, in conjunction with coexpression of a third plasmid encoding AAT1, a post-Golgi secretory pathway marker (18) that served as a control protein. As shown in Figure 3, B and C, using the anti-HA antibody that specifically detects the WT species, we observed a small but significant decrease in the secretion of the WT protein in the presence of the N222D mutant protein (P < .05; n = 3). Although we also observed a small effect of the mutant on enhancing the intracellular accumulation of WT protein (Figure 3, C and D), this was not statistically significant due to well-to-well variation; repetitions of the experiment yielded similar results. Secretion of the control protein AAT1 was unaffected under all conditions tested, indicating that retention of the N222D variant in the ER does not globally affect trafficking of secretory proteins. We observed similar results using HEK cells (data not shown).
Increased degradation of N222D PC1/3 occurs via the ubiquitination/proteasome system
As shown above, the PC1/3-N222D variant interacts with WT PC1/3 enzyme but does not profoundly affect its secretion. We therefore asked what happens to the N222D mutant within cells, expecting that its ER retention might result in targeting to degradative pathways (17). Neuro2a cells expressing PC1/3-WT-HA and PC1/3-N222D-HA constructs were incubated with the proteasomal inhibitor MG132 to prevent degradation of ubiquitinated protein. We observed an increased accumulation of both WT and N222D PC1/3 in the presence of MG132 (Figure 4A). These data are consistent with proteasomal degradation of ER-retained PC1/3. The amount of PC1/3 WT enzyme increased by 1.6-fold after MG132 treatment, whereas the amount of the PC1/3 N222D mutant increased by 1.9-fold (Figure 4B); similar results were found in 2 other experiments. These data collectively suggest that at least half of the ER-retained N222D pool of PC1/3 can be degraded via the ubiquitin- proteasome system.
Figure 4.

Increased degradation of PC1/3 N222D via the ubiquitination/proteasome system and increased intracellular retention in the presence of MG132. A, Neuro2a cells and HEK cells were transiently transfected with PC1/3-WT-HA or PC1/3-N222D-HA alone or coexpressed with Flag-tagged ubiquitin and cultured in the presence of 5μM lactacystin overnight. Cells were harvested, lysed, and immunoblotted with anti-HA antiserum to detect the immunoprecipitated proteins, and anti-Flag-HRP was then used to detect the ubiquitinated PC1/3. Upper panels show IP, and the lower panel shows ubiquitinated proteins. B, Neuro2a cells were transfected with PC1/3-WT-HA or PC1/3-N222D-HA in triplicate and cultured in the presence of 5μM of the proteasomal inhibitor MG132 for 12 hours. Cells were harvested, lysed, and analyzed for the expression of PC1/3 using an anti-HA antibody. An actin blot was carried out as a loading control. C, Quantification of PC1/3 vs actin expression using GraphPad Prism; data are given as the mean + SD, n = 3. *, P < .05 using an unpaired Student's t test. IP, immunoprecipitation; WB, Western blot.
We then compared the degradation of WT and N222D PC1/3 by carrying out direct ubiquitination assays in Neuro2a cells after transient transfection with either PC1/3-WT-HA or PC1/3-N222D-HA in the presence or absence of Flag-tagged ubiquitin constructs and in the presence of 5μM lactacystin (a proteasomal inhibitor, to block the degradation of ubiquitinated protein) (Figure 4C, left panel). Cells were then lysed, and PC1/3-WT-HA and PC1/3-N222D-HA were immunoprecipitated with anti-2B5 polyclonal antiserum and analyzed for ubiquitination with the anti-Flag HRP antibody. Both WT and N222D PC1/3 were found to be similarly ubiquitinated in Neuro2A cells. Interestingly, however, a similar experiment conducted using HEK cells (Figure 4C, right panel) revealed a dramatic increase in the ubiquitination of the N222D variant, suggesting differing degradation patterns in cells possessing solely constitutive and/or partially regulated secretory pathways, and increased susceptibility of the N222D variant to ubiquitination.
The PC1/3 N222D protein exhibits defective secretion and a longer intracellular half-life than WT PC1/3
Although a significant portion of the PC1/3-N222D mutant pool is targeted for degradation, as seen in Figure 4, there is also increased intracellular accumulation of PC1/3-N222D, which localizes to the ER (Figure 2). We carried out a pulse-chase experiment in Neuro2a cells to address potential differences in the biosynthesis of WT and N222D PC1/3. Transiently-transfected cells expressing PC1/3-WT-HA or PC1/3-N222D-HA constructs were metabolically labeled with 35S-Cys/Met and chased for 2 hours in medium containing cold methionine. Media were harvested and cells harvested, lysed, and analyzed as described. Consistent with the blotting results (Figure 3, B and C), we observed an increased intracellular content of the N222D variant and decreased secretion as compared with the WT protein after 2 hours of chase (Figure 5A). We then determined the intracellular half-life of the 2 proteins using CHX, which inhibits protein synthesis and is routinely used for monitoring protein half-life (17). Transiently transfected PC1/3-WT-HA and PC1/3-N222D-HA constructs were chased in plating media containing CHX for the indicated time points (Figure 5B). The N222D mutant exhibited a longer half-life than the WT protein (Figure 5B). No PC1/3-immunoreactive protein was detectable in the chase media by Western blotting during the course of this experiment (data not shown). Quantification of the density of the bands in Figure 5B revealed that PC1/3-WT-HA levels were significantly reduced at time points greater than 2 hours when compared with the mutant protein (Figure 5C). Linear regression analysis of the decay in band intensity over time also showed a significant difference in the decay of the WT (507 ± 37 AU/h) and mutant (403 ± 30 AU/h) proteins (P < .05). These data agree well with the strong intracellular accumulation and inefficient secretion observed for the PC1/3-N222D mutant. In agreement with its intracellular retention, no enzymatic activity of PC1/3-N222D was detected in secretion media from transfected HEK cells; this was the case whether the cleavable or the uncleavable form was transfected (data not shown).
Figure 5.

The PC1/3 N222D protein exhibits defective secretion and a longer intracellular half-life than WT PC1/3. A, Pulse-chase assay to assess the biosynthesis of the WT and N222D variant of PC1/3. Neuro2a cells transiently transfected PC1/3-WT-HA and PC1/3-N222D-HA were metabolically labeled with 35S-Cys/Met and chased for 2 hours in medium containing cold methionine. Media were harvested, and cells were harvested, lysed, and immunoprecipitated using anti-2B5 antibody to recover PC1/3 and analyzed as described in Materials and Methods. B, CHX experiment revealing differential degradation profiles of WT PC1/3 and PC1/3-N222D. Transiently transfected PC1/3-WT-HA and PC1/3-N222D-HA constructs were cultured in plating media overnight. Cells were chased in plating media containing 0.1 mg/mL CHX for the indicated time points and analyzed by immunoblotting using anti-HA antibody to detect PC1/3. C, Quantification of B was carried out with ImageJ software, and the data were analyzed using GraphPad Prism, as indicated in the Materials and Methods. (*, P < .05; n = 3; mean ± SD).
Discussion
The prohormone convertase PC1/3 has been overwhelmingly linked to human obesity in many independent genome-wide association studies as well as by clinical data in patients lacking function in both PC1/3 alleles (27). Interestingly, although mice haplodeficient in PC1/3 do not develop obesity (14), a recently identified mouse model with an N222D point mutation, obtained by chemical mutagenesis, does indeed resemble human obesity phenotypes caused by PC1/3 loss of function (15). In the present study, we aimed at elucidating the cell biology of the N222D PC1/3 mutation. Therefore, we first focused our effort on identifying possible differences in insulin processing in pancreatic islets in the mutant mice that may result in the release of mutated or misprocessed insulin forms that may potentially modify the insulin-receptor interaction. Previous studies have suggested that WT PC1/3 first cleaves the B-chain/C-peptide junction of proinsulin, whereas PC2 preferentially cleaves the C-peptide/A-chain junction (23, 24). We found that PC1/3N222D produces increased amounts of proinsulin processing intermediates and specifically attacks the C-peptide/A-chain junction while leaving the B-chain uncleaved (intermediate 1) (Figure 1A), supporting the idea that proinsulin cleavage is severely compromised in the N222D mutant mouse. Our pulse-chase data confirm an earlier finding from a static metabolic labeling experiment (15) and further reveal that in the pancreatic islets from PC1/3N222D mutant mice, there is accumulation of proinsulin and its processing intermediates, again indicating a major proinsulin processing defect with inefficient insulin production.
Prohormone processing defects can theoretically be either due to impaired catalytic activity of a convertase, impaired convertase intermolecular processing, and/or mislocalization of processing enzymes. Thus far, studies on the PC1/3-N222D mutant (9) and many other PC1/3 SNPs (11, 28) have attributed enzyme malfunction solely to defects in enzyme maturation. In the present study, by using an uncleavable form of the enzyme, we were able to follow the localization, biosynthesis, secretion, and degradation profile of PC1/3-N222D in neuroendocrine cells. Because the WT and mutant PC1/3 display different maturation rates, this type of tracking would be impossible if full maturation occurred (15). Using this approach, we observed that PC1/3-N222D-transfected cells displayed aberrant transport through the secretory pathway, with the mutant protein accumulating in the ER. Interestingly, although the activity of the mutant enzyme was eliminated, processing of the zymogen was unaffected, indicating that the mutation does not affect the initial step of enzyme maturation, the removal of propeptide in the ER.
It has been demonstrated that certain ER-retained proteins can continue to interact with their WT assembly partners and thereby affect the function of WT proteins, inducing pathological consequences (29, 30). Given potential misfolding of the mutant protein, the strong ER retention, and the known oligomerization of PC1/3 (26), we tested whether the interaction of PC1/3-N222D with WT PC1/3 could affect transit of the WT protein through the secretory pathway. Our immunoprecipitation data show clear association of the WT enzyme with the mutant enzyme and a modestly small dominant-negative effect on intracellular retention of the WT enzyme by the mutant.
The ER accumulation of the PC1/3-N222D mutant could be due to misfolding of the protein within the ER, either because of aberrant glycosylation, aberrant oligomerization of the mutant enzyme (26), or a local misfolding event (26). Although the migration properties of the WT and mutant proteins did not differ upon Western blotting, small changes in glycosylation cannot be detected by this type of analysis. Intriguingly, even though the PC1/3-N222D mutant accumulated in the ER, its overexpression did not generally compromise the secretory machinery, because we observed efficient secretion of AAT1 in the presence of PC1/3-N222D (18).
Misfolded proteins in the ER are commonly disposed by ER-associated degradation, which includes recognition of misfolded proteins, retrotranslocation from the ER to the cytosol, ubiquitination by various ubiquitination enzymes, targeting of substrates to the proteasome, and degradation by the proteasome (31). We first analyzed the degradation characteristics of the mutant enzyme by inhibiting the proteasomal degradation. In 3 experiments, the PC1/3-N222D protein accumulated to a greater extent in comparison with the WT enzyme in the presence of the proteasomal inhibitor MG132. Although increased ubiquitination of PC1/3-N222D was not obvious in Neuro2a cells, a dramatic increase in the accumulation of the ubiquitinated form of the PC1/3-N222D mutant over WT PC1/3 was detected in HEK cells, demonstrating cell-dependent differential susceptibility to degradation. This discrepancy can likely be ascribed to biochemical differences in the early secretory pathways of cells possessing constitutive and/or regulated secretory pathways. Differential effects of PC1/3 mutations in varying cell lines have been attributed to differential expression, secretion, and maturation pathways (11). We propose that a substantial portion of the N222D mutant protein is disposed of by efficient targeting to degradative pathways, ie, the ubiquitin-proteasome system.
In addition to the ER retention phenotype and increased degradation, we found that the PC1/3-N222D mutant was not synthesized and transported normally through the secretory pathway. A dynamic metabolic labeling experiment showed defective secretion of the PC1/3-N222D mutant and confirmed increased intracellular accumulation. A CHX-chase experiment further showed that cellular PC1/3 N222D exhibits a longer half-life as compared with WT PC1/3. Consistent with our finding of major ER retention, we were unable to detect significant quantities of mutant PC1/3 secreted into conditioned media. Taken together, these data confirm the lack of the ability of PC1/3-N222D mutant to efficiently traverse the secretory pathway. We speculate that a portion of the ER-retained PC1/3-N222D mutant may interact with WT PC1/3 and target the complex for degradation via the ubiquitin-proteasome system. Although this must represent a small population of the WT protein, because the majority seems to traffic independently of the mutant protein, it may be sufficient to cause functional impairment in terms of hormone processing.
Enzyme activity assays of conditioned media from cells expressing PC1/3-N222D confirmed that the mutant enzyme is not only secreted defectively but also that the nominal amount of the enzyme that was secreted was completely inactive when tested at several different concentrations, even after concentration; this was the case for both the cleavable and uncleavable versions of this mutant (unpublished data). These results contrast with previous data showing that secreted PC1/3-N222D exhibits only a 36%–45% reduction in enzymatic activity as compared with WT enzyme (15, 32). This disparity is likely due to large differences in assays and constructs between groups, which include our routine inclusion of general protease inhibitors, the use of different WT backbone sequences (32, 33), and the use of Flag-immunopurified human enzyme (11, 32) or baculovirus-expressed His-tagged mouse enzyme (15).
The N222D mutation is located in a side chain loop in a conserved calcium-binding site necessary for the structural stabilization of the protein. Interestingly, this mutation is also located adjacent to a known common human polymorphism, N221D (rs6232) (11, 34). Previous studies of this polymorphism showed clear association with a small increase in the risk for obesity (9, 32). An early report indicated that the N221D mutation results in only a 10% reduction in total activity (15, 32), whereas data from our group showed a 36% decrease in the specific activity of the N221D variant (35); thus, the N221D mutation may not be as deleterious as the neighboring N222D substitution. (These contrasting activity data again highlight the need for standardization of assays and constructs.) It is intriguing that most the heterozygous PC1/3 mutants recently reported are located either within or adjacent to a major calcium-binding site (11). It could therefore be interesting to determine whether the N221D mutant enzyme exhibits altered calcium requirements for either activity or stability.
In conclusion, our data concerning the intracellular behavior of the N222D PC1/3 variant suggest a possible mechanistic insight into its dysfunction. Although the N222D mutant is synthesized normally and the proprotein processed correctly to the 87-kDa form, the mutant protein is not able to traverse the secretory pathway distal to the ER and is likely subjected to increased ER retrotranslocation and to ubiquitin/proteasomal-mediated degradation. Previous work has shown that native N222D proteins are also subjected to impaired processing to the more active smaller forms (15); the ER retention we demonstrate here likely contributes to this reduced processing. We speculate that these profound differences in protein trafficking and degradation result in a much smaller amount of secretory granule enzyme available for normal proinsulin processing, hence the defective proinsulin processing observed in mutant mouse islets. Increasing numbers of studies show that PC1/3 SNPs and/or deficiency states are associated with risk for obesity, diabetes, and, in certain cases, severe hormonal and metabolic defects (9–13, 32, 36–42). Although the N222D mutation is itself not found in humans, our data highlight the potential contribution of incorrect PC1/3 enzyme protein trafficking through the secretory pathway in these disease states.
Acknowledgments
We thank Lindsay Pickett for early studies and for support with the generation of PC1/3-WT-Flag and PC1/3-N222D-Flag constructs, Valeria Albornoz for assistance with the MG132 experiments, and Kevin Li and Wei Gao for help with Western blottings. We also thank Juan S. Bonifacino (National Institutes of Health, Bethesda, MD) for providing us with plasmids encoding Ubiquitin-Flag and HA-BACE1 constructs.
Present address for J.R.P.: Department of Medical Sciences, University of Castilla-La Mancha, 13071 Ciudad Real, Spain.
Present address for N.G.: BioScience Center, San Diego State University, San Diego, CA 92182.
This work was supported by National Institutes of Health Grants DA05084 (to I.L.), DK48280 (to P.A.), and DK079925 (to N.G.).
Disclosure Summary: The authors have nothing to disclose.
For News & Views see page 2343
- AAT1
- α-1 antitrypsin
- BACE1
- β-secretase 1
- CHX
- cycloheximide
- ER
- endoplasmic reticulum
- FBS
- fetal bovine serum
- HA
- influenza hemagglutinin antigen
- HRP
- horseradish peroxidase
- PC1/3
- prohormone convertase 1/3
- SNP
- single nucleotide polymorphism
- TGN
- trans-Golgi network
- WT
- wild type.
References
- 1. Steiner DF. On the discovery of precursor processing. Methods Mol Biol. 2011;768:3–11 [DOI] [PubMed] [Google Scholar]
- 2. Zhou Y, Lindberg I. Enzymatic properties of carboxyl-terminally truncated prohormone convertase 1 (PC1/SPC3) and evidence for autocatalytic conversion. J Biol Chem. 1994;269:18408–18413 [PubMed] [Google Scholar]
- 3. Milgram SL, Mains RE. Differential effects of temperature blockade on the proteolytic processing of three secretory granule-associated proteins. J Cell Sci. 1994;107:737–745 [DOI] [PubMed] [Google Scholar]
- 4. Benjannet S, Rondeau N, Paquet L, et al. Comparative biosynthesis, covalent post-translational modifications and efficiency of prosegment cleavage of the prohormone convertases PC1 and PC2: glycosylation, sulphation and identification of the intracellular site of prosegment cleavage of PC1 and PC2. Biochem J. 1993;294(pt 3):735–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Coates LC, Birch NP. Posttranslational maturation of the prohormone convertase sPC3 in vitro. J Neurochem. 1997;68:828–836 [DOI] [PubMed] [Google Scholar]
- 6. Zhou Y, Rovere C, Kitabgi P, Lindberg I. Mutational analysis of PC1 (SPC3) in PC12 cells. 66-kDa PC1 is fully functional. J Biol Chem. 1995;270:24702–24706 [DOI] [PubMed] [Google Scholar]
- 7. Zhou A, Webb G, Zhu X, Steiner DF. Proteolytic processing in the secretory pathway. J Biol Chem. 1999;274:20745–20748 [DOI] [PubMed] [Google Scholar]
- 8. Jackson RS, Creemers JW, Farooqi IS, et al. Small-intestinal dysfunction accompanies the complex endocrinopathy of human proprotein convertase 1 deficiency. J Clin Invest. 2003;112:1550–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Farooqi IS, Volders K, Stanhope R, et al. Hyperphagia and early-onset obesity due to a novel homozygous missense mutation in prohormone convertase 1/3. J Clin Endocrinol Metab. 2007;92:3369–3373 [DOI] [PubMed] [Google Scholar]
- 10. Jackson RS, Creemers JW, Ohagi S, et al. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat Genet. 1997;16:303–306 [DOI] [PubMed] [Google Scholar]
- 11. Creemers JW, Choquet H, Stijnen P, et al. Heterozygous mutations causing partial prohormone convertase 1 deficiency contribute to human obesity. Diabetes. 2012;61:383–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Choquet H, Kasberger J, Hamidovic A, Jorgenson E. Contribution of common PCSK1 genetic variants to obesity in 8,359 subjects from multi-ethnic American population. PLoS One. 2013;8:e57857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Villalobos-Comparan M, Villamil-Ramirez H, Villarreal-Molina T, et al. PCSK1 rs6232 is associated with childhood and adult class III obesity in the Mexican population. PLoS One. 2012;7:e39037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhu X, Zhou A, Dey A, et al. Disruption of PC1/3 expression in mice causes dwarfism and multiple neuroendocrine peptide processing defects. Proc Natl Acad Sci USA. 2002;99:10293–10298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lloyd DJ, Bohan S, Gekakis N. Obesity, hyperphagia and increased metabolic efficiency in Pc1 mutant mice. Hum Mol Genet. 2006;15:1884–1893 [DOI] [PubMed] [Google Scholar]
- 16. Liu M, Hodish I, Rhodes CJ, Arvan P. Proinsulin maturation, misfolding, and proteotoxicity. Proc Natl Acad Sci USA. 2007;104:15841–15846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ishikura S, Weissman AM, Bonifacino JS. Serine residues in the cytosolic tail of the T-cell antigen receptor α-chain mediate ubiquitination and endoplasmic reticulum-associated degradation of the unassembled protein. J Biol Chem. 2010;285:23916–23924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Feng L, Arvan P. The trafficking of α 1-antitrypsin, a post-Golgi secretory pathway marker, in INS-1 pancreatic β cells. J Biol Chem. 2003;278:31486–31494 [DOI] [PubMed] [Google Scholar]
- 19. Prabhu Y, Burgos PV, Schindler C, Farías GG, Magadan JG, Magadán JG, Bonifacino JS. Adaptor protein 2-mediated endocytosis of the β-secretase BACE1 is dispensable for amyloid precursor protein processing. Mol Biol Cell. 2012;23:2339–2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232 [DOI] [PubMed] [Google Scholar]
- 21. Muller L, Zhu P, Juliano MA, Juliano L, Lindberg I. A 36-residue peptide contains all of the information required for 7B2-mediated activation of prohormone convertase 2. J Biol Chem. 1999;274:21471–21477 [DOI] [PubMed] [Google Scholar]
- 22. Vindrola O, Lindberg I. Biosynthesis of the prohormone convertase mPC1 in AtT-20 cells. Mol Endocrinol. 1992;6:1088–1094 [DOI] [PubMed] [Google Scholar]
- 23. Irminger JC, Meyer K, Halban P. Proinsulin processing in the rat insulinoma cell line INS after overexpression of the endoproteases PC2 or PC3 by recombinant adenovirus. Biochem J. 1996;320(pt 1):11–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rhodes CJ, Lincoln B, Shoelson SE. Preferential cleavage of des-31,32 proinsulin over intact proinsulin by the insulin secretory granule type II endopeptidase. J Biol Chem. 1992;267:22719–22727 [PubMed] [Google Scholar]
- 25. Hornby PJ, Rosenthal SD, Mathis JP, Vindrola O, Lindberg I. Immunocytochemical localization of the neuropeptide-synthesizing enzyme PC1 in AtT-20 cells. Neuroendocrinology. 1993;58:555–563 [DOI] [PubMed] [Google Scholar]
- 26. Hoshino A, Kowalska D, Jean F, Lazure C, Lindberg I. Modulation of PC1/3 activity by self-interaction and substrate binding. Endocrinology. 2011;152:1402–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Creemers JW, Khatib AM. Knock-out mouse models of proprotein convertases: unique functions or redundancy? Front Biosci. 2008;13:4960–4971 [DOI] [PubMed] [Google Scholar]
- 28. Mbikay M, Sirois F, Nkongolo KK, Basak A, Chrétien M. Effects of rs6234/rs6235 and rs6232/rs6234/rs6235 PCSK1 single-nucleotide polymorphism clusters on proprotein convertase 1/3 biosynthesis and activity. Mol Genet Metab. 2011;104:682–687 [DOI] [PubMed] [Google Scholar]
- 29. Park SY, Ye H, Steiner DF, Bell GI. Mutant proinsulin proteins associated with neonatal diabetes are retained in the endoplasmic reticulum and not efficiently secreted. Biochem Biophys Res Commun. 2010;391:1449–1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wright J, Wang X, Haataja L, et al. Dominant protein interactions that influence the pathogenesis of conformational diseases. J Clin Invest. 2013;123:3124–3134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brodsky JL. Cleaning up: ER-associated degradation to the rescue. Cell. 2012;151:1163–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Benzinou M, Creemers JW, Choquet H, et al. Common nonsynonymous variants in PCSK1 confer risk of obesity. Nat Genet. 2008;40:943–945 [DOI] [PubMed] [Google Scholar]
- 33. Creemers JW, Roebroek AJ, Van de Ven WJ. Expression in human lung tumor cells of the proprotein processing enzyme PC1/PC3. Cloning and primary sequence of a 5 kb cDNA. FEBS Lett. 1992;300:82–88 [DOI] [PubMed] [Google Scholar]
- 34. Henrich S, Lindberg I, Bode W, Than ME. Proprotein convertase models based on the crystal structures of furin and kexin: explanation of their specificity. J Mol Biol. 2005;345:211–227 [DOI] [PubMed] [Google Scholar]
- 35. Pickett LA, Yourshaw M, Albornoz V, et al. Functional consequences of a novel variant of PCSK1. PLoS One. 2013;8:e55065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martín MG, Lindberg I, Solorzano-Vargas RS, et al. Congenital proprotein convertase 1/3 deficiency causes malabsorptive diarrhea and other endocrinopathies in a pediatric cohort. Gastroenterology 2013;145:138–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bandsma RH, Sokollik C, Chami R, et al. From diarrhea to obesity in prohormone convertase 1/3 deficiency: age-dependent clinical, pathologic, and enteroendocrine characteristics. J Clin Gastroenterol. 2013;47:834–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yourshaw M, Solorzano-Vargas RS, Pickett LA, et al. Exome sequencing finds a novel PCSK1 mutation in a child with generalized malabsorptive diarrhea and diabetes insipidus. J Pediatr Gastroenterol Nutr. 2013;57:759–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wei X, Ma X, Lu R, et al. Genetic Variants in PCSK1 gene are associated with the risk of coronary artery disease in type 2 diabetes in a Chinese han population: a case control study. PLoS One. 2014;9:e87168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sirois F, Kaefer N, Currie KA, Chrétien M, Nkongolo KK, Mbikay M. Allelic clustering and ancestry-dependent frequencies of rs6232, rs6234, and rs6235 PCSK1 SNPs in a Northern Ontario population sample. J Community Genet. 2012;3:319–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gjesing A, Vestmar M, Jorgensen T, et al. The effect of PCSK1 variants on waist, waist-hip ratio and glucose metabolism is modified by sex and glucose tolerance status. PLoS One. 2011;6:e23907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Strawbridge RJ, Dupuis J, Prokopenko I, et al. Genome-wide association identifies nine common variants associated with fasting proinsulin levels and provides new insights into the pathophysiology of type 2 diabetes. Diabetes. 2011;60:2624–2634 [DOI] [PMC free article] [PubMed] [Google Scholar]