

Abstract
Adverse experiences during gestation such as maternal stress and infection are known risk factors for neurodevelopmental disorders, including schizophrenia, autism, and attention deficit/hyperactivity disorder. The mechanisms by which these distinct exposures may confer similar psychiatric vulnerability remain unclear, although likely involve pathways common to both stress and immune responses at the maternal-fetal interface. We hypothesized that maternal stress-induced activation of immune pathways within the placenta, the sex-specific maternal-fetal intermediary, may contribute to prenatal stress programming effects on the offspring. Therefore, we assessed for markers indicative of stress-induced placental inflammation, and examined the ability of maternal nonsteroidal antiinflammatory drug (NSAID) treatment to ameliorate placental effects and thereby rescue the stress-dysregulation phenotype observed in our established mouse model of early prenatal stress (EPS). As expected, placental gene expression analyses revealed increased levels of immune response genes, including the proinflammatory cytokines IL-6 and IL-1β, specifically in male placentas. NSAID treatment partially ameliorated these EPS effects. Similarly, in adult offspring, males displayed stress-induced locomotor hyperactivity, a hallmark of dopaminergic dysregulation, which was ameliorated by maternal NSAID treatment. Fitting with these outcomes and supportive of dopamine pathway involvement, expression of dopamine D1 and D2 receptors was altered by EPS in males. These studies support an important interaction between maternal stress and a proinflammatory state in the long-term programming effects of maternal stress.
Epidemiological studies establish gestation as a period of vulnerability to environmental perturbations. Fetal antecedents such as maternal stress or infection are associated with an increased risk for neurodevelopmental disorders, including schizophrenia, autism spectrum disorder (ASD), and attention deficit hyperactivity disorder (ADHD) (1–7). Gender also appears to be a contributing factor, as such disorders present with known sex biases in onset, symptomology, and treatment. For instance, boys present with ASD at 4–5 times the rate of girls, and ADHD at 2–3 times the rate of girls (8–11). The contributing mechanisms by which distinct exposures such as stress and infection program disease vulnerability are not clear, although likely involve pathways common to both at the maternal-fetal interface.
Studies in humans and animal models support the critical involvement of the developing placenta in the long-term programming effects and transmission of the maternal milieu to the fetus (12–14). Derived from the blastocyst, the placenta reflects the fetal genetic sex, and sex differences in patterns of gene expression, signaling, and responses to maternal stimuli have been established in human and rodent tissue (15–24). Differential placental responses to prenatal perturbations such as maternal stress may lead to sex-specific fetal programming ultimately contributing to disease risk.
In our early prenatal stress (EPS) mouse model, we have previously shown that male offspring present with heightened stress sensitivity and impaired cognitive function (25–27). Similar outcomes have been reported in guinea pigs as well (28, 29). Further, we have established that the mouse placenta responds to EPS with sex-specific changes in gene expression and function, supporting a placental mechanism whereby stress experienced early in gestation can influence embryo development throughout gestation (18, 26).
Reciprocal interactions between neuroendocrine stress response pathways and the immune system are well documented, and evidence suggests that inflammation occurs downstream of stress in pregnancy (30–37). Furthermore, maternal immune activation has been similarly linked to neurodevelopmental disorder risk, and in animal models, leads to offspring outcomes that are analogous to maternal stress effects (1, 2, 12, 38–42). Thus, examination of placental inflammation as a potential contributing mechanism is warranted. Therefore, we hypothesized that offspring programming effects of maternal stress early in pregnancy involve, in part, sex-specific changes in placental immune pathway activation. To test this, we examined markers indicative of placental inflammation in our EPS model and the potential amelioration of the EPS offspring phenotype by maternal antiinflammatory treatment concomitant with prenatal stress.
Materials and Methods
Animals
Male and nulliparous female C57/Bl6:129 F1 hybrid mice generated in house were bred at 10–12 weeks of age as described elsewhere (18). Detection of a copulation plug denoted embryonic day (E) 1. Females were individually housed and randomly assigned to stress or control groups, with or without antiinflammatory treatment. Following completion of maternal treatments, dams were either sacrificed at E12.5 for placental analyses (6–8 dams/ group) or allowed to give birth (8–10 dams/group) and remain with litters until weaned on postnatal day (P) 28. For all embryonic and postnatal studies, only 1 male and female per litter were used in each assay to avoid litter effects. Mice were maintained on a 12-hour light, 12-hour dark cycle (lights on at 7:00 am) with ambient temperature of 22°C and relative humidity of 42%. Food (Purina Rodent Chow; 28.1% protein, 59.8% carbohydrate, 12.1% fat) and water were provided ad libitum. All animal procedures were conducted in agreement with the Guide for the Care and Use of Laboratory Animals in accordance with NIH guidelines and were approved by the University of Pennsylvania Institutional Animal Care and Use Committee.
Maternal treatments
EPS.
Chronic variable stress was administered to dams on embryonic days E1–E7 as described previously (43). Dams experienced 1 novel stressor per day as follows: multiple cage changes, 100 dB white noise overnight (Sleep Machine; Brookstone), 15-minute restraint in a 50-mL conical tube, 36-hour constant light, novel objects in the home cage overnight (8 marbles similar in size and color), 1-hour exposure to fox odor (1:5000; 2,4,5-trimethylthiazole; Acros Organics), and wet bedding overnight. Nonstressed controls remained undisturbed with the exception of weekly cage changes.
Nonsteroidal antiinflammatory drug (NSAID) administration.
Dams assigned to receive antiinflammatory treatment were administered acetylsalicylic acid (aspirin; 81-mg tablet; CVS Pharmacy) at 162 mg/L in drinking water on E1–E7 as described previously (44). NSAID-treated water was replaced every 48 hours. Normal drinking water was resumed upon light-phase onset on E8. Acetylsalicylic acid was selected due to its cyclooxygenase-inhibitory action, oral bioavailability, known efficacy across diverse species, and previously established dosing paradigm (45, 46). Duration of NSAID treatment was selected in order to encompass the period of maternal stress and avoid the dysmasculinizing effects resulting from cyclooxygenase inhibition during late gestation (45). NSAID treatment did not alter maternal food or water intake; however, the potential influence on postnatal maternal care was not examined.
Embryonic analyses
Placenta and embryonic tissue collection.
To determine the effects of EPS on placental gene expression, dams designated for embryonic analyses were killed on E12.5 as described previously (26). The E12.5 placenta is used for analysis because the mouse chorioallantoic placenta is fully differentiated at this time (21). All dissections were completed between 10:00 am and 2:00 pm. Conceptuses were isolated from the uterine wall, rinsed in dH2O, blotted dry, and placed on Parafilm (Fisher Scientific). The amniotic sac was pierced with a needle and discarded. The umbilical cord was removed using forceps. Placentas were placed fetal side down on a petri dish, hemisected in the transverse plane, submerged in 500 μL RNA Later (Ambion), and stored at −80 C until RNA isolation. Intrauterine position and observed resorptions were recorded. Embryonic tissue was retained for determination of sex by genotyping using primers specific to Jarid1 (5′-TGAAGCTTTTGGCTTTGAG-3′ and 5′-CCGCTGCCAAATTCTTTGG-3′) as described elsewhere (47). One male and one female placenta per litter were selected for gene expression analyses. To limit effects of intrauterine position, we selected placentas from the first third of the embryos from the cervical end and excluded female placentas flanked by two males (2M females) (48). Because the steroidogenic cells of the fetal gonad do not differentiate until E12.5, placentas have not yet been influenced by gonadal hormones of neighboring embryos (49).
TaqMan low-density quantitative RT-PCR immune array.
Placentas (n = 6–8 per group) were submerged in 2 mL TRIzol reagent (Invitrogen), probe sonicated, and RNA isolated. Total RNA (150 ng) was reverse transcribed to cDNA using the High Capacity cDNA Reverse Transcriptase Kit (Applied Biosystems). Expression levels of 90 immune response genes and 6 candidate endogenous controls were determined using preconfigured 384-well microfluidic cards (catalog no. 4367786; Applied Biosystems) run on a 7900-HT fast real-time PCR Machine (Applied Biosystems). Data were analyzed using the comparative Ct method (50). For each sample, raw Ct values for each target gene were normalized to the average of the endogenous controls, β-actin (Actb) and glyceraldehyde-3-phosphate dehydrogenase (Gapdh). Gene expression relative to nonstressed, vehicle-treated controls was determined. Ct values greater than 35 and genes with expression in less than 50% of samples were excluded from analysis.
Offspring behavior analyses
Offspring were weaned on P28 and housed 3–4 per cage by sex and treatment. Littermates were distributed into cohorts for testing as follows: 1) light-dark exploration test; 2) prepulse inhibition of the acoustic startle response; 3) Barnes maze test of spatial learning; and 4) postmortem brain analyses. Cohorts comprised 1 male and 1 female per litter in order to avoid litter effects. Mice were habituated to handling 1 week prior to testing. The potential impact of estrous cycle stage was not examined.
Light-dark exploration test.
The light-dark exploration test was performed as described previously (51). The apparatus comprised a Plexiglas enclosure divided into a brightly illuminated chamber (300 lux) and a dark chamber (5 lux). Experimentally naïve adult offspring (10–12 weeks old; n = 8–9 per group) were placed in the center of the light chamber, and their activity was video monitored for the 10-minute test duration. Testing was conducted between 9:00 pm and 2:00 am, 2–5 hours after lights off. Time spent in the light compartment, total distance traveled, and light-dark transitions were quantified by ANY-maze version 4.75 (Stoelting Co).
Prepulse inhibition (PPI) of acoustic startle.
PPI was recorded in SR-LAB startle chambers (San Diego Instruments) as described previously (52, 53). All test sessions were conducted 2–6 hours after lights off. A session began with 5 minutes acclimation to background noise (65 dB) followed by 5 consecutive acute habituation tones (40 milliseconds duration; 120 dB). Ten repetitions of each of the following types were then presented pseudorandomly: startle pulse only (40 milliseconds; 120 dB), no stimulus (65 dB background), and prepulses (20 milliseconds; +4, +8, or +16 dB above background) preceding the startle pulse (40 milliseconds; 120 dB) by 100 milliseconds. Variable intertrial intervals averaged 15 seconds. Acoustic startle response was defined as the peak startle magnitude recorded during 65 consecutive 1-millisecond readings following the startle pulse onset. The percentage PPI for each prepulse intensity was calculated for each animal as [1 − (average response to prepulse + startle)/(average response to startle only)] × 100 before group averages were compared. Animals with average response in startle-only trials of less than or equal to 50 mV were excluded from analysis.
Barnes maze test.
The Barnes maze test of spatial learning and memory was performed as described previously (25). The apparatus consisted of a white circular platform with 24 holes evenly dispersed around the perimeter. A target escape box was located in the same hole throughout the test. Distinct visual cues remained in a fixed position around the maze perimeter. Mice (11–14 weeks old; n = 7–9 per group) were trained for 2 trials per day for 3 days. A trial ended upon entry into the target box or after 4 minutes of elapsed time. If the mouse failed to locate the target, the investigator guided the mouse into the target box, and a latency of 240 seconds was assigned. Trials were video recorded and scored by a trained experimenter blinded to treatment. Latency to nose-poke into the target box was determined for each trial. Mice that failed to locate the target in at least 1 of the 6 trials were excluded from analysis (n = 15).
Adult brain tissue collection.
Experimentally naïve male offspring (n = 7–9/group for mRNA; n = 4/group for protein) were anesthetized and decapitated at 4 months of age. Brains were removed, rapidly frozen on dry ice, and stored at −80 °C. Whole brains were cryosectioned at −20 °C and micropunched using 0.75-mm Harris Uni-Core tissue punchers (Ted Pella), according to the Paxinos and Franklin mouse brain atlas (54). For the prefrontal cortex (PFC), bilateral punches were collected from 2 successive 300-μm sections from +1.98 to +1.38 mm anterior to bregma. For the nucleus accumbens (NAc), bilateral punches from 2 successive 300-μm sections from +1.68 to +1.08 anterior to bregma, with punchers placed immediately ventral to the anterior commissure tracts. Micropunches for gene expression assays were immediately submerged in 100 uL TRIzol reagent (Invitrogen), frozen on dry ice, and stored at −80 °C until RNA isolation. Micropunches for Western blots were submerged in 50 μL radioimmune precipitation assay buffer, protein extracted, and stored at −80 °C.
Prefrontal cortical, nucleus accumbens, and ventral tegmental area quantitative RT-PCR.
Total RNA (150 ng) was reversed transcribed to cDNA as described above. PFC and NAc gene expression was analyzed by TaqMan Gene Expression Assays (Life Technologies) for the target genes glutamic acid decarboxylase 1 (Gad1; Mm00725661_s1), dopamine receptor type I (Drd1a; Mm01353211_m1), and dopamine receptor type II (Drd2; Mm00438545_m1). Raw Ct values for each sample were normalized to glyceraldehyde-3-phosphate dehydrogenase (Gapdh; Mm99999915_g1) and analyzed using the comparative Ct method as described above.
Western blot.
Following quantification by Pierce BCA Protein Assay (Thermo Scientific), 20 μg total protein was loaded per lane of NuPAGE 10% Bis-Tris gels (Life Technologies). Western blots were completed as described (55). Blots were probed for tyrosine hydroxylase (TH) (1:1000; Pel-Freeze), stripped, and reprobed for β-actin (1:1000; EMD Millipore). Densitometric analyses were performed in ImageJ (NIH). Within each sample, TH values were normalized to β-actin for quantitative analysis.
Statistical analyses
Statistical analyses were performed using JMP10 (SAS Institute) and R for Mac version 3.0.1 (R Foundation for Statistical Computing). Statistical significance was set at P < .05. Data are shown as mean ± SEM. Data were analyzed by two-way ANOVA within sex for prenatal stress (control, EPS) and antiinflammatory treatment (vehicle, NSAID), with repeated measures where appropriate (trials 1–6 in the Barnes maze test; prepulse intensity for PPI). Upon detection of significant EPS × NSAID interactions, the ability of NSAID treatment to rescue EPS effects was assessed by post hoc Tukey HSD test. Statistically significant rescue was defined a priori by the co-occurrence of an EPS effect (control/vehicle vs EPS/vehicle, P < .05) that was no longer present in offspring administered concomitant NSAID (control/vehicle vs EPS/NSAID; P > .05). Western blots were analyzed by two-tailed Student's t test.
Results
Litter size, sex ratio, and resorption rate
Because early gestation manipulations can result in fetal lethality, we assessed for treatment effects on litter size and sex ratio (56). Consistent with our previous studies using this mild-stress paradigm, EPS did not alter litter size [F(1, 33) = 0.0027, P = .96] or sex ratio [F(1, 33) = 0.032, P = .86] (43). There were no effects of maternal NSAID treatment on litter size [F(1, 33) = 0.14, P = .72] or sex ratio [F(1, 33) = 0.09, P = .77]. In agreement with these results, neither EPS [F(1, 33) = 1.7; P = .20] nor maternal NSAID [F(1, 33) = 0.035; P = .85] altered the number of fetal resorptions observed on E12.5. No significant interactions between EPS or NSAID treatment were detected. Mean litter size, male percentage, and resorption rates are summarized in Supplemental Table 1.
EPS induces sex-dependent changes in placental gene expression
To test the hypothesis that early-gestation maternal stress induces placental inflammation, expression of immune response genes in placentas from E12.5 males and females was analyzed by TaqMan Low Density Mouse Immune Array. Of the 90 target genes assayed, 81 were detectable in the placenta. Only one gene, the proapoptotic factor, Fas ligand (FASL), was affected in placentas of both sexes (Figure 1, A and B). EPS significantly increased FASL [males: F(1, 24) = 8.19, P = .0086; females: F(1, 26) = 5.18, P = .031], an effect that did not interact with NSAID treatment [males: F(1, 24) = 0.001, P = .97; females: F(1, 26) = 0.19; P = .67].
Figure 1.

EPS altered expression of immune response genes in E12.5 placentas. Analyses of TaqMan Low Density Mouse Immune Array revealed significant main effects of EPS. Differentially expressed genes in males (A) and females (B) are organized into functional clusters, and their relative expression values are shown. The proapoptotic factor, FASL, was up-regulated in prenatally stressed males and females. In males, 11 additional genes were up-regulated by prenatal stress, including genes involved in cytokine signaling, chemokine signaling, cell surface antigens, and endothelial molecules. Significant interactions between EPS and NSAID treatment were detected for IL-6 and CCR7. Post hoc comparisons revealed increased IL-6 and CCR7 in stressed male placentas relative to nonstressed males. Maternal NSAID treatment rescued these EPS effects, as placental IL6 and CCR7 of EPS/NSAID males did not differ from controls. EPS decreased CCL2 in females (B). No main effects of NSAID were observed. Data are mean ± SEM. *, P < .05 main effect of EPS by two-way ANOVA; #, P < .05 vs CON/VEH by Tukey HSD. CON, control; EDN1, endothelin 1; PTPRC, protein tyrosine phosphatase receptor C; SELP, P-selectin; VEH, vehicle.
In male placentas, 12 genes were up-regulated and ranged in magnitude from 1.25- to 1.75-fold relative to nonstressed, vehicle-treated controls (Figure 1A). EPS increased proinflammatory cytokines, [IL-6; F(1, 24) = 4.3; P = .049] and IL-1β [F(1, 24) = 4.71; P = .04], cytokine receptor IL-2 receptor α [F(1, 24) = 5.51; P = .027], and the cyclooxygenase, prostaglandin-endoperoxide synthase 2 [F(1, 24) = 4.53, P = .044]. EPS also up-regulated genes involved in chemokine signaling, including chemokine ligand 5 [CCL5; F(1, 24) = 5.86; P = .023] and chemokine ligand 10 [CXCL10; F(1, 24) = 9.19; P = .0058]. Cell surface antigens, histocompatibility 2 class II antigen E β [F(1, 24) = 8.2; P = .0086] and protein tyrosine phosphatase receptor C [F(1, 24) = 7.56; P = .011], were significantly increased. Finally, EPS increased expression of the endothelial molecules, endothelin 1 [F(1, 24) = 5.21; P = .032] and P-selectin [F(1, 24) = 4.93; P = .036]. Although the magnitude of change often appears decreased by concomitant maternal antiinflammatory treatment, NSAID rescued EPS effects on IL-6 and chemokine receptor 7 (CCR7) only, as evidenced by significant interactions between EPS and NSAID [IL-6: F(1, 24) = 6.11, P = .021; CCR7: F(1, 24) = 4.64; P = .041]. Post hoc comparisons revealed increased IL-6 (P = .019) and CCR7 (P = .048) in stressed male placentas relative to nonstressed males (Figure 2, B and C). Maternal NSAID treatment rescued these EPS effects, because placental IL6 (P = .64) and CCR7 (P = .73) of EPS/NSAID males did not differ from controls. No main effects of NSAID treatment were detected. ANOVAs for all expressed genes are detailed in Supplemental Tables 2 and 3.
Figure 2.

Prenatal stress elicited locomotor hyperactivity in adult males that was prevented by maternal antiinflammatory treatment. In the light-dark exploration test, time in light, distance traveled, and zone transitions were assessed in male (A–C) and female (D–F) offspring. Prenatally stressed males exhibited increased time in the light compartment (A), due to general hyperactivity as assessed by distance traveled (B) and zone transitions (C). NSAID treatment concomitant with maternal stress prevented hyperactivity in males, because exploration of EPS/NSAID males did not differ from controls. Light-dark box exploration in females was unaffected by EPS and NSAID (D–F). *, P < .05; **, P < .01. sec, seconds.
In females (Figure 1B), EPS decreased chemokine ligand 2 [CCL2; F(1, 26) = 4.41; P = .046], an effect that did not interact with maternal NSAID treatment [F(1, 26) = 0.002; P = .96]. Notably, proinflammatory cytokine expression was unaffected in female placentas (Supplemental Table 3).
Offspring behavioral assessment
Maternal antiinflammatory treatment rescues locomotor hyperactivity in prenatally stressed males
To test the hypothesis that inflammatory processes mediate behavioral dysregulation by EPS, offspring were examined for the potential rescue of disease-relevant endophenotypes. We identified significant EPS effects in the light-dark exploration test that interacted with NSAID treatment. In males (Figure 2, A–C), there was a significant effect of EPS [F(1, 30) = 6.94; P = .013] and a significant interaction between the effects of EPS and NSAID on time in light [F(1, 30) = 6.64; P = .015]. As shown (Figure 2A), prenatally stressed males spent more time in the light chamber relative to nonstressed controls (P = .0047). Maternal NSAID treatment rescued EPS effects on light time, as offspring of stressed dams treated with concomitant NSAID did not differ from controls (P = .093). To determine whether changes in activity accounted for the increased light time, distance traveled and number of chamber transitions were analyzed. There was a significant effect of EPS on distance traveled [F(1, 30) = 5.27; P = .023], and a significant interaction between the effects of EPS and NSAID on distance traveled [F(1, 30) = 7.96; P = .0084] and number of light-dark transitions [F(1, 30) = 6.75; P = .014]. Post hoc comparisons revealed that prenatally stressed males exhibited increased distance traveled (P = .0056) and transitions (P = .048) relative to nonstressed males (Figure 2, B and C). Maternal NSAID treatment rescued these EPS effects, because distance traveled (P = .17) and transitions (P = .1) of EPS/NSAID males did not differ from controls. There were no main effects of NSAID on light time [F(1, 30) = 0.82; P = .37], distance traveled [F(1, 30) = 0.023; P = .87], or transitions [F(1, 30) = 0.73; P = .4]. In females (Figure 2, D–F), neither EPS nor NSAID affected time spent in light [EPS: F(1, 31) = 0.54; P = .47; NSAID: F(1, 31) = 0.44; P = .51; EPS*NSAID: F(1, 31) = 0.0043; P = .95], distance traveled [EPS: F(1, 31) = 0.26; P = .61; NSAID: F(1, 31) = 0.0057; P = .94; EPS*NSAID: F(1, 31) = 1.38; P = .25], or light-dark transitions [EPS: F(1, 31) = 0.29; P = .59; NSAID: F(1, 31) = 1.42; P = .24; EPS*NSAID: F(1, 31) = 0.72; P = .40].
EPS does not alter sensorimotor gating or spatial learning
ASR and PPI.
Acoustic startle to 120 dB was unaffected in males (Supplemental Table 4). As shown (Figure 3, A and B), expected main effects of prepulse intensity on PPI were observed in males [F(2, 27) = 3.63; P < .0001] and females [F(2, 20) = 5.54; P < .0001]. In males, PPI was not significantly altered by EPS [F(1, 28) = 0.57; P = .46] or NSAID [F(1, 28) = 1.69; P = .20]. In females, there was a significant effect of NSAID on PPI [F(1, 21) = 4.45; P = .047] that did not interact with prepulse intensity [F(2, 20) = 0.063; P = .54] or EPS [F(1, 21) = 1.73; P = .20], and there was no effect of EPS [F(1, 21) = 2.1; P = .16].
Figure 3.

EPS did not alter sensorimotor gating or spatial learning. Neither EPS nor NSAID altered PPI of the acoustic startle response in male (A) or female (B) offspring. In the Barnes maze test of spatial learning, the latency to find the target improved for all groups. No effects of EPS or NSAIDs on target latency were observed (C and D). Data are mean ± SEM. CON, control; VEH, vehicle.
Barnes maze.
Spatial learning in the Barnes maze was unaffected by EPS (Figure 3, C and D). Although latency to find the target decreased for all groups [males: F(5, 19) = 2.38; P = .0002; females: F(5, 30) = 3,74; P < .0001], there were no effects of EPS [males: F(5, 19) = 0.34; P = .30; females: F(5, 30) = 0.022; P = .98] or NSAID [males: F(5, 19) = 0.11; P = .83; females F(5, 30) = 0.37; P = .078], and no interactions [males: F(5, 19) = 0.49; P = .15; females: F(5, 30) = 0.15; P = .46].
Mechanistic evaluation of male-specific hyperactivity
To identify neural circuits mediating male-specific hyperactivity in the light-dark exploration test, we examined gene expression of experimentally naïve male littermates in brain regions related to locomotor activity and novelty responsiveness. Specifically, we assessed dopamine receptor type 1 (D1), dopamine receptor type 2 (D2), and glutamic acid decarboxylase 1 (GAD1) expression in the PFC and NAc, circuits known to mediate locomotor activity and that are disrupted after prenatal stress and prenatal infection (57–68). In the NAc (Figure 4, A–C), there was no effect of EPS on D1 expression [F(1, 25) = 3.019; P = .095] and no interaction with NSAID treatment [F(1, 25) = 1.16; P = .29]. There was a significant effect of EPS on NAc D2 [F(1, 24) = 8.53; P = .0075], but no effect of NSAID [F(1, 24) = 2.044; P = .17] and no interaction [F(1, 24) = 0.12; P = .73]. NAc D2 was decreased in prenatally stressed males relative to nonstressed controls with (P = .0052) and without (P = .029) maternal NSAID treatment (Figure 4C). In the PFC (Figure 4, D–F), there was a main effect of EPS on D1 [F(1, 24) = 9.211; P = .0057], that did not interact with NSAID [F(1, 24) = 1.22, P = .28]. Post hoc comparisons revealed that PFC D1 was significantly increased only in EPS/NSAID males relative to nonstressed controls (P = .035). PFC D1 in EPS/vehicle offspring did not differ from controls (P = .53). There was no effect of EPS [F(1, 23) = 2.28; P = .14] or NSAID [F(1, 23) = 0.068; P = .80] on PFC D2, and no interaction [F(1, 23) = 0.084; P = .77]. No main effects of NSAID on dopamine receptor expression were observed. GAD1 was not affected by EPS [NAc: F(1, 25) = 0.071; P = .79; PFC: F(1, 24) = 0.12; P = .72] or NSAIDs [NAc: F(1, 25) = 1.098; P = .95; PFC: F(1, 24) = 0.22; P = .65].
Figure 4.

EPS programmed changes in dopamine receptor gene expression in males. Real-time PCR performed on NAc (A–C) and PFC (D–F) micropunches from experimentally naïve male offspring revealed significant main effects of EPS on D1 and D2 dopamine receptor expression. EPS significantly decreased D2 in the NAc (B). Post hoc comparisons revealed a significant reduction of NAc D2 in prenatally stressed males with and without NSAID treatment. In the PFC, a main effect was observed for EPS on D1 (D). Post hoc comparisons revealed significantly increased D1 only in offspring administered NSAID concomitant with EPS. Neither EPS nor NSAID treatment affected GAD1 expression (C and F). Bar graphs are mean expression relative control ± SEM. *, P < .05; and **, P < .01 vs control (CON)/vehicle (VEH).
Based on EPS effects on dopamine receptor expression, we quantified TH protein, the rate-limiting enzyme for dopamine synthesis, in the NAc and PFC by Western blot. EPS did not impact TH, because there was no difference between control/vehicle and EPS/vehicle [NAc: t(6) = 0.19, P = 85; PFC: t(6) = 0.16; P = .88] (Figure 5, A and C). In prenatally stressed males (Figure 5, B and D), NSAID treatment did not impact TH [NAc: t(6) = 1.14; P = .30; PFC: t(6) = 0.24; P = .82].
Figure 5.
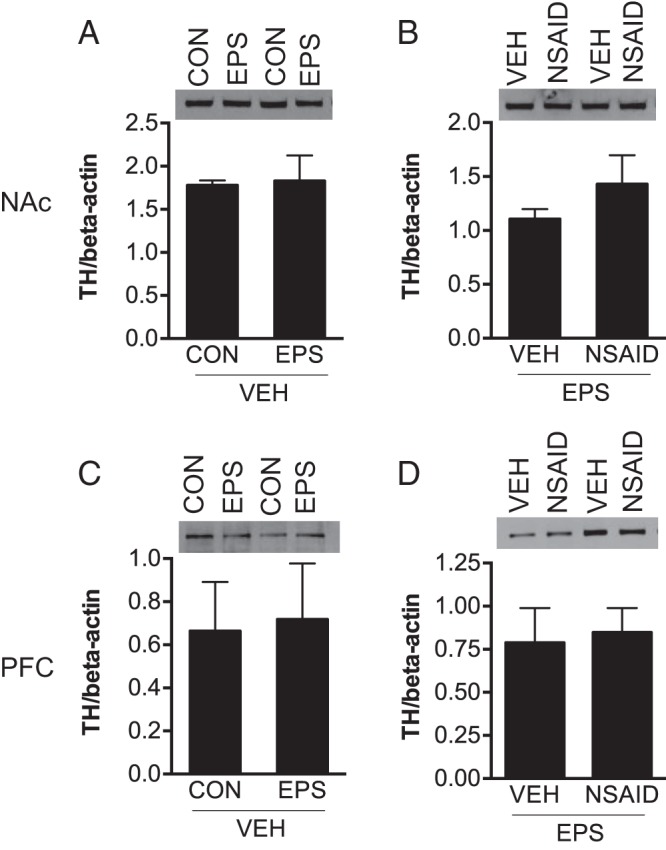
Mesocorticolimbic TH protein content was unaffected in males. In vehicle-treated offspring, prenatal stress did not alter TH content in the NAc (A) or PFC (C). Maternal NSAID treatment did not impact TH levels in prenatally stressed males (B and D). Representative Western blots for TH are shown in the upper panels. Protein levels were determined by densitometric analyses, normalized to β-actin, analyzed by Student's t test, and expressed as mean ± SEM in lower panels.
Discussion
Maternal stress during pregnancy is associated with increased risk of neurodevelopmental disorders, including schizophrenia, ASD, and ADHD (5–7, 69–72). Although substantial evidence from animal models demonstrates that prenatal stress elicits sex-dependent endophenotypes relevant to these disorders, the mechanisms by which these programming effects occur are not clear (25, 26, 64, 73, 74). Similar outcomes arise following maternal immune activation, suggesting that the detrimental processes of maternal stress and infection may converge upon common pathways (2, 12, 38–40, 42). Certainly, elevated immune system activation has been increasingly identified as a risk factor for neuropsychiatric disease (75–78). To investigate the hypothesis that stress-induced immune activation contributes, in part, to prenatal stress programming, pregnant dams were NSAID-treated during chronic stress exposure early in gestation (EPS), and offspring were examined for the potential rescue of disease-relevant endophenotypes.
Because the placenta is the sex-specific intermediary at the maternal-fetal interface that is responsive to a changing maternal milieu, we hypothesized that sex-dependent EPS effects may arise via actions on the developing placenta (13, 17). Prenatal stress produced significant changes in gene expression patterns, where immune-related genes including proinflammatory cytokines (IL-1β, IL-6) and chemokines (CCL5, chemokine ligand 10) were up-regulated by EPS specifically in males. In addition, EPS increased expression of genes that are positively regulated by inflammatory stimuli (IL-2 receptor α, prostaglandin-endoperoxide synthase 2, protein tyrosine phosphatase receptor C, P-selectin, and endothelin 1) in males. Such changes are indicative of a placental proinflammatory state in prenatally stressed males (79).
As predicted, maternal NSAID treatment during EPS ameliorated, in part, these placental changes, including normalizing IL-6 levels in male tissue. These data suggest that cyclooxygenase-dependent processes occurring during chronic stress exposure contribute to placental effects. These data are in agreement with previous studies indicating that early gestation psychogenic stress in mice triggers inflammatory responses in the decidua (56, 80). It is well established that maternal immune stimulation in rodents elevates proinflammatory cytokines, especially IL-6, in the placenta (81–83). Moreover, this cytokine elevation is necessary and sufficient to induce the long-term programming effects of maternal immune activation on sensorimotor gating, open field exploration, social preference, and frontal cortex gene expression in mice (84). Although maternal NSAID treatment appeared to reduce most EPS effects, a statistically significant rescue was limited to IL-6 and CCR7. Because these genes had the greatest magnitude of up-regulation by EPS, it is conceivable that statistical power to detect significant interactions upon more modestly affected genes was insufficient. Future studies may benefit from increased NSAID dose or greater specificity in immune system targeting.
Consistent with our hypothesis, the proinflammatory gene expression pattern occurred in prenatally stressed males, but not females. In addition, EPS up-regulated the proapoptotic factor, FASL, in males and females and decreased the monocyte chemoattractant, CCL2, in females. Within the placenta, FASL is expressed by maternal macrophages and placenta trophoblasts, where it mediates immune tolerance of the embryo, vascular remodeling, and trophoblast turnover (85, 86). Increased FASL has also been associated with trophoblast apoptosis following potent immune challenge in E13 placentas (87). Thus, identifying cell type specificity in future immunohisotologic studies may help to elucidate downstream effects of increased FASL.
Although treatment effects were detected for a limited number of targets analyzed (15%), the relatively specific up-regulation of proinflammatory genes is in agreement with our hypothesis that maternal stress induces placental inflammation. Placentas were collected on E12.5 (upon full differentiation), 5 days following stress cessation. We have previously described dynamic patterns of placental gene expression across gestation; therefore, it is likely that longitudinal analyses would reveal a spatiotemporal pattern of differentially expressed genes and effect magnitudes (18). Dramatic transcriptome changes would not be expected using this mild, time-limited stress paradigm, because it does not alter fetal survival (43). Furthermore, placental gene expression was assessed using preconfigured arrays for a broad set of immune-related genes, including a subset undetectable in E12.5 placentas (as described above) and genes for which the extraembryonic function is unclear. Notably, RNA was extracted from whole-placenta homogenates comprised of the labyrinth zone, junctional zone, spongiotrophoblast giant cells, as well as maternal and fetal vasculature. Because these cell layers vary in origin (fetal or maternal) and function, region-specific treatment effects are likely but were not examined in the present study. This methodological limitation may account for the modest relative expression changes observed.
To determine whether the normalization of placental proinflammatory gene expression in males correlated with a rescue of long-term behavioral outcomes after EPS, we examined adult offspring for evidence of behavioral stress dysregulation. In the light-dark exploration test, we observed a significant increase in overall activity of EPS males, with elevated distance traveled and light-dark transitions, suggestive of a hyperactive phenotype. As hypothesized, maternal NSAID treatment rescued this phenotype in males. Related to the increased activity of these animals, their time spent in the light compartment was also increased. Although the light-dark box task has been pharmacologically validated to assess anxiety-like behaviors, locomotor activity is a necessary control, and thus a clear interpretation of time spent in the light and dark compartments in our mice is not possible (88). However, robust effects on distance and transitions suggest that EPS influences spontaneous or stress-induced activity, in part, through activation of proinflammatory signaling, specifically in developing males. Notably, previous studies have shown that both prenatal stress and prenatal immune activation induce hyperactivity, although such locomotor effects are dependent on strain/species, gestational timing, and stressor type or duration (89–96). Of translational importance, in humans, maternal stress has been associated with behavioral hyperactivity and ADHD (6, 7).
To further assess potential phenotypic changes in these offspring relevant to neurodevelopmental disorders, mice were examined in tests for sensorimotor gating (PPI) and hippocampal-dependent spatial learning (Barnes maze). Acoustic startle appeared reduced by EPS and rescued by NSAID treatment; however, this effect did not reach significance. It is likely that a more challenging environment (eg, chronic stress) would reveal more maladaptive responses in these animals. In examination of cognitive performance in the Barnes maze, no effects of treatment were found. However, we noted that, similar to our previous reports, EPS males were slower to learn the task, and EPS females were faster than their same-sex controls (25). This outcome has been shown across many species in which maternal stress has an interaction with offspring sex for direction of impact on cognition (64, 97). These data are in agreement with our previous investigations demonstrating increased susceptibility of male offspring to EPS (25, 26). Although similar male vulnerability has also been reported in mouse, rat, and guinea pig models of maternal stress, sex-specific effects may depend on stressor types, gestational timing, and duration (28, 29, 98). In the present study, maternal stress elicited hyperlocomotion in male offspring without significantly impacting sensorimotor gating or spatial learning. Substantive evidence supports behavioral domain specificity of maternal stress effects, for which gestational stage and stressor(s) are key factors (99). Moreover, such specificity aids in identifying underlying mechanisms of EPS because neural circuits known to mediate hyperlocomotion are well delineated (57–59).
To identify neural circuits mediating locomotor hyperactivity after EPS, we examined mesocorticolimbic dopaminergic circuitry, because it is implicated in the pathophysiology of neurodevelopmental disorders, is perturbed in models of prenatal stress and infection, and is involved in spontaneous and novelty-stimulated locomotion (57–68). In males, we detected main effects of EPS on dopamine receptor gene expression in the NAc (decreased D2) and PFC (increased D1). Because EPS affected both D1 and D2 receptors, we hypothesized that these changes may be secondary to differences in upstream dopamine synthesis and availability. However, TH levels were unaffected by EPS in both PFC and NAc. Consistent with the present work, multiple lines of evidence demonstrate that the dopamine system is sensitive to perturbation by adverse prenatal events, including prenatal stress and infection, thereby supporting the link between these early insults and neurodevelopmental disorders in which dopamine dysregulation has been strongly implicated (60, 63, 68, 100).
In the present study, aspirin was selected as a tool by which to assess the involvement of proinflammatory processes in our mouse model of prenatal stress. We are not suggesting the use of NSAIDs during pregnancy to prevent neurodevelopmental abnormalities, because adverse effects on platelet function, heart and lung development, and miscarriage incidence have been reported, and the long-term effects on fetal development have not been fully investigated (101).
In summary, our findings demonstrate that maternal stress induces placental inflammation, specifically in males, and identify inflammation as a likely contributor to the sex-specific effects of maternal stress on offspring neurobehavioral outcome. Together, these data reveal a novel mechanism underlying the heightened susceptibility of males to early developmental insult and identify inflammation as one common factor mediating neuropsychiatric disease risk across diverse fetal antecedents known to challenge the immune system.
Acknowledgments
This work was supported by National Institutes of Health Grants MH087597 and MH091258.
Disclosure Summary: The authors of this manuscript have nothing to declare.
Footnotes
- ADHD
- attention deficit hyperactivity disorder
- ASD
- autism spectrum disorder
- CCL2
- chemokine ligand 2
- CCR7
- chemokine receptor 7
- D1 and D2
- dopamine receptors type 1 and type 2
- EPS
- early prenatal stress
- FASL
- Fas ligand
- GAD1
- glutamic acid decarboxylase 1
- Nac
- nucleus accumbens
- NSAID
- nonsteroidal antiinflammatory drug
- PFC
- prefrontal cortex
- PPI
- prepulse inhibition
- TH
- tyrosine hydroxylase.
References
- 1. Brown AS. Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev Neurobiol. 2012;72:1272–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Howerton CL, Bale TL. Prenatal programing: at the intersection of maternal stress and immune activation. Horm Behav. 2012;62:237–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Khashan AS, Abel KM, McNamee R, et al. Higher risk of offspring schizophrenia following antenatal maternal exposure to severe adverse life events. Arch Gen Psychiatry. 2008;65:146–152 [DOI] [PubMed] [Google Scholar]
- 4. Kinney DK, Munir KM, Crowley DJ, Miller AM. Prenatal stress and risk for autism. Neurosci Biobehav Rev. 2008;32:1519–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Koenig JI, Kirkpatrick B, Lee P. Glucocorticoid hormones and early brain development in schizophrenia. Neuropsychopharmacology. 2002;27:309–318 [DOI] [PubMed] [Google Scholar]
- 6. Li J, Olsen J, Vestergaard M, Obel C. Attention-deficit/hyperactivity disorder in the offspring following prenatal maternal bereavement: a nationwide follow-up study in Denmark. Eur Child Adolesc Psychiatry. 2010;19:747–753 [DOI] [PubMed] [Google Scholar]
- 7. Ronald A, Pennell CE, Whitehouse AJ. Prenatal maternal stress associated with ADHD and autistic traits in early childhood. Front Psychol. 2011;1:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Autism and Developmental Disabilities Monitoring Network Surveillance Year 2008 Principal Investigators. Centers for Disease Control and Prevention 2012 Prevalence of autism spectrum disorders–Autism and Developmental Disabilities Monitoring Network, 14 sites, United States, 2008. MMWR Surveill Summ. 2012;61:1–19 [PubMed] [Google Scholar]
- 9. Bloom B, Cohen RA, Freeman G. Summary health statistics for U.S. children: National Health Interview Survey, 2010. Vital Health Stat. 2011;10(250):1–80 [PubMed] [Google Scholar]
- 10. Fombonne E. Epidemiological surveys of autism and other pervasive developmental disorders: an update. J Autism Dev Disord. 2003;33:365–382 [DOI] [PubMed] [Google Scholar]
- 11. Froehlich TE, Lanphear BP, Epstein JN, Barbaresi WJ, Katusic SK, Kahn RS. Prevalence, recognition, and treatment of attention-deficit/hyperactivity disorder in a national sample of US children. Arch Pediatr Adolesc Med. 2007;161:857–864 [DOI] [PubMed] [Google Scholar]
- 12. Hsiao EY, Patterson PH. Placental regulation of maternal-fetal interactions and brain development. Dev Neurobiol. 2012;72:1317–1326 [DOI] [PubMed] [Google Scholar]
- 13. Jansson T, Powell TL. Role of the placenta in fetal programming: underlying mechanisms and potential interventional approaches. Clin Sci (Lond). 2007;113:1–13 [DOI] [PubMed] [Google Scholar]
- 14. Myatt L. Placental adaptive responses and fetal programming. J Physiol. 2006;572:25–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Clifton VL. Review: Sex and the human placenta: mediating differential strategies of fetal growth and survival. Placenta. 2010;31(Suppl):S33–S39 [DOI] [PubMed] [Google Scholar]
- 16. Cvitic S, Longtine MS, Hackl H, et al. The human placental sexome differs between trophoblast epithelium and villous vessel endothelium. PLoS One. 2013;8:e79233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gabory A, Roseboom TJ, Moore T, Moore LG, Junien C. Placental contribution to the origins of sexual dimorphism in health and diseases: sex chromosomes and epigenetics. Biol Sex Differ. 2013;4:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Howerton CL, Morgan CP, Fischer DB, Bale TL. O-GlcNAc transferase (OGT) as a placental biomarker of maternal stress and reprogramming of CNS gene transcription in development. Proc Natl Acad Sci USA. 2013;110:5169–5174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mao J, Zhang X, Sieli PT, Falduto MT, Torres KE, Rosenfeld CS. Contrasting effects of different maternal diets on sexually dimorphic gene expression in the murine placenta. Proc Natl Acad Sci USA. 2010;107:5557–5562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Muralimanoharan S, Maloyan A, Myatt L. Evidence of sexual dimorphism in the placental function with severe preeclampsia. Placenta. 2013;34:1183–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rossant J, Cross JC. Placental development: lessons from mouse mutants. Nat Rev Genet. 2001;2:538–548 [DOI] [PubMed] [Google Scholar]
- 22. Sood R, Zehnder JL, Druzin ML, Brown PO. Gene expression patterns in human placenta. Proc Natl Acad Sci USA. 2006;103:5478–5483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stark MJ, Wright IM, Clifton VL. Sex-specific alterations in placental 11β-hydroxysteroid dehydrogenase 2 activity and early postnatal clinical course following antenatal betamethasone. Am J Physiol Regul Integr Comp Physiol. 2009;297:R510–R514 [DOI] [PubMed] [Google Scholar]
- 24. Yeganegi M, Watson CS, Martins A, et al. , Effect of Lactobacillus rhamnosus GR-1 supernatant and fetal sex on lipopolysaccharide-induced cytokine and prostaglandin-regulating enzymes in human placental trophoblast cells: implications for treatment of bacterial vaginosis and prevention of preterm labor. Am J Obstet Gynecol. 2009; 200:532.e1–8. [DOI] [PubMed] [Google Scholar]
- 25. Mueller BR, Bale TL. Early prenatal stress impact on coping strategies and learning performance is sex dependent. Physiol Behav. 2007;91:55–65 [DOI] [PubMed] [Google Scholar]
- 26. Mueller BR, Bale TL. Sex-specific programming of offspring emotionality after stress early in pregnancy. J Neurosci. 2008;28:9055–9065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Morgan CP, Bale TL. Early prenatal stress epigenetically programs dysmasculinization in second-generation offspring via the paternal lineage. J Neurosci. 2011;31:11748–11755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kapoor A, Matthews SG. Short periods of prenatal stress affect growth, behaviour and hypothalamo-pituitary-adrenal axis activity in male guinea pig offspring. J Physiol. 2005;566:967–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kapoor A, Kostaki A, Janus C, Matthews SG. The effects of prenatal stress on learning in adult offspring is dependent on the timing of the stressor. Behav Brain Res. 2009;197:144–149 [DOI] [PubMed] [Google Scholar]
- 30. Arck PC. Stress and pregnancy loss: role of immune mediators, hormones and neurotransmitters. Am J Reprod Immunol. 2001;46:117–123 [DOI] [PubMed] [Google Scholar]
- 31. Blois SM, Joachim R, Kandil J, et al. Depletion of CD8+ cells abolishes the pregnancy protective effect of progesterone substitution with dydrogesterone in mice by altering the Th1/Th2 cytokine profile. J Immunol. 2004;172:5893–5899 [DOI] [PubMed] [Google Scholar]
- 32. Coussons-Read ME, Okun ML, Schmitt MP, Giese S. Prenatal stress alters cytokine levels in a manner that may endanger human pregnancy. Psychosom Med. 2005;67:625–631 [DOI] [PubMed] [Google Scholar]
- 33. Coussons-Read ME, Okun ML, Nettles CD. Psychosocial stress increases inflammatory markers and alters cytokine production across pregnancy. Brain Behav Immun. 2007;21:343–350 [DOI] [PubMed] [Google Scholar]
- 34. Butts CL, Sternberg EM. Neuroendocrine factors alter host defense by modulating immune function. Cell Immunol. 2008;252:7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Merlot E, Moze E, Dantzer R, Neveu PJ. Suppression of restraint-induced plasma cytokines in mice pretreated with LPS. Stress. 2002;5:131–135 [DOI] [PubMed] [Google Scholar]
- 36. Parker VJ, Douglas AJ. Stress in early pregnancy: maternal neuro-endocrine-immune responses and effects. J Reprod Immunol. 2010;85:86–92 [DOI] [PubMed] [Google Scholar]
- 37. Zhou D, Kusnecov AW, Shurin MR, DePaoli M, Rabin BS. Exposure to physical and psychological stressors elevates plasma interleukin 6: relationship to the activation of hypothalamic-pituitary-adrenal axis. Endocrinology. 1993;133:2523–2530 [DOI] [PubMed] [Google Scholar]
- 38. Boksa P. Effects of prenatal infection on brain development and behavior: a review of findings from animal models. Brain Behav Immun. 2010;24:881–897 [DOI] [PubMed] [Google Scholar]
- 39. Bronson SL, Richtand NM. Developmental consequences of prenatal exposure to maternal immune activation. In: Ritsner MS, ed. Handbook of Schizophrenia Spectrum Disorders. Vol 1 New York: Springer Science-Business Media B.V; 2011;263–286 [Google Scholar]
- 40. Holloway T, Moreno JL, Umali A, et al. Prenatal stress induces schizophrenia-like alterations of serotonin 2A and metabotropic glutamate 2 receptors in the adult offspring: role of maternal immune system. J Neurosci. 2013;33:1088–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Markham JA, Koenig JI. Prenatal stress: role in psychotic and depressive diseases. Psychopharmacology (Berl). 2011;214:89–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Meyer U, Feldon J, Fatemi SH. In-vivo rodent models for the experimental investigation of prenatal immune activation effects in neurodevelopmental brain disorders. Neurosci Biobehav Rev. 2009;33:1061–1079 [DOI] [PubMed] [Google Scholar]
- 43. Mueller BR, Bale TL. Impact of prenatal stress on long term body weight is dependent on timing and maternal sensitivity. Physiol Behav. 2006;88:605–614 [DOI] [PubMed] [Google Scholar]
- 44. Gerber AR, Bale TL. Antiinflammatory treatment ameliorates HPA stress axis dysfunction in a mouse model of stress sensitivity. Endocrinology. 2012;153:4830–4837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Amateau SK, McCarthy MM. Induction of PGE2 by estradiol mediates developmental masculinization of sex behavior. Nat Neurosci. 2004;7:643–650 [DOI] [PubMed] [Google Scholar]
- 46. Vane JR, Botting RM. The mechanism of action of aspirin. Thromb Res. 2003;110:255–258 [DOI] [PubMed] [Google Scholar]
- 47. Clapcote SJ, Roder JC. Simplex PCR assay for sex determination in mice. Biotechniques. 2005;38:702, 704,, 706 [DOI] [PubMed] [Google Scholar]
- 48. Ryan BC, Vandenbergh JG. Intrauterine position effects. Neurosci Biobehav Rev. 2002;26:665–678 [DOI] [PubMed] [Google Scholar]
- 49. O'Shaughnessy PJ, Baker PJ, Johnston H. The foetal Leydig cell– differentiation, function and regulation. Int J Androl. 2006;29:90–105; discussion 105–108 [DOI] [PubMed] [Google Scholar]
- 50. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108 [DOI] [PubMed] [Google Scholar]
- 51. McEuen JG, Semsar KA, Lim MA, Bale TL. Influence of sex and corticotropin-releasing factor pathways as determinants in serotonin sensitivity. Endocrinology. 2009;150:3709–3716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Geyer MA, Dulawa SC. Assessment of murine startle reactivity, prepulse inhibition, and habituation. Curr Protoc Neurosci 2003. Chapter 8:Unit 8.17 [DOI] [PubMed] [Google Scholar]
- 53. Rodgers AB, Morgan CP, Bronson SL, Revello S, Bale TL. Paternal stress exposure alters sperm microRNA content and reprograms offspring HPA stress axis regulation. J Neurosci. 2013;33:9003–9012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Paxinos G FK. The Mouse Brain In Stereotaxic Coordinates. San Diego, CA: Academic Press; 2001 [Google Scholar]
- 55. Teegarden SL, Bale TL. Decreases in dietary preference produce increased emotionality and risk for dietary relapse. Biol Psychiatry. 2007;61:1021–1029 [DOI] [PubMed] [Google Scholar]
- 56. Arck PC, Merali FS, Stanisz AM, et al. Stress-induced murine abortion associated with substance P-dependent alteration in cytokines in maternal uterine decidua. Biol Reprod. 1995;53:814–819 [DOI] [PubMed] [Google Scholar]
- 57. Canales JJ, Iversen SD. Psychomotor-activating effects mediated by dopamine D(2) and D(3) receptors in the nucleus accumbens. Pharmacol Biochem Behav. 2000;67:161–168 [DOI] [PubMed] [Google Scholar]
- 58. Hooks MS, Kalivas PW. The role of mesoaccumbens–pallidal circuitry in novelty-induced behavioral activation. Neuroscience. 1995;64:587–597 [DOI] [PubMed] [Google Scholar]
- 59. Gruen RJ, Wenberg K, Selim M, Friedhoff AJ, Bradberry CW. Novelty-associated locomotion: correlation with cortical and sub-cortical GABAA receptor binding. Eur J Pharmacol. 1996;309:115–120 [DOI] [PubMed] [Google Scholar]
- 60. Baier CJ, Katunar MR, Adrover E, Pallarés ME, Antonelli MC. Gestational restraint stress and the developing dopaminergic system: an overview. Neurotox Res. 2012;22:16–32 [DOI] [PubMed] [Google Scholar]
- 61. Markham JA, Mullins SE, Koenig JI. Peri-adolescent maturation of the prefrontal cortex is sex-specific and disrupted by prenatal stress J Comp Neurol. 2013;521:1828–1843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Muhammad A, Carroll C, Kolb B. Stress during development alters dendritic morphology in the nucleus accumbens and prefrontal cortex. Neuroscience. 2012;216:103–109 [DOI] [PubMed] [Google Scholar]
- 63. Son GH, Chung S, Geum D, et al. Hyperactivity and alteration of the midbrain dopaminergic system in maternally stressed male mice offspring. Biochem Biophys Res Commun. 2007;352:823–829 [DOI] [PubMed] [Google Scholar]
- 64. Weinstock M. Sex-dependent changes induced by prenatal stress in cortical and hippocampal morphology and behaviour in rats: an update. Stress. 2011;14:604–613 [DOI] [PubMed] [Google Scholar]
- 65. Baharnoori M, Bhardwaj SK, Srivastava LK. Effect of maternal lipopolysaccharide administration on the development of dopaminergic receptors and transporter in the rat offspring. PLoS One. 2013;8:e54439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bitanihirwe BK, Peleg-Raibstein D, Mouttet F, Feldon J, Meyer U. Late prenatal immune activation in mice leads to behavioral and neurochemical abnormalities relevant to the negative symptoms of schizophrenia. Neuropsychopharmacology. 2010;35:2462–2478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Piontkewitz Y, Arad M, Weiner I. Abnormal trajectories of neurodevelopment and behavior following in utero insult in the rat. Biol Psychiatry. 2011;70:842–851 [DOI] [PubMed] [Google Scholar]
- 68. Vuillermot S, Weber L, Feldon J, Meyer U. A longitudinal examination of the neurodevelopmental impact of prenatal immune activation in mice reveals primary defects in dopaminergic development relevant to schizophrenia. J Neurosci. 2010;30:1270–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Khashan AS, McNamee R, Henriksen TB, et al. Risk of affective disorders following prenatal exposure to severe life events: a Danish population-based cohort study. J Psychiatr Res. 2011;45:879–885 [DOI] [PubMed] [Google Scholar]
- 70. Goel N, Bale TL. Sex differences in the serotonergic influence on the hypothalamic-pituitary-adrenal stress axis. Endocrinology. 2010;151:1784–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. van Os J, Selten JP. Prenatal exposure to maternal stress and subsequent schizophrenia. The May 1940 invasion of The Netherlands. Br J Psychiatry. 1998;172:324–326 [DOI] [PubMed] [Google Scholar]
- 72. Watson JB, Mednick SA, Huttunen M, Wang X. Prenatal teratogens and the development of adult mental illness. Dev Psychopathol. 1999;11:457–466 [DOI] [PubMed] [Google Scholar]
- 73. Brummelte S, Lieblich SE, Galea LA. Gestational and postpartum corticosterone exposure to the dam affects behavioral and endocrine outcome of the offspring in a sexually-dimorphic manner. Neuropharmacology. 2012;62:406–418 [DOI] [PubMed] [Google Scholar]
- 74. Markham JA, Taylor AR, Taylor SB, Bell DB, Koenig JI. Characterization of the cognitive impairments induced by prenatal exposure to stress in the rat. Front Behav Neurosci. 2010;4:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bilbo SD, Schwarz JM. The immune system and developmental programming of brain and behavior. Front Neuroendocrinol. 2012;33:267–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Fineberg AM, Ellman LM. Inflammatory cytokines and neurological and neurocognitive alterations in the course of schizophrenia. Biol Psychiatry. 2013;73:951–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gesundheit B, Rosenzweig JP, Naor D, et al. Immunological and autoimmune considerations of autism spectrum disorders. J Autoimmun. 2013;44:1–7 [DOI] [PubMed] [Google Scholar]
- 78. Najjar S, Pearlman DM, Alper K, Najjar A, Devinsky O. Neuroinflammation and psychiatric illness. J Neuroinflammation. 2013;10:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hauguel-de Mouzon S, Guerre-Millo M. The placenta cytokine network and inflammatory signals. Placenta. 2006;27:794–798 [DOI] [PubMed] [Google Scholar]
- 80. Markert UR, Arck PC, McBey BA, et al. Stress triggered abortions are associated with alterations of granulated cells into the decidua. Am J Reprod Immunol. 1997;37:94–100 [DOI] [PubMed] [Google Scholar]
- 81. Gilmore JH, Jarskog LF, Vadlamudi S. Maternal poly I:C exposure during pregnancy regulates TNF α, BDNF, and NGF expression in neonatal brain and the maternal-fetal unit of the rat. J Neuroimmunol. 2005;159:106–112 [DOI] [PubMed] [Google Scholar]
- 82. Hsiao EY, Patterson PH. Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav Immun. 2011;25:604–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Urakubo A, Jarskog LF, Lieberman JA, Gilmore JH. Prenatal exposure to maternal infection alters cytokine expression in the placenta, amniotic fluid, and fetal brain. Schizophr Res. 2001;47:27–36 [DOI] [PubMed] [Google Scholar]
- 84. Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27:10695–10702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Guller S, LaChapelle L. The role of placental Fas ligand in maintaining immune privilege at maternal-fetal interfaces. Semin Reprod Endocrinol. 1999;17:39–44 [DOI] [PubMed] [Google Scholar]
- 86. Harris LK, Keogh RJ, Wareing M, et al. Invasive trophoblasts stimulate vascular smooth muscle cell apoptosis by a fas ligand-dependent mechanism. Am J Pathol. 2006;169:1863–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ejima K, Koji T, Tsuruta D, Nanri H, Kashimura M, Ikeda M. Induction of apoptosis in placentas of pregnant mice exposed to lipopolysaccharides: possible involvement of Fas/Fas ligand system. Biol Reprod. 2000;62:178–185 [DOI] [PubMed] [Google Scholar]
- 88. Crawley JN. Neuropharmacologic specificity of a simple animal model for the behavioral actions of benzodiazepines. Pharmacol Biochem Behav. 1981;15:695–699 [DOI] [PubMed] [Google Scholar]
- 89. Golan H, Stilman M, Lev V, Huleihel M. Normal aging of offspring mice of mothers with induced inflammation during pregnancy. Neuroscience. 2006;141:1909–1918 [DOI] [PubMed] [Google Scholar]
- 90. Huizink AC, Mulder EJ, Buitelaar JK. Prenatal stress and risk for psychopathology: specific effects or induction of general susceptibility? Psychol Bull. 2004;130:115–142 [DOI] [PubMed] [Google Scholar]
- 91. Martínez-Téllez RI, Hernández-Torres E, Gamboa C, Flores G. Prenatal stress alters spine density and dendritic length of nucleus accumbens and hippocampus neurons in rat offspring. Synapse. 2009;63:794–804 [DOI] [PubMed] [Google Scholar]
- 92. Matrisciano F, Tueting P, Dalal I, et al. Epigenetic modifications of GABAergic interneurons are associated with the schizophrenia-like phenotype induced by prenatal stress in mice. Neuropharmacology. 2013;68:184–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Owen D, Matthews SG. Repeated maternal glucocorticoid treatment affects activity and hippocampal NMDA receptor expression in juvenile guinea pigs. J Physiol. 2007;578:249–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Stöhr T, Schulte Wermeling D, Szuran T, et al. Differential effects of prenatal stress in two inbred strains of rats. Pharmacol Biochem Behav. 1998;59:799–805 [DOI] [PubMed] [Google Scholar]
- 95. Weller A, Glaubman H, Yehuda S, Caspy T, Ben-Uria Y. Acute and repeated gestational stress affect offspring learning and activity in rats. Physiol Behav. 1988;43:139–143 [DOI] [PubMed] [Google Scholar]
- 96. Wilson CA, Vazdarjanova A, Terry AV., Jr Exposure to variable prenatal stress in rats: effects on anxiety-related behaviors, innate and contextual fear, and fear extinction. Behav Brain Res. 2013;238:279–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Bowman RE, MacLusky NJ, Sarmiento Y, Frankfurt M, Gordon M, Luine VN. Sexually dimorphic effects of prenatal stress on cognition, hormonal responses, and central neurotransmitters. Endocrinology. 2004;145:3778–3787 [DOI] [PubMed] [Google Scholar]
- 98. Weinstock M. Gender differences in the effects of prenatal stress on brain development and behaviour. Neurochem Res. 2007;32:1730–1740 [DOI] [PubMed] [Google Scholar]
- 99. Weinstock M. The long-term behavioural consequences of prenatal stress. Neurosci Biobehav Rev. 2008;32:1073–1086 [DOI] [PubMed] [Google Scholar]
- 100. Palmer AA, Brown AS, Keegan D, et al. Prenatal protein deprivation alters dopamine-mediated behaviors and dopaminergic and glutamatergic receptor binding. Brain Res. 2008;1237:62–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. James AH, Brancazio LR, Price T. Aspirin and reproductive outcomes. Obstet Gynecol Surv. 2008;63:49–57 [DOI] [PubMed] [Google Scholar]