

Abstract
Germline haploinsufficiency of human or mouse Sim1 is associated with hyperphagic obesity. Sim1 encodes a transcription factor required for proper formation of the paraventricular (PVN), supraoptic, and anterior periventricular hypothalamic nuclei. Sim1 expression persists in these neurons in adult mice, raising the question of whether it plays a physiologic role in regulation of energy balance. We previously showed that Sim1 heterozygous mice had normal numbers of PVN neurons that were hyporesponsive to melanocortin 4 receptor agonism and showed reduced oxytocin expression. Furthermore, conditional postnatal neuronal inactivation of Sim1 also caused hyperphagic obesity and decreased hypothalamic oxytocin expression. PVN projections to the hindbrain, where oxytocin is thought to act to modulate satiety, were anatomically intact in both Sim1 heterozygous and conditional knockout mice. These experiments provided evidence that Sim1 functions in energy balance apart from its role in hypothalamic development but did not rule out effects of Sim1 deficiency on postnatal hypothalamic maturation. To address this possibility, we used a tamoxifen-inducible, neural-specific Cre transgene to conditionally inactivate Sim1 in adult mice with mature hypothalamic circuitry. Induced Sim1 inactivation caused increased food and water intake and decreased expression of PVN neuropeptides, especially oxytocin and vasopressin, with no change in energy expenditure. Sim1 expression was not required for survival of PVN neurons. The results corroborate previous evidence that Sim1 acts physiologically as well as developmentally to regulate body weight. Inducible knockout mice provide a system for studying Sim1's physiologic function in energy balance and identifying its relevant transcriptional targets in the hypothalamus.
The mouse Single-minded homolog 1 (Sim1) gene encodes a member of the bHLH-PAS family of nuclear transcription factors and is expressed during embryogenesis in developing somites, nephrogenic cord, foregut, and forebrain (1, 2). Sim1 expression is required for terminal migration and differentiation of the neurons of the paraventricular (PVN), supraoptic (SON), and anterior periventricular (aPV) nuclei of the hypothalamus, which produce the neuroendocrine peptides oxytocin, arginine vasopressin, corticotropin-releasing hormone, thyrotropin-releasing hormone, and somatostatin. Sim1 continues to be expressed in neurons of these nuclei, as well as in the amygdala, in adult mice. Its transcriptional target genes are not known. We hypothesized a role for Sim1 in energy homeostasis after discovering that cytogenetic abnormalities disrupting one allele of the orthologous human SIM1 gene were associated with early-onset, severe obesity (3). This hypothesis was confirmed by the phenotype of Sim1 heterozygous knockout mice, which includes hyperphagic obesity exacerbated by high fat (HF) diet (4, 5), as well as the finding of rare human SIM1 loss-of-function missense mutations associated with hyperphagic obesity (6, 7).
The mechanism of hyperphagia associated with Sim1 deficiency remains to be clarified. Sim1 homozygous knockout mice fail to form the PVN, SON, and aPV and die perinatally (8). Michaud et al (5) proposed that hyperphagia is due to a modest decrease in the number of Nissl-stained PVN cells in Sim1 heterozygotes. By contrast, we found no decrease in Sim1-expressing PVN neurons in Sim1 heterozygous mice carrying a Sim1-GFP reporter transgene. Instead, we observed defective activation of these neurons in response to melanocortin 4 receptor (Mc4r) agonism (9). Furthermore, transgenic overexpression of Sim1 conferred resistance to obesity induced either by HF diet or by the Ay mutation that abrogates CNS Mc3r and Mc4r signaling (10). Moreover, Yang et al (11) showed that viral-mediated increase or decrease in Sim1 expression in the hypothalamus of adult mice acutely reduced or increased food intake, respectively. Finally, we used a neural-specific Cre transgene whose expression begins postnatally to conditionally inactivate Sim1 after birth, and showed that these mice develop hyperphagic obesity without loss of PVN neurons or gross alteration of hindbrain projections that are thought to mediate satiety (12).
Whereas the preponderance of evidence thus favors physiologic rather than developmental perturbation as the cause of hyperphagic obesity associated with Sim1 deficiency, we wished to exclude the possibility that reduced Sim1 expression alters late hypothalamic development/maturation occurring during early postnatal life. In addition, we sought to develop a system for studying Sim1 deficiency in vivo without confounding effects of developmental compensation. We therefore used a tamoxifen-inducible, neural-specific Cre transgene (CaMK-CreERT2) to conditionally inactivate Sim1 in hypothalamic neurons of adult mice and measured the resulting effects on energy homeostasis.
Materials and Methods
Animal care and use
All experimental protocols were approved by the UT Southwestern Institutional Animal Care and Use Committee. Animals were housed in a specific-pathogen-free vivarium under a standard 12-hour light,12-hour dark cycle with ad libitum access to water and food except for metabolic chamber studies. Mice were fed standard low-fat, low phytoestrogen chow diet (Teklad Global Diet 2016) or high fat (HF) diet (Research Diets D12492; 5.24 kcal/g, with 26.3% (by weight) available carbohydrate, 34.9% available fat, and 26.2% available protein). Mice carrying floxed Sim1 (Sim1fl, JAX Stock #7570) (12), CaMK-CreERT2 (European Mutant Mouse Archive ID 2125) (13), or Rosa26-eYFP (JAX Stock #6148) alleles (14) were previously described. Sim1fl mice were maintained in the homozygous state on a mixed 129 × 1/SvJ;C57Bl/6 genetic background. CaMK-CreERT2 mice were of a mixed FVB/N;C57Bl/6 background. Littermate controls were used for all experiments. Sim1 and Rosa26-eYFP alleles were genotyped by PCR using tail biopsy DNA as previously described (12). The CaMK-CreERT2 allele was genotyped by PCR using primers A (CAMK-1), 5′-GGTTCTCCGTTTGCACTCAGGA-3′, B (CAMK-17), 5′-GCTTGCAGGTACAGGAGGTAGT-3′ and C (CAMK-5), 5′-CTGCATGCACGGGACAGCTCT-3′ (provided by Stefan Berger, German Cancer Research Center).
Drug administration
Tamoxifen was dissolved in sunflower seed oil (Sigma) to a final concentration of 30 mg/ml. Eight-week-old mice were injected ip with 180 mg/kg tamoxifen daily for 2–5 days and allowed to recover for 1–2 weeks. Initial studies were done with 5 daily injections, as described (13). To reduce associated morbidity and mortality, later studies used fewer injections after a time course showed that as few as 2 daily injections yielded maximal Sim1 inactivation (data not shown).
Metabolic cages
Oxygen consumption, carbon dioxide production, and water intake were measured in metabolic cages (TSE Systems) after 5 days of habituation. Heat production (H2[kcal/h/kg] = (3.815 + 1.232 × RER) × VO2[ml/h/kg]/1000) was calculated by TSE Calo software using the Weir equation (15).
Statistical analysis
Data were analyzed using Microsoft Excel and GraphPad Prism and are shown as mean ± SEM. Statistical comparisons were made using unpaired two-sample Student's t test with Welch's correction if variance was unequal (F test) or 2-way ANOVA. Growth curves were compared using 2-way repeated measures ANOVA. One control female mouse in the growth and feeding studies on chow diet had grossly aberrant weight compared with all other mice and for that reason was excluded. Excluding this outlier decreased the reported statistical significance of the ANOVA but did not change the conclusions. All tests were two-sided and P ≤ .05 was considered significant.
Histology
Procedures for hypothalamic sectioning (35 μM) and Nissl staining and antibodies for eYFP immunostaining were described previously (12), except that eYFP was detected colorimetrically using DAB. Growth and feeding studies, hypothalamic tissue dissection, RNA isolation, and real-time PCR measurements of hypothalamic Sim1, Mc4r, and neuropeptide gene expression were performed as described previously (12).
Results
Induced Sim1 heterozygotes
The CaMKCreERT2 transgene expresses a Cre recombinase/estrogen receptor fusion protein in forebrain neurons that translocates from the cytoplasm to the nucleus upon tamoxifen binding. It was previously shown to efficiently recombine a floxed glucocorticoid receptor allele in the PVN (16). We verified that the CaMK-CreERT2 transgene robustly recombined a floxed Rosa26-eYFP reporter transgene in sites of hypothalamic Sim1 expression, eg, PVN and SON, after tamoxifen treatment (14) (Figure 1).
Figure 1.
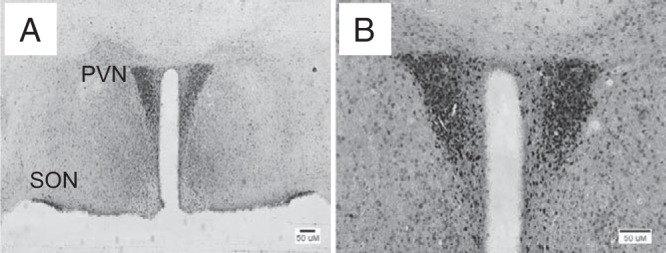
CaMK-CreERT2 recombines loxP sequences in hypothalamic Sim1 neurons. CamK-CreERT2 transgenic mice were crossed with mice expressing eYFP under the control of a Cre recombinase-activatable Rosa26 promoter, and doubly transgenic progeny were treated with tamoxifen. Animals were killed and their brains sectioned and immunostained for eYFP using colorimetric detection. A, Low magnification image showing staining of the paraventricular (PVN) and supraoptic (SON) hypothalamic nuclei, indicating Cre expression in Sim1 neurons. B, Higher magnification image of another section showing PVN staining.
We next crossed the CaMKCreERT2 transgenic mice to mice that were heterozygous for a floxed Sim1 allele and analyzed the progeny. Mice carrying both the Cre transgene and the single copy of the floxed Sim1 allele, designated iHet, were predicted to become heterozygous for Sim1 in Cre-expressing cells upon tamoxifen treatment. Neither the floxed Sim1 allele (12) nor this Cre transgene alone (data not shown) affected body weight; therefore we used littermates lacking the Cre transgene, with or without the floxed Sim1 allele, as controls. Eight-week-old control or iHet mice were treated with tamoxifen and allowed to recover for 2 weeks, then killed and their hypothalamic tissues collected. In the absence of an antibody that reliably detects Sim1 protein, we assessed Sim1 expression at the level of RNA, as in previous studies (4, 5, 9–12). Quantitative real-time PCR showed that neither tamoxifen treatment nor the Cre transgene by itself affected Sim1 expression, but Sim1 transcript abundance in hypothalamus was decreased by approximately 50% in tamoxifen-treated iHet males (Figure 2A) or females (Figure 2B) compared with controls, consistent with efficient inactivation of the floxed Sim1 allele in Sim1-expressing neurons. The fact that the Cre transgene did not affect body weight or Sim1 expression in the absence of tamoxifen indicated that recombinase activity was not significantly “leaky”.
Figure 2.

Induced hypothalamic Sim1 deficiency in adult mice increases weight gain on chow diet. A and B, Inducible Sim1 heterozygouss (CamK-CreERT2, Sim1fl/+) male (A) or female (B) mice or same-sex littermate controls (CaMKCreERT2 transgene negative and/or Sim1+/+) treated with tamoxifen or vehicle at age 8 weeks and killed ≥ 10 weeks later. *, P < .05; ***, P < .001; ****, P < .0001; 2-way ANOVA, Tukey's test. C and D, Growth curves of iKO, iHet or control male (C) or female (D) mice, weaned and maintained on chow diet. **, P < .01; ****, P < .0001 versus control, repeated measures 2-way ANOVA. E and F, Weekly food intake of iKO, iHet, or control male (E) or female (F) mice, weaned and maintained on chow diet. **, P < .01; ****, P < .0001 versus control, repeated measures 2-way ANOVA.
We next examined the effect of tamoxifen treatment on weight curves of iHet and iKO mice fed a low fat chow diet. We crossed mice that were heterozygous for the floxed Sim1 allele with mice that were doubly heterozygous for the floxed Sim1 allele and the Cre transgene in order to obtain progeny that were either wild-type, heterozygous, or homozygous for the floxed Sim1 allele, approximately half of which carried the Cre transgene. Progeny lacking either the Cre transgene or the floxed Sim1 allele were used as controls. Animals were weaned and maintained on chow diet, treated with tamoxifen at age eight weeks, and then individually housed for measurement of weekly food intake and body weight. Over the next seven weeks, iKO males (Figure 2C) and iHet and iKO females (Figure 2D) showed modest but statistically significant increases in the rate of body weight gain compared with controls; iHet males (Figure 2C) did not show an increase, but the number of iHet male progeny from the cross was very low for unknown reasons. iKO mice of either sex showed increased food intake compared with controls (Figure 2, E and F).
Because previous studies showed that hyperphagic obesity associated with Sim1 inactivation was exacerbated by HF diet (4, 12), we tested whether tamoxifen-treated iHet versus control mice weaned and maintained on HF diet showed a more robust phenotype. For these experiments we crossed mice heterozygous for the Cre transgene with mice homozygous for the floxed Sim1 allele in order to generate iHets and controls in equal proportion. Control and iHet mice gained weight on HF diet at the same rate prior to tamoxifen treatment, but over the ensuing 8 weeks after treatment the iHet males (Figure 3A) and females (Figure 3B) gained significantly more weight than controls.
Figure 3.

Induced hypothalamic Sim1 deficiency in adult mice increases weight gain and linear growth on high fat diet. A and B, Growth curves of tmx-iHet or control male (G) or female (H) mice, weaned and maintained on high fat diet. **, P < .01; ****, P < .0001; repeated measures 2-way ANOVA. C and D, Weight curves of male (C) or female (D) iKO or control mice weaned and maintained on high-fat diet and treated with tamoxifen (tmx) at eight weeks of age. **, P < .01; ****, P < .0001; repeated measures 2-way ANOVA. E, Nose-to-anus length of 18-week-old iKO or control mice, weaned and maintained on a high-fat diet and treated with tamoxifen at age 8 weeks. **, P < .01; ***, P < .001; t test. F, Hypothalamic Sim1 expression in control or iKO mice treated with tamoxifen at eight weeks of age and killed at age 18 weeks. ****, P < .0001, t test.
We previously showed that the hyperphagic obesity phenotype of postnatal conditional Sim1 knockouts was more robust for homozygotes than heterozygotes (12). We therefore crossed mice that were homozygous for the floxed Sim1 allele to mice doubly heterozygous for the floxed Sim1 allele and the CaMK-CreERT2 transgene and studied progeny that were homozygous for the floxed Sim1 allele and heterozygous for the inducible Cre transgene, designated iKO. Controls from the same cross were homozygous for the floxed Sim1 allele but lacked the Cre transgene. Male and female iKO or control mice were treated with tamoxifen and allowed to recover as before. Their weights were indistinguishable prior to tamoxifen treatment at eight weeks of age (Figure 3, C and D). Within one to two weeks after tamoxifen administration, both male (Figure 3C) and female (Figure 3D) iKO mice gained more weight than same-sex littermate controls. The magnitude of the difference in weight gain was greater in females. The onset of increased weight gain after tamoxifen treatment appeared to be somewhat sooner in iKO versus iHet mice, although the different breeding strategies used for the HF diet experiments did not allow us to directly compare heterozygotes and homozygotes from a single cross. Tamoxifen-treated iKO mice also showed increased linear growth (Figure 3E), as previously observed for germline Sim1 heterozygotes (4) and for conditional postnatal CNS Sim1 homozygotes (12). Quantitative real-time PCR confirmed that Sim1 transcripts were ablated in tamoxifen-treated iKO mice (Figure 3F).
Energy homeostasis
We measured food intake and energy expenditure of tamoxifen-treated iKO mice in order to determine the cause of their increased weight gain. A cohort of iKO and control mice maintained on chow diet was treated with tamoxifen at eight weeks of age as previously described. After a week of recovery, daily food intake was measured for 7 days. The mice were then challenged with HF diet and their daily food consumption measured for seven days. Mice were then returned to chow diet for three weeks, and the HF diet challenge was then repeated. Control and iKO mice weighed the same at the onset of the initial HF diet challenge. However, both male and female iKO mice gained more weight than same-sex controls during this challenge, and the iKO mice remained heavier thereafter (Figure 4, A and B). There was no significant difference in baseline consumption of chow diet (Figure 4, C and D). However, iKO males ate significantly more than controls during both HF diet challenges, and iKO females ate significantly more than controls during the second challenge (Figure 4, C and D). Because iKO mice weighed more than controls after the first HF diet challenge, we also analyzed food intake as the percent increase in mean daily caloric consumption on HF versus chow diet for each animal. During the initial HF diet challenge, iKO males showed a trend toward a greater increase in caloric consumption than controls; this difference reached statistical significance in females (Figure 4E). Both male and female iKO mice showed a greater increase in consumption than controls during the second HF diet challenge (Figure 4E). Body composition studies at the end of the study showed that iKO mice had greater lean and fat mass than controls (Figure 4F), as previously observed for germline Sim1 heterozygotes (4) and conditional postnatal CNS Sim1 homozygotes (12).
Figure 4.

Induced hypothalamic Sim1 homozygosity in adult mice causes hyperphagia and increased lean and fat mass on a high fat diet. Mice were weaned and maintained on chow diet, treated with tamoxifen (tmx) at age eight weeks, and challenged with high fat (HF) diet for one week at ages 10 and 14 weeks. A and B, Weight curves of male (A) or female (B) mice. C and D, Mean daily consumption of chow (age nine weeks) or HF diet by male (C) or female (D) mice. E, Percent increase in caloric intake on HF versus chow diet. F, Lean and fat mass of mice shown in (A) and (B) at age 16 weeks. Numbers of mice are the same for C–F as in A and B. *, P < .05; **, P < .01, t test.
Another cohort of iKO and control mice was bred and maintained on chow diet, treated with tamoxifen at 8 weeks of age, allowed to recover for 2 weeks, then placed in metabolic cages. The iKO mice showed no significant differences from controls in rates of oxygen consumption or carbon dioxide production (Figure 5, A and B), respiratory quotient or rate of heat production (Figure 5, C and D), or measures of locomotor activity (not shown). Another cohort of tamoxifen-treated male iKO and control mice showed no difference in HF diet-induced thermogenesis (Figure 6, A and B). In the metabolic cages, iKO mice did not consume more chow diet than controls, but they did drink significantly more water (Figure 6C). HF diet consumption could not be accurately measured in metabolic cages for technical reasons related to the soft consistency of the diet formulation.
Figure 5.

Energy expenditure of induced hypothalamic Sim1 homozygous mice was normal. Control or inducible Sim1 homozygotes (iKO) were treated with tamoxifen, allowed to recover two weeks, and placed in metabolic cages. Shown are rates of (A and B) oxygen consumption and carbon dioxide production or (C and D) respiratory quotient and rate of heat production during light or dark phase. There were no statistically significant differences between control and iKO mice for any parameter measured. (A and C) males; (B and D) females.
Figure 6.

Induced hypothalamic Sim1 homozygous mice in metabolic cages show normal diet-induced thermogenesis and normal consumption of chow diet but increased water intake. A, and B, Hourly rate of oxygen consumption and response to shift from chow to high fat diet (A) was measured in a cohort of 10-week-old male control or iKO mice treated with tamoxifen at age 8 weeks. B, Increase in overall mean hourly oxygen consumption in response to shift to high fat diet did not differ between control and iKO groups. C, In metabolic cages, iKO mice did not consume more chow but drank significantly more water than controls. *, P < .05; **, P < .01; ***, P < .001, n.s. indicates not significant.
Hypothalamic neuropeptide expression and histology
We next measured the expression of PVN neuropeptide and melanocortin 4 receptor (Mc4r) in female iKO or control mice, weaned and maintained on chow diet, 2 weeks after tamoxifen treatment at age 8 weeks. Steady-state mRNA levels of oxytocin, vasopressin, corticotropin-releasing hormone, and thyrotropin-releasing hormone were significantly decreased in iKO mice versus controls (Figure 7). Oxytocin and vasopressin transcripts showed the largest reductions, as previously seen for Sim1 heterozygotes (17) and conditional postnatal CNS Sim1 homozygotes (12).
Figure 7.
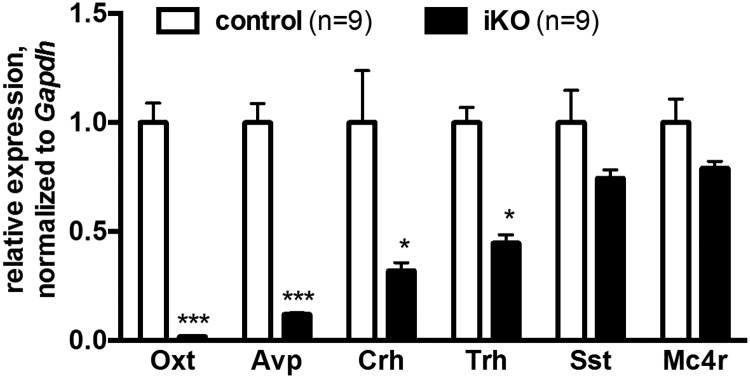
Expression of multiple hypothalamic neuroendocrine genes is decreased in induced Sim1 homozygotes. Hypothalamic expression of oxytocin (Oxt), vasopressin (Avp), corticotropin-releasing hormone (Crh), thyrotropin-releasing hormone (Trh), somatostatin (Sst), and melanocortin 4 receptor (Mc4r) genes measured by quantitative RT-PCR of 10-week old control or iKO female mice weaned and maintained on chow diet and treated with tamoxifen at age eight weeks. *, P < .05; ***, P < .001, t test.
To determine whether the observed changes in HF food intake, body weight and PVN neuropeptide expression in tamoxifen-treated iKO mice were due to gross loss of PVN neurons, we examined control (Figure 8A) and iKO animals at 2 weeks (Figure 8B) or 6 months (Figure 8C) following tamoxifen administration. Nissl staining showed no gross PVN histologic changes after Sim1 inactivation, either at the level of midnuclei shown or at other rostral and caudal levels (not shown). Because these findings were consistent with previous findings in postnatal conditional Sim1 homozygotes in which Sim1 was ablated even earlier in life (12), we did not pursue detailed stereologic investigations.
Figure 8.

PVN histology is grossly intact acutely and chronically after abrogation of Sim1 expression. Representative Nissl-stained sections through the PVN (37) of control (A) or iKO mice (B and C) 2 weeks (A and B) or 6 months (C) after tamoxifen treatment. Mice were weaned and maintained on chow diet.
Discussion
SIM1 is one of only a handful of genes associated with human monogenic obesity. Initially implicated on the basis of cytogenetic abnormalities (3), its role in human energy balance was corroborated by the identification of loss-of-function point mutations associated with early-onset obesity (6, 7). Prior studies indicate that mouse Sim1 acts in the hypothalamic melanocortin signaling pathway (9, 10) to principally regulate food intake rather than energy expenditure (4, 5, 18). SIM1 thus provides a potential pharmacologic target for treatment of obesity that may not have undesired effects associated with melanocortin agonists, such as increased blood pressure (19–22). However, a key issue is whether Sim1, which was originally identified as a transcription factor gene essential for proper formation of the hypothalamus, acts solely during embryonic and/or early postnatal development and maturation of hypothalamic neurons and circuits, or whether it also acts physiologically as a regulator of energy homeostasis in adults. If the hyperphagic phenotype of Sim1 haploinsufficiency is due to a fixed developmental defect, as has been proposed (5), then modulating the activity of Sim1 or its transcriptional targets would not be a useful strategy for altering food intake in adults. By contrast, if Sim1 acts physiologically, it then becomes a potential target for modulating food intake.
We previously showed that germline Sim1 haploinsufficiency does not reduce PVN cellularity, as measured either using a Sim1-eGFP reporter (9) or by stereologic counting of Nissl-stained cells (12). We obtained further evidence for a physiologic role of Sim1 in regulating food intake by showing that chronic Sim1 transgenic overexpression confers resistance to obesity induced by either HF diet or defective melanocortin signaling. Furthermore, Yang et al (11) showed that adenoviral knockdown or overexpression of Sim1 in the hypothalamus of adult mice caused acute increase or decrease in food intake, respectively. Last, using a CamK-Cre transgene whose expression begins during late prenatal/early postnatal life, we showed that conditional CNS Sim1 inactivation recapitulated the hyperphagic obesity phenotype of germline Sim1 heterozygosity (12) in a gene dosage-dependent manner with no obvious effect on PVN cellularity or hindbrain projections, consistent with a physiologic effect of Sim1 on food intake. However, Sim1 inactivation in these experiments was concomitant with postnatal hypothalamic development, and we, therefore, could not rule out an effect of Sim1 deficiency on functional maturation of hypothalamic circuits.
In the present study, we extended our previous findings, taking advantage of a tamoxifen-inducible CaMK-Cre transgene to conditionally inactivate Sim1 in adult mice with fully formed, mature hypothalamic circuits. Inactivation of Sim1 in adult mice resulted in increased weight gain, particularly in homozygotes on HF diet, with no measurable effect on energy expenditure. PVN neuropeptides in the induced homozygotes showed changes similar to those previously observed in germline heterozygous (17) and conditional postnatal Sim1 homozygous (12) knockout mice. Sim1 expression was not required for gross survival of PVN neurons. Taken together, these results strongly corroborate previous evidence for a physiologic function for Sim1 in regulating food intake.
As with Sim1 heterozygotes and conditional postnatal knockout mice (4, 12), the magnitude of the effect of induced Sim1 knockout on body weight was greater in females than in males. The reason for this sex difference is unclear, as Sim1 inactivation was similarly efficient in males and females. Sex differences are seen in numerous models of obesity and could be due to hormonal (23) and/or genetic (24) effects.
Our results with Sim1 inactivation contrast with those reported by Xi et al (25), who showed that postnatal ablation of Sim1 neurons causes both hyperphagia and reduced energy expenditure. Taken together with the present study, these data suggest that Sim1 neurons are important in both food intake and energy expenditure, consistent with neuroanatomic studies showing projections to other regions of the hypothalamus, hindbrain satiety centers, and the sympathetic nervous system (26–30). However, once Sim1 neurons have properly differentiated and matured, continued Sim1 expression appears to be important only for regulation of food intake, not energy expenditure. The pathway of non-Sim1 dependent regulation of energy expenditure is unknown but appears to involve Mc4r signaling outside the PVN and/or amygdala (18).
The magnitude of hyperphagia and rate of weight gain following tamoxifen treatment in induced Sim1 deficient mice appeared to be somewhat less than that seen in previous studies of germline or conditional postnatal Sim1 deficiency. One explanation may be genetic background differences due to the transgenes used. In addition, Sim1 deficiency during early postnatal development could increase vulnerability to secondary changes from early hyperphagia that could exacerbate dysregulation of energy homeostasis, such as alterations in hypothalamic leptin circuits (31, 32). Increased water intake of induced Sim1 homozygotes may interfere with their food consumption. Indeed, poor feeding is a frequent symptom of pediatric diabetes insipidus (33). Finally, the feeding behavior of mice with impaired melanocortin signaling has been shown to be exquisitely sensitive to environmental stresses such as housing conditions (13); some of our previous studies were performed in another vivarium with different cages. It is even possible that environmental factors such as gut microbiota could vary between vivaria and contribute to the differences in the severity of hyperphagic obesity in the different studies (34). The reason that female iKO mice on chow diet were hyperphagic but did not gain more weight than controls is unknown; metabolic cage studies did not show significant differences in energy expenditure.
The relative importance of Sim1 during development versus adulthood is difficult to determine. Data from the Allen Brain Atlas (35) indicate that Sim1 is expressed at lower levels in hypothalamic neurons during adulthood than it is up through postnatal day 14, suggesting that Sim1 expression may play a relatively minor role in adult neurons. Additionally, Sim1 protein levels throughout development have never been quantified due to the lack of a good antibody. However, addressing this issue definitively would require conditionally inactivating Sim1 at varying times pre- and postnatally. Such experiments are feasible using a Cre transgene that is regulated by tetracycline rather than tamoxifen.
Increased water intake in induced Sim1 homozygotes is likely central diabetes insipidus due to the markedly decreased vasopressin expression. Although Sim1 itself is expressed in kidney, the neural-specific transgene we used did not affect this expression (data not shown). Furthermore, Duplan et al (36) reported increased susceptibility of Sim1 germline heterozygous mice to chronic dehydration. Additional studies are in progress to characterize the pathophysiology of increased water intake in animals with reduced Sim1 dosage.
Our results show that conditional inactivation of Sim1 in the central nervous system of adult mice with fully formed, mature hypothalamic circuits rapidly causes increased weight gain on a HF diet. These findings corroborate previous evidence that Sim1 functions not only in hypothalamic development but also in physiologic regulation of energy balance. In particular, Sim1 acts in a dosage-dependent manner in the PVN or possibly amygdala to regulate food intake. Understanding the molecular function of Sim1 in physiologic regulation of feeding will require the identification of its upstream regulators and downstream transcriptional targets in hypothalamic neurons, which has been confounded by the lack of suitable cell culture models for Sim1-expressing hypothalamic neurons. The availability of hypothalamic cells and tissues with or without Sim1 expression affords an in vivo system for such studies.
Acknowledgments
We thank Viren Amin for his skilled mouse husbandry and technical assistance and the University of Texas Southwestern Mouse Metabolism Core, Joel Elmquist, and members of the University of Texas Southwestern Center for Hypothalamic Studies for their help with various aspects of these studies.
Present Affiliation: Department of Reproductive Medicine, Division of Reproductive Endocrinology and Infertility, University of California San Diego (KPT).
This work was supported by National Institutes of Health grants R01DK079986, RL1DK081185, and UL1DE019584.
Disclosure Summary: The authors declare that they have no conflicts of interest.
Footnotes
- aPV
- anterior periventricular
- HF
- high fat
- Mc4r
- melanocortin 4 receptor
- PVN
- paraventricular
- Sim1
- Single-minded homolog 1
- SON
- supraoptic.
References
- 1. Ema M, Morita M, Ikawa S, et al. Two new members of the murine Sim gene family are transcriptional repressors and show different expression patterns during mouse embryogenesis. Mol Cell Biol. 1996;16:5865–5875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fan CM, Kuwana E, Bulfone A, et al. Expression patterns of two murine homologs of Drosophila single-minded suggest possible roles in embryonic patterning and in the pathogenesis of Down syndrome. Mol Cell Neurosci. 1996;7:1–16 [DOI] [PubMed] [Google Scholar]
- 3. Holder JL, Jr, Butte NF, Zinn AR. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum Mol Genet. 2000;9:101–108 [DOI] [PubMed] [Google Scholar]
- 4. Holder JL, Jr, Zhang L, Kublaoui BM, et al. Sim1 gene dosage modulates the homeostatic feeding response to increased dietary fat in mice. Am J Physiol Endocrinol Metab. 2004;287:E105–E113 [DOI] [PubMed] [Google Scholar]
- 5. Michaud JL, Boucher F, Melnyk A, et al. Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus. Hum Mol Genet. 2001;10:1465–1473 [DOI] [PubMed] [Google Scholar]
- 6. Ramachandrappa S, Raimondo A, Cali AM, et al. Rare variants in single-minded 1 (SIM1) are associated with severe obesity. J Clin Invest. 2013;123:3042–3050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bonnefond A, Raimondo A, Stutzmann F, et al. Loss-of-function mutations in SIM1 contribute to obesity and Prader-Willi-like features. J Clin Invest. 2013;123:3037–3041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Michaud JL, Rosenquist T, May NR, Fan CM. Development of neuroendocrine lineages requires the bHLH-PAS transcription factor SIM1. Genes Dev. 1998;12:3264–3275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kublaoui BM, Holder JL, Jr, Gemelli T, Zinn AR. Sim1 haploinsufficiency impairs melanocortin-mediated anorexia and activation of paraventricular nucleus neurons. Mol Endocrinol. 2006;20:2483–2492 [DOI] [PubMed] [Google Scholar]
- 10. Kublaoui BM, Holder JL, Jr, Tolson KP, Gemelli T, Zinn AR. SIM1 overexpression partially rescues agouti yellow and diet-induced obesity by normalizing food intake. Endocrinology. 2006;147:4542–4549 [DOI] [PubMed] [Google Scholar]
- 11. Yang C, Gagnon D, Vachon P, et al. Adenoviral-mediated modulation of Sim1 expression in the paraventricular nucleus affects food intake. J Neurosci. 2006;26:7116–7120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tolson KP, Gemelli T, Gautron L, Elmquist JK, Zinn AR, Kublaoui BM. Postnatal Sim1 deficiency causes hyperphagic obesity and reduced Mc4r and oxytocin expression. J Neurosci. 2010;30:3803–3812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. De Souza J, Butler AA, Cone RD. Disproportionate inhibition of feeding in A(y) mice by certain stressors: a cautionary note. Neuroendocrinology. 2000;72:126–132 [DOI] [PubMed] [Google Scholar]
- 14. Srinivas S, Watanabe T, Lin CS, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weir JB. New methods for calculating metabolic rate with special reference to protein metabolism. J Physiol. 1949;109:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Erdmann G, Schutz G, Berger S. Inducible gene inactivation in neurons of the adult mouse forebrain. BMC Neurosci. 2007;8:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kublaoui BM, Gemelli T, Tolson KP, Wang Y, Zinn AR. Oxytocin deficiency mediates hyperphagic obesity of Sim1 haploinsufficient mice. Mol Endocrinol. 2008;22:1723–1734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Balthasar N, Dalgaard LT, Lee CE, et al. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123:493–505 [DOI] [PubMed] [Google Scholar]
- 19. da Silva AA, do Carmo JM, Kanyicska B, Dubinion J, Brandon E, Hall JE. Endogenous melanocortin system activity contributes to the elevated arterial pressure in spontaneously hypertensive rats. Hypertension. 2008;51:884–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. da Silva AA, Kuo JJ, Hall JE. Role of hypothalamic melanocortin 3/4-receptors in mediating chronic cardiovascular, renal, and metabolic actions of leptin. Hypertension. 2004;43:1312–1317 [DOI] [PubMed] [Google Scholar]
- 21. Greenfield JR, Miller JW, Keogh JM, et al. Modulation of blood pressure by central melanocortinergic pathways. N Engl J Med. 2009;360:44–52 [DOI] [PubMed] [Google Scholar]
- 22. Kuo JJ, da Silva AA, Tallam LS, Hall JE. Role of adrenergic activity in pressor responses to chronic melanocortin receptor activation. Hypertension. 2004;43:370–375 [DOI] [PubMed] [Google Scholar]
- 23. Brown LM, Gent L, Davis K, Clegg DJ. Metabolic impact of sex hormones on obesity. Brain Res. 2010;1350:77–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen X, McClusky R, Chen J, et al. The number of x chromosomes causes sex differences in adiposity in mice. PLoS Genet. 2012;8:e1002709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xi D, Gandhi N, Lai M, Kublaoui BM. Ablation of Sim1 Neurons Causes Obesity through Hyperphagia and Reduced Energy Expenditure. PloS One. 2012;7:e36453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Blevins JE, Schwartz MW, Baskin DG. Evidence that paraventricular nucleus oxytocin neurons link hypothalamic leptin action to caudal brain stem nuclei controlling meal size. Am J Physiol Regul Integr Comp Physiol. 2004;287:R87–R96 [DOI] [PubMed] [Google Scholar]
- 27. Badoer E. Hypothalamic paraventricular nucleus and cardiovascular regulation. Clin Exp Pharmacol Physiol. 2001;28:95–99 [DOI] [PubMed] [Google Scholar]
- 28. Kirchgessner AL, Sclafani A, Nilaver G. Histochemical identification of a PVN-hindbrain feeding pathway. Physiol Behav. 1988;42:529–543 [DOI] [PubMed] [Google Scholar]
- 29. Sawchenko PE, Swanson LW. Immunohistochemical identification of neurons in the paraventricular nucleus of the hypothalamus that project to the medulla or to the spinal cord in the rat. J Comp Neurol. 1982;205:260–272 [DOI] [PubMed] [Google Scholar]
- 30. Swanson LW, Sawchenko PE. Hypothalamic integration: organization of the paraventricular and supraoptic nuclei. Ann Rev Neurosci. 1983;6:269–324 [DOI] [PubMed] [Google Scholar]
- 31. Bouret SG, Gorski JN, Patterson CM, Chen S, Levin BE, Simerly RB. Hypothalamic neural projections are permanently disrupted in diet-induced obese rats. Cell Metab. 2008;7:179–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bouret SG, Simerly RB. Developmental programming of hypothalamic feeding circuits. Clin Genet. 2006;70:295–301 [DOI] [PubMed] [Google Scholar]
- 33. Saborio P, Tipton GA, Chan JC. Diabetes insipidus. Pediatr Rev. 2000;21:122–129 [DOI] [PubMed] [Google Scholar]
- 34. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031 [DOI] [PubMed] [Google Scholar]
- 35. Allen Brain Atlas. http://www.brain-map.org
- 36. Duplan SM, Boucher F, Alexandrov L, Michaud JL. Impact of Sim1 gene dosage on the development of the paraventricular and supraoptic nuclei of the hypothalamus. Eur J Neurosci. 2009;30:2239–2249 [DOI] [PubMed] [Google Scholar]
- 37. Paxinos G, Franklin KBJ. 2001 The Mouse Brain in Stereotaxic Coordinates. 2nd ed San Diego, CA: Academic Press [Google Scholar]