

During the last decade, the mechanistic (or mammalian) target of rapamycin (mTOR) kinase has emerged as a critical regulator of cell size and metabolism because of its ability to couple nutrients, growth factors and oxygen availability with the regulation of protein and lipid synthesis, lysosome biogenesis and neuronal morphology and activity among other processes [1,2].
mTOR forms two structurally and functionally distinct complexes in cells called mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) (Figure 1). The characteristic components of mTORC1 are Raptor (regulatory-associated protein of mTOR) and PRAS40 (proline-rich Akt substrate 40 kDa); while defining components of the more recently discovered mTORC2 include Rictor (rapamycin-insensitive companion of TOR), mSin1 (mammalian stress-activated MAP kinase-interacting protein 1) and Protor-1 and 2 (protein observed with Rictor 1 and 2) [1,2].
Figure 1.
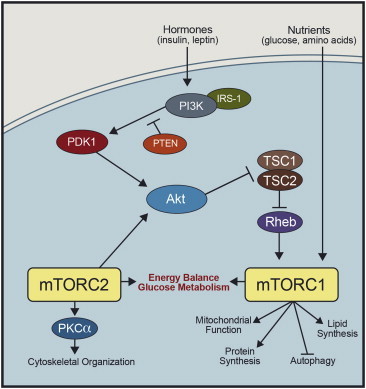
Simplified scheme illustrating the regulation of the mTOR pathway in the brain. For further details on upstream regulators of mTORC1 and mTORC2 and their downstream molecular targets the reader should refer to Ref. [1,2]. IRS-1, insulin receptor substrate 1; PTEN, phosphatase and tensin homolog; PDK1, phosphatidylinositol-dependent kinase 1; Rheb, Ras homolog enriched in brain.
Since the initial findings describing the role of mTORC1 and its downstream targets S6 kinase 1 (S6K1) and S6 ribosomal protein (S6) in the central regulation of food intake [3], subsequent studies in the field have mainly focused on dissecting the function of this signaling pathway in the hypothalamus (reviewed in Ref. [4]). In this issue of Molecular Metabolism, Kocalis et al. investigate the role of mTORC2 in energy balance regulation by using mice with conditional deletion of Rictor in Nestin-positive cells (Neuronal Rictor KO or NRic-KO), or in either POMC- or AgRP-expressing neurons [5]. NRic-KO mice were smaller and weighed less than controls at weaning [5]. However, once adults, they showed catch-up growth and altered body composition (decreased lean mass and increased fat mass) with profound obesity mostly due to decreased energy utilization [5]. Apart from the presence of peripheral insulin resistance and alterations in fuel partitioning, NRic-KO mice had increased circadian plasma corticosterone at the zenith and in response to stress [5]. Thus, the authors suggest that glucocorticoid excess could explain the fat accumulation, decreased lean mass and metabolic derangements observed in this animal model [5]. While deletion of Rictor in POMC neurons led to late onset obesity, mainly caused by a mild hyperphagia, and glucose intolerance, which was comparable to that described in NRic-KO mice [5]. Finally, lack of Rictor in AgRP neurons did not affect energy balance and only modestly impaired glucose tolerance [5]. Thus, this study reveals that Rictor and appropriate mTORC2 activity are important players in the CNS regulation of energy balance. However, it does not provide any information on the neuronal mechanism(s) that are under the control of mTORC2 and that might lead to the behavioral and metabolic alterations described above.
Interestingly, mTORC2 has been implicated in the control of neuron size, neuron morphology and synaptic plasticity [2,6,7], functions that up until recently were thought to be under the exclusive control of mTORC1. It should be noted that Kocalis et al. used a nestin-cre mouse to delete Rictor in neurons. This cre mouse induces recombination in all neural tube-derived cells [8], implying that this genetic strategy deleted Rictor in neural progenitor cells. Thomanetz et al. recently showed that conditional deletion of Rictor in nestin-positive cells has drastic effects on neuronal development and function [6]. In their study, mice without Rictor in nestin-positive cells were smaller at a young age, as also found by Kocalis et al., and had microcephaly [6]. This was due to the decreased number and size of neurons, which had defective synaptic connectivity and activity [6]. These pathological changes were observed in the hippocampus and cerebellum [6]. It is, however, likely that they may be found in other brain structures, including the hypothalamus, and that they may help to explain the NRic-KO phenotype as described by Kocalis et al.
As for the potential molecular mechanism involved in such changes, Thomanetz et al. found decreased brain expression of the protein kinase C isoform α (PKCα), among other PKC isoforms [6]. Activation of PKCα by mTORC2 regulates cell shape by affecting the actin cytoskeleton (reviewed in Ref. [1]). Similarly, Kocalis et al. observed decreased levels of PKCα and its activity in the hypothalamus of NRic-KO mice [5], a finding that further implies that this animal model might have morphological and functional neuronal alterations. In this context, it will be paramount to investigate in POMC Ric-KO and AgRP Ric-KO mice whether the lack of Rictor affects morphology and synaptic activity of POMC and AgRP neurons differently, thus clarifying the underlying reasons of the phenotypic differences observed between these 2 mutant mouse lines.
Moreover, it should be mentioned that defective neuronal function results not only from reduced, but also from increased neuronal cell size. In particular, overactivity of mTORC1 in POMC neurons, due to the overexpression of its upstream regulatory protein Tuberous sclerosis complex 1 (TSC1) in mice, cell-autonomously increases POMC neurons' somatic size, while reducing axonal projections to the hypothalamic paraventricular nucleus [9]. Notably, these morphological changes are associated with hyperphagia and obesity [9].
Finally, an open question remains whether an interaction exists between mTORC2 and mTORC1, particularly in the context of central leptin and insulin sensing. The phosphopinositide 3′OH-kinase (PI3K)/Akt pathway is known to be a major player in determining the actions of leptin and insulin in hypothalamic neurons. mTORC2 phosphorylates the kinase Akt at ser 473 and induces its full activation [10]. But PI3K and Akt are also upstream activators of mTORC1 [1]. Kocalis et al. did not observe any changes in Raptor and mTOR protein levels within the hypothalamus of NRic-KO mice. However, phosphorylation levels of mTORC1 downstream targets, which are routinely used as markers of mTORC1 activity, were not evaluated, leaving open the possibility that hypothalamic mTORC1 may be altered.
By pointing out the implication of Rictor in energy balance regulation, the study of Kocalis et al. has led to a series of stimulating questions that currently remain unanswered and whose investigation will certainly provide new vistas on how mTORC1 and mTORC2 control neuronal function and associated behavior. The hope is that these new insights will lead to the identification of novel molecular targets for the treatment of obesity and its associated metabolic disorders.
Footnotes
This commentary refers to “Rictor/mTORC2 facilitates central regulation of energy and glucose homeostasis by Heidi E. Kocalis et al.”, http://dx.doi.org/10.1016/j.molmet.2014.01.014.
References
- 1.Laplante M., Sabatini D.M. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Costa-Mattioli M., Monteggia L.M. mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nature Neuroscience. 2013;16(11):1537–1543. doi: 10.1038/nn.3546. [DOI] [PubMed] [Google Scholar]
- 3.Cota D., Proulx K., Smith K.A., Kozma S.C., Thomas G., Woods S.C. Hypothalamic mTOR signaling regulates food intake. Science. 2006;312(5775):927–930. doi: 10.1126/science.1124147. [DOI] [PubMed] [Google Scholar]
- 4.Martínez de Morentin P.B., Martinez-Sanchez N., Roa J., Ferno J., Nogueiras R., Tena-Sempere M. Hypothalamic mTOR: the rookie energy sensor. Current Molecular Medicine. 2014;14(1):3–21. doi: 10.2174/1566524013666131118103706. [DOI] [PubMed] [Google Scholar]
- 5.Kocalis H.E., Hagan S.L., George L., Turney M.K., Siuta M.A., Laryea G.N. Rictor/mTORC2 facilitates central regulation of energy and glucose homeostasis. Molecular Metabolism. 2014 doi: 10.1016/j.molmet.2014.01.014. [in press] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomanetz V., Angliker N., Cloëtta D., Lustenberger R.M., Schweighauser M., Oliveri F. Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. The Journal of Cell Biology. 2013;201(2):293–308. doi: 10.1083/jcb.201205030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Urbanska M., Gozdz A., Swiech L.J., Jaworski J. Mammalian target of rapamycin complex 1 (mTORC1) and 2 (mTORC2) control the dendritic arbor morphology of hippocampal neurons. The Journal of Biological Chemistry. 2012;287(36):30240–30256. doi: 10.1074/jbc.M112.374405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graus-Porta D., Blaess S., Senften M., Littlewood-Evans A., Damsky C., Huang Z. Beta1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron. 2001;31(3):367–379. doi: 10.1016/s0896-6273(01)00374-9. [DOI] [PubMed] [Google Scholar]
- 9.Mori H., Inoki K., Münzberg H., Opland D., Faouzi M., Villanueva E.C. Critical role for hypothalamic mTOR activity in energy balance. Cell Metabolism. 2009;9(4):362–374. doi: 10.1016/j.cmet.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sarbassov D.D., Guertin D.A., Ali S.M., Sabatini D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]