

Abstract
We recently reported that local overexpression of VEGF-A in white adipose tissue (WAT) protects against diet-induced obesity and metabolic dysfunction. The observation that VEGF-A induces a “brown adipose tissue (BAT)-like” phenotype in WAT prompted us to further explore the direct function of VEGF-A in BAT. We utilized a doxycycline (Dox)-inducible, brown adipocyte-specific VEGF-A transgenic overexpression model to assess direct effects of VEGF-A in BAT in vivo. We observed that BAT-specific VEGF-A expression increases vascularization and up-regulates expression of both UCP1 and PGC-1α in BAT. As a result, the transgenic mice show increased thermogenesis during chronic cold exposure. In diet-induced obese mice, introducing VEGF-A locally in BAT rescues capillary rarefaction, ameliorates brown adipocyte dysfunction, and improves deleterious effects on glucose and lipid metabolism caused by a high-fat diet challenge. These results demonstrate a direct positive role of VEGF-A in the activation and expansion of BAT.
Keywords: VEGF-A, BAT, Cold tolerance, Energy expenditure
Abbreviations: BAT, brown adipose tissue; WAT, white adipose tissue; UCP1, uncoupling protein1; Dox, doxycycline; HFD, high-fat diet; PGC-1α, PPARγ co-activator-1α; OCR, oxygen consumption rate; OGTT, oral glucose tolerance test; HIF1, hypoxia-induced factor1
1. Introduction
In the lean state, adipose tissue is a well-vascularized tissue [1]. In addition to providing access to nutritional components in accordance with the metabolic demands of the tissue, the adipose tissue vasculature plays multiple roles to ensure that the tissue has appropriate access to oxygen [2–4]. Recent studies on vascular function of adipose tissue have focused on VEGF-A, a bona fide endothelial growth factor [2,5]. As a potent angiogenic factor, VEGF-A functions by specifically stimulating vascular endothelial cell activation, proliferation, migration, and vessel permeability [6]. Subsequent studies indicated that local overexpression of VEGF-A in a physiological range in white adipose tissue (WAT) protects against diet-induced obesity and metabolic dysfunction [2,5,7]. These findings further suggest that the ultimate consequence of VEGF-A modulated angiogenesis in controlling adipose tissue expansion and overall metabolic health are context dependent: in the early stages of diet-induced obesity development, VEGF-A overexpression facilitates healthy adipose tissue expansion and confers protection from metabolic insults, whereas in the context of preexisting adipose tissue dysfunction, anti-angiogenic action by blocking VEGF-A signaling leads to improved insulin sensitivity and ameliorated metabolic functions [2,5,8]. More importantly, we found that at the early stages of diet-induced obesity, specific overexpression of VEGF-A in WAT leads to a “beigeing” phenotype as indicated by marked induction of UCP1 and PGC1α [2,9]. Additionally, the mice showed a lean phenotype when challenged with HFD [2,5,7]. This beigeing phenotype in WAT is of particular importance for VEGF-A functions leading to the effects of increased energy expenditure and resistance to diet-induced metabolic insults [2,5,7]. However, recent studies suggest that the beige cells themselves might not be sufficient to affect whole-body physiology under ambient conditions [10]. This raises the question whether VEGF-A has a more profound functional impact on classical brown adipose tissue (BAT).
Even though the vasculature in WAT and BAT has common features and functions [11], BAT is metabolically more active and therefore displays a higher vascular density [1,12]. BAT is composed by brown adipocytes, characterized by multilocular lipid droplets with a central nucleus, and a high density of mitochondria. Upon stimulation, brown adipose tissue exerts enhanced energy expenditure and increased glucose and fatty acid metabolism [13,14]. BAT is the major site for adaptive non-shivering thermogenesis in rodents. As a thermogenic organ, BAT activation can counteract phenotypes associated with obesity [15]. Thus, we reasoned that an enhanced local development of angiogenesis by VEGF-A in metabolically active BAT might lead to metabolically beneficial phenotypes.
The mitochondria in brown fat cells express high levels of UCP1, a proton transporter localized in the inner mitochondrial membrane [13,16]. When activated, UCP1 increases the permeability of the inner mitochondrial membrane by allowing free fatty acids (co-factors for UCP1) to flip-flop across inner and outer leaflets of the membrane, bypassing the ATP-synthase and effectively uncoupling the electron transport chain, thereby allowing the electrochemical energy to dissipate as heat, resulting in thermogenesis [13,17,18]. Interestingly, UCP1 is highly enriched in BAT and is not expressed in regular white adipocytes, though beige fat cells in WAT also display UCP1 induction. The unique features of BAT that enable the tissue to remove a large amount of lipids from circulation to activate thermogenesis and produce heat affect systemic energy expenditure and mark it as a potential therapeutic target in obese subjects.
Interestingly, cold acclimation dramatically up-regulates VEGF-A levels in BAT [12]. Stimulated VEGF-A-induced VEGFR2 signaling further initiates cold-induced adipose tissue angiogenesis [12]. This suggests that angiogenesis in BAT plays an important role in regulating energy expenditure [12]. In the current study, we locally supplied VEGF-A using a novel doxycycline-inducible BAT-specific transgenic mouse model to better define the role of VEGF-A in BAT. Our findings suggest that VEGF-A can activate brown fat tissue. It can up-regulate both PGC-1α and UCP1 expression, thus increasing thermogenesis and energy expenditure. Moreover, in a diet-induced obese model, VEGF-A mediated angiogenesis further facilitates healthy expansion of BAT. The mice retained metabolic flexibility on a HFD, with improved glucose tolerance, lipid clearance and energy expenditure. These results highlight the importance of VEGF-A action for energy homeostasis and metabolism of BAT.
2. Materials and methods
2.1. Animals
To construct the UCP1-rtTA plasmid, a 3.1-kb UCP1 promoter from its original pGL vector [19] was cloned into a pBluescript vector containing an rtTA cassette and a rabbit β-globin 3′ UTR [2]. The UCP1-rtTA transgenic mice were generated by the transgenic core facility at UTSW. BAT-specific VEGF-A transgenic mice were obtained by crossing UCP1-rtTA and TRE-VEGF-A [2] lines. Age-matched UCP1-rtTA but lacking the TRE-VEGF-A transgenic mice were used as littermate controls. All the mice were on a pure C57BL/6 background. Mice were maintained on a 12 h dark/light cycle and fed a normal chow diet, unless otherwise indicated. All animals were 6 weeks old at the time of experiments. Animals were bred in house in the UTSW Medical Center. The Institutional Animal Care and Use Committee of the University of Texas Southwestern Medical Center has approved all animal experimental protocols.
2.2. Blood vessel stains
This method has been described in detail previously [20]. In brief, to stain functional blood vessels, mice were injected with 100 μg of Rhodamine Griffonia (Bandeiraea) simplicifolia lectin (Vector Laboratories, Burlingame, CA, USA) through the tail vein. Three minutes after the injection, the animal was perfused with 1% paraformaldehyde through the left ventricle to fix the tissues. The brown and subcutaneous adipose tissues were then excised for further fixation overnight in 10% PBS-buffered formalin. Tissues were rinsed with 50% ethanol for 3 times and kept in 50% ethanol at 4 °C. Lectin-stained capillaries were visualized by confocal microscopy (Leica TCS SP5 confocal microscope).
2.3. Dox containing high-fat diet
For all the experiments with HFD, mice were fed with a diet containing 60% calories from fat (Cat No. D12492; Research Diets). For low dose Dox treatment, the Dox powder (Sigma) was mixed with HFD paste (Research Diets) to a final concentration of 60 mg/kg.
2.4. Measurement of body composition
The fat mass and the bone-free lean body composition were measured in non-anesthetized mice using an Echo 3-in-1 nuclear magnetic resonance (MRI) mini Spec instrument (Bruker, Germany) [2].
2.5. Indirect calorimetric measurements
Before the metabolic cage studies, the mice were housed individually in metabolic chambers for 1 week for acclimation. For the indirect calorimetric cage studies, the mice were maintained on a 12 h dark/light cycle with lights on from 7:00 am to 7:00 pm at room temperature (20 °C∼22 °C). Metabolic parameters were obtained continuously using TSA metabolic chambers in an open circuit indirect calorimetric system (TSA System, Germany) [2,21]. During the whole measuring process, all VEGF-A transgenic mice and their littermate controls were provided with HFD paste containing 60 mg/kg Dox and water ad libitum.
2.6. Oral glucose tolerance tests and blood chemistry
For the OGTTs, mice were fasted for 5 h prior to administration of glucose (2.5 g/kg body weight) by gastric gavage. At each indicated time point, blood samples from the tail veins were collected in heparin-coated capillary tubes. Glucose levels were measured using an oxidase–peroxidase assay (Sigma–Aldrich). Mice did not have access to food throughout the experiment. The tests were performed 5 weeks after VEGF-A induction under HFD. Serum TG levels were measured following a 3-h fast with the kit from Infinity (Infinity; Thermo Fisher Scientific, Waltham, MA, USA).
2.7. Cold tolerance test
To perform the cold tolerance test, biocompatible and sterile microchip transponders (IPTT-300 Extended Accuracy Calibration; Bio Medic Data Systems, Seaford, DE, USA) were implanted subcutaneously over the shoulder blades as suggested by the manufacturer. The VEGF-A transgenic mice and their littermate controls were pre-induced with normal chow plus 60 mg/kg Dox for 5 days. The mice were then transferred to cold room at 6 °C. For the acute assay, food was removed from cages at the time of transfer to the cold room. The body temperature was measured at each of the indicated time points; for the chronic assay, the mice were kept in the cold room at 6 °C for 3 more weeks with regular chow plus Dox. Then food was removed and the temperature was measured at each of the indicated time points.
2.8. Isolation of mitochondria from BAT
Brown adipose tissues were homogenized using a motorized Dounce homogenizer in ice-cold MSHE buffer (70 mM sucrose, 210 mM mannitol, 5 mM HEPES, and 1 mM EDTA) containing 0.5% fatty acid-free BSA. Homogenates then underwent low centrifugation (800 g for 10 min) to remove nuclei and cell debris, followed by high centrifugation (8,000 g for 10 min) to obtain the mitochondrial pellets, which were washed once in ice-cold MSHE buffer and were resuspended in a minimal amount of MSHE buffer prior to determination of protein concentrations using a BCA assay (Pierce).
2.9. Mitochondrial functional assays
Oxygen consumption rates (OCRs) were determined using the XF24 Extracellular Flux Analyzer (Seahorse Bioscience, MA, USA) following the manufacturers' protocols. For the electron-flow (EF) measurements, isolated mitochondria were seeded at 5 μg of protein per well in XF24 V7 cell-culture microplates (Seahorse Bioscience), then pelleted by centrifugation (2,000 g for 20 min at 6 °C) in 1X MAS buffer (70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, and 1 mM EGTA in 0.2% FA-free BSA; pH 7.2) supplemented with 10 mM pyruvate, 10 mM malate, and 4 μM carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) (for EF experiments), with a final volume of 500 μl per well. The XF24 plate was then transferred to a temperature-controlled (37 °C) Seahorse analyzer and subjected to a 10-min equilibration period and 2 assay cycles to measure the basal rate, comprising of a 30-s mix, and a 3-min measure period each; and compounds were added by automatic pneumatic injection followed by a single assay cycle after each; comprising of a 30-s mix and a 3-min measure period. For EF experiments, OCR measurements were obtained following sequential additions of rotenone (2 μM final concentration), succinate (10 mM), antimycin A (4 μM), and ascorbate (10 mM) (the latter containing 1 mM N,N,N',N'-tetra methyl-p-phenylenediamine (TMPD)). OCR measurements were recorded at set interval time points. All compounds and materials above were obtained from Sigma–Aldrich.
2.10. Histology (H&E staining)
A portion of adipose and liver tissues were fixed in 10% formalin and embedded in paraffin. General morphology was visualized by hematoxylin and eosin (H&E) staining.
2.11. Quantitative real-time PCR
Adipose and liver tissues were excised and quickly frozen in liquid nitrogen for future assays. For the Q-PCR analyses, total RNAs were extracted from tissues in Trizol (Invitrogen, Carlsbad, CA, USA) using a TissueLyser (Qiagen, Valenica, CA, USA) and then isolated using the RNeasy RNA extraction kit (Qiagen) following the protocol from the company. The quality and quantity of the RNA were determined by absorbance at 260/280 nm. cDNAs were prepared by reverse transcribing 1 μg of total RNA with Superscript III reverse transcriptase and oligo (dT)20 (Invitrogen). The mRNA levels were calculated by using the comparative threshold cycle (CT) method. All the primer sequences have been published previously [2,20]. GAPDH was used as the invariant control. Q-PCR reactions were carried out on an ABI Prism 7900 HT sequence detection system (Applied Biosystems).
2.12. Statistical analysis
Experimental results are presented as means ± SD. Differences between two groups were determined for statistical significance by a standard two-tailed Student's t-test. Significance was accepted at a p value of <0.05.
3. Results
3.1. Generation of transgenic mice with BAT-specific expression of VEGF-A
To achieve inducible expression of VEGF-A specifically in BAT, we established a double transgenic mouse strain employing a tetracycline-inducible system. The TRE driven VEGF-A transgenic model has been described previously [22]. In our model, the TRE is regulated by the reverse tetracycline-dependent transcriptional activator (rtTA), whose expression is under the control of the BAT-specific UCP1 promoter [19]. As this UCP1 promoter driven rtTA is a newly generated mouse model, we wanted to verify the brown adipose tissue specificity of rtTA expression. The levels of rtTA in a number of tissues were measured by both Q-PCR and regular PCR 5 days after normal chow diet plus 60 mg/kg Dox induction. The results indicate that rtTA mRNAs were expressed with high levels in BAT, while the expression was not detectable in other tissues, including other fat pads (Figure 1A left). The results were further verified by regular PCR by which rtTA band was only detected in BAT (Figure 1A right). We further confirmed that VEGF-A in the double transgenic mouse is induced in the presence of Dox. To effectively express rtTA in BAT, we used double transgenic mice, i.e., homozygous versions for UCP1-rtTA. We used a lower dose of Dox (60 mg/kg) to achieve a VEGF-A level within a physiological range, without causing edema formation [2]. Expression of VEGF-A in a number of fat pads was assayed in the presence or at the absence of 60 mg/kg Dox treatment. The results indicate that VEGF-A was predominantly induced in BAT (Figure 1B), while only moderate induction was seen in SWAT (probably due to low level UCP1 induction in local beige cells). No induction was observed in other tissues, such as the liver (data not shown). Taken together, our system allows inducible expression of VEGF-A, with expression restricted to BAT.
Figure 1.
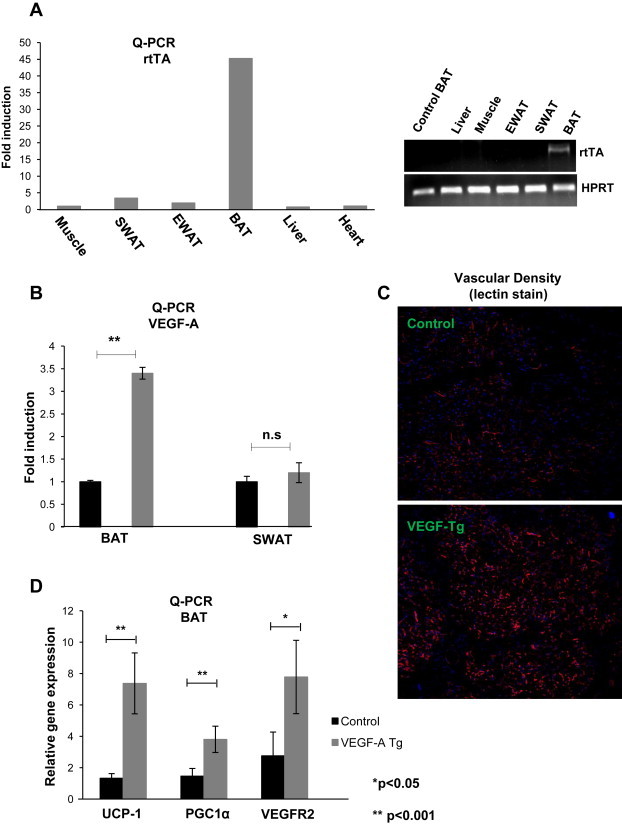
Overexpression of VEGF-A in BAT stimulates vascularization and up-regulates mitochondrial genes. (A) Left: Q-PCR analysis for tissue distribution of rtTA mRNAs in a UCP1 promoter driven rtTA overexpression model; Right: regular PCR analysis of rtTA overexpression in different fat pads and other organs in the UCP1-rtTA transgenic model. (B) Q-PCR analysis of VEGF-A overexpression in BAT and SWAT in UCP1-rtTA and TRE-VEGF-A double transgenic mouse stain (n = 4 per group, Student's t-test, **p < 0.001). (C) Representative sections of functional blood vessels in BAT labeled by tail-injected Rhodamine fluorine dye tagged lectin-1 in VEGF-A Tg and their littermate control mice. Blood vessels are shown in red, while the nuclei are in blue (DAPI staining). The images were visualized with a confocal microscope. (D) Q-PCR analysis of vascular endothelial cell marker VEGFR2 and mitochondrial functional proteins UCP1 and PGC1α in BAT of VEGF-A Tg and control mice (n = 4 in controls; n = 5 in VEGF-A Tg). The difference was analyzed by Student's t-test. *p < 0.05, **p < 0.001 vs. controls.
3.2. Locally derived VEGF-A in BAT stimulates angiogenesis and triggers up-regulation of mitochondrial genes
To functionally assess the consequences of VEGF-A induction in BAT, we examined the local angiogenic response in BAT. Confocal fluorescent imaging reveals that compared with the control group, a higher vascular density is seen in the BAT of VEGF-A transgenic mice (Figure 1C). As expected, genes affected by VEGF-A expression included VEGFR2, which was significantly up-regulated in BAT of the VEGF-A transgenic mice (Figure 1D). Interestingly, mRNA levels of mitochondrial proteins, such as UCP1 and PGC1α, were also markedly up-regulated in VEGF-A transgenic BAT (Figure 1D). Short-term induction of VEGF-A in BAT, therefore, effectively stimulates angiogenesis and triggers an up-regulation of mitochondrial genes.
3.3. VEGF-A overexpression in BAT is sufficient to trigger enhanced thermogenesis
Five days of induction with regular chow plus Dox not only stimulated the up-regulation of VEGFR2 in BAT, but also the key thermogenic proteins, such as UCP1 and PGC1α (Figure 1D). Consistent with these changes in thermogenic gene expression, VEGF-A transgenic mice defended their body temperature modestly better during an acute cold stress in the absence of food during the experiment (Figure 2A). More importantly, these mice were significantly better at defending their body temperature after 3 weeks of cold exposure. Baseline core body temperatures were different to start out with. Upon removal of food, body temperatures of both wildtype and transgenic mice decreased as expected. However, temperatures diverged further towards the end of the experiment, as the wildtype mice started to drop their body temperature more rapidly. (Figure 2B). These results suggest that VEGF-A in and by itself in BAT is sufficient to enhance thermogenesis during cold exposure.
Figure 2.
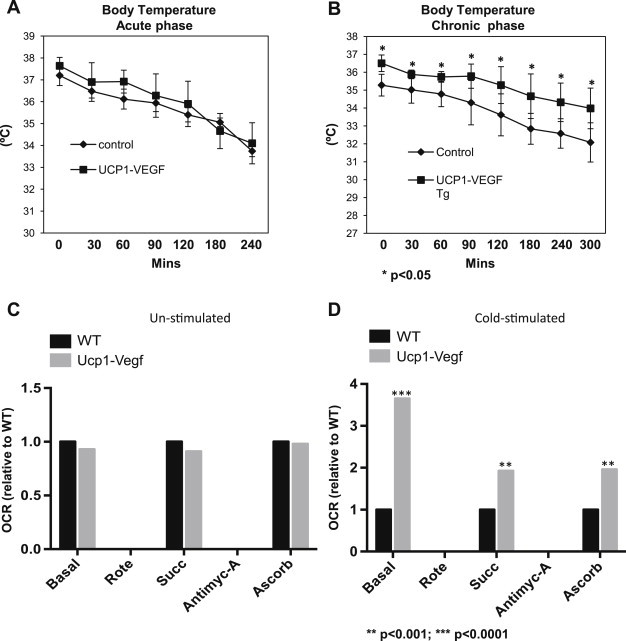
Overexpression of VEGF-A in BAT stimulates themogenesis and enhances mitochondrial function upon cold exposure. (A) Measurements of body temperature in VEGF-A transgenic mice and their littermate controls (n = 6 per group) in an acute cold exposure setting (n = 6 per group, Student's t-test, no significance). (B) Measurements of body temperature in VEGF-A transgenic mice and their littermate controls (n = 5 per group, Student's t-test, *p < 0.05 vs. controls) in after chronic phase cold exposure for 3 weeks. Mice did not have access to food during the experiments in (A) and (B). (C) Mitochondrial electron-flow analysis and oxygen consumption rates in response to the substrates pyruvate, malate, succinate, and ascorbate in unstimulated BAT from VEGF-A transgenic and their littermate control groups (n = 2 per group, Student's t-test indicates no difference vs. controls). (D) Mitochondrial electron-flow analysis and oxygen consumption rates in cold-stimulated BAT (3 weeks) from VEGF-A transgenic and their littermate control groups. The readings represent relative folds to wildtype (n = 2 per group, Student's t-test, **p < 0.01; ***p < 0.0001 vs. controls). Wildtype levels were in each case set to 1 to allow for a relative comparison.
3.4. Overexpression of VEGF-A in BAT enhances cold-stimulated mitochondrial respiration
To determine whether BAT-specific overexpression of VEGF-A alters BAT mitochondrial activity, we performed a more detailed analysis of mitochondrial activity and examined oxygen consumption rates (OCRs), using well-established methods to assess mitochondrial respiratory capacity [23,24]. More specifically, we performed sequential electron-flow analyses on mitochondria isolated from BAT tissues. This is an experiment that assesses OCRs simultaneously while examining whether any defects or dysfunction exist within the mitochondrial electron transport chain (ETC) through sequential use of specific inhibitors and substrates of the various complexes of the ETC. Interestingly, while no marked differences were observed in BAT mitochondria isolated from WT mice and UCP1-VEGF mice that were housed under baseline room temperature conditions (20 °C–22 °C) (Figure 2C), we observed a significant increase in mitochondrial OCRs in transgenic BAT mitochondria derived from mice housed in cold-stimulated environment (6 °C), when compared with WT BAT mitochondria (Figure 2D). Such marked differences in OCRs were evident in response to several different substrates; in particular BAT mitochondria isolated from transgenic mice exhibited higher mitochondrial respiration rates under basal conditions (media contained the substrates pyruvate and malate, in addition to the chemical uncoupler FCCP), in response to the complex II substrate succinate, in addition to the complex IV substrate, ascorbate. Of note, OCR values were normalized to total amount of mitochondrial protein utilized per well (5 μg BAT mitochondrial protein loaded per well). In light of these observations, given that the VEGF-A transgenic mice harbor more BAT in general, this suggests that the enhanced BAT mitochondrial respiration rates in transgenic mice under cold-conditions may exert a major impact on BAT function and whole-body energy homeostasis, particularly under metabolically challenging conditions. Given the increased mitochondrial respiration rates of BAT mitochondria from transgenic mice, we further note that no defects in ETC activity or mitochondrial dysfunction can be measured in BAT of transgenic mice. Taken together, these data clearly demonstrate that an increase in VEGF-A action and signaling, specifically in BAT, profoundly enhances cold-induced mitochondrial oxidative capacity, which ultimately may exert a positive impact on cold-induced BAT function and whole-body energy homeostasis; a key element under times of nutrient excess.
3.5. BAT-specific VEGF-A expression triggers reduced body weight gain upon exposure to an HFD-challenge
We investigated the impact of local VEGF-A in BAT on the gradual manifestation of metabolic dysfunction over the course of a HFD exposure. To address this question, we challenged 6-week-old mice with HFD in the presence of 60 mg/kg DOX for 8 weeks. During the 8-week treatment regimen, VEGF-A transgenic mice gained significantly less body weight than the control littermates (Figure 3A). The difference in the body weight on the HFD in the VEGF-A transgenic mice is due to a difference in fat mass (Figure 3B). Particularly, the size of the subcutaneous fat pads in VEGF-A transgenic mice was significantly smaller (Figure 3C, middle panel). In contrast, the epididymal fat pads had no obvious weight difference between the groups (Figure 3C, bottom panel).
Figure 3.
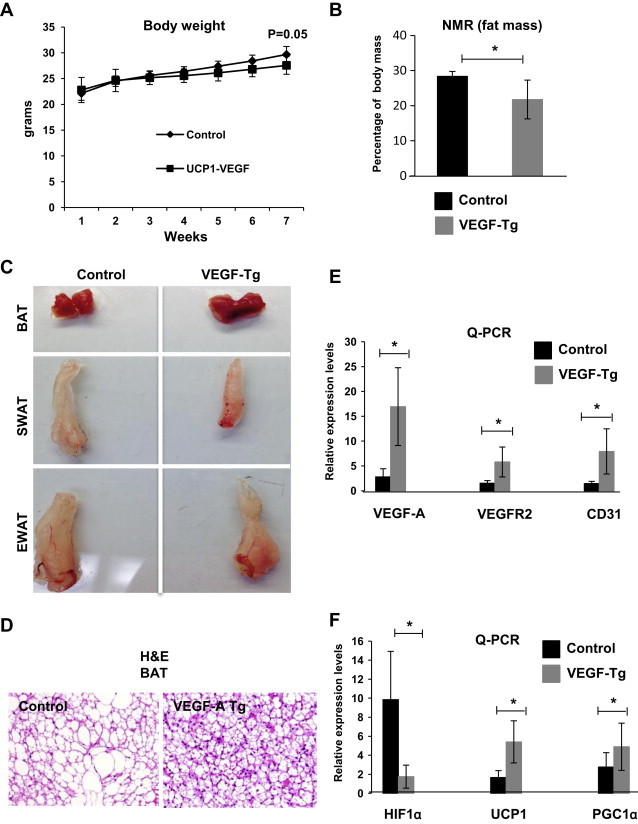
BAT-specific overexpression of VEGF-A triggers brown adipose tissue angiogenesis, healthy expansion and functional improvement upon HFD exposure. (A) Body weight gain in VEGF-A transgenic mice and their littermate controls during an HFD plus Dox feeding for 7 weeks (n = 6 per group, Student's t-test, no difference vs. controls). (B) Percentage of fat mass in VEGF-A mice and their littermate controls measured by NMR (n = 6 per group, Student's t-test, *p < 0.05 vs. controls). (C) Representative fat tissues excised from VEGF-A transgenic mice and their littermate controls after a HFD-challenge (n = 6 per group). (D) Representative of H&E staining of BAT from VEGF-A transgenic and their littermate control mice after a HFD-challenge. (E) Q-PCR analysis of angiogenic genes, including VEGF-A, VEGFR2, and CD31 in BAT of VEGF-A transgenic and their littermate control mice under HFD challenging (n = 5 per group, Student's t-test, *p < 0.05 vs. controls). (F) Q-PCR analysis of BAT function-related genes, including HIF1α, UCP1, and PGC1α in BAT of VEGF-A transgenic and their littermate control mice (n = 5 per group, Student's t-test, *p < 0.05 vs. controls).
3.6. Local VEGF-A overexpression improves brown adipose tissue function under an HFD-challenge
Diet-induced obesity leads to reduced vascular network and enhanced hypoxia in the BAT [7]. To determine whether local overexpression of VEGF-A rescues the prevailing conditions under HFD, we performed IHC analysis on BAT. The H&E stains indicate that compared with the control group, VEGF-A transgenic BAT has smaller adipocytes with more multi-lobular lipid droplets, even though the whole brown fat size is larger in the transgenic mice (Figure 3C, top panel and D). This includes a reduced lipid accumulation in brown adipose tissue. Q-PCR analysis indicates the angiogenic markers, including VEGFR2 and CD31, were significantly increased in the BAT of transgenic mice (Figure 3E), suggesting enhanced vascularization in the BAT of these mice. As a result, upon induced VEGF-A overexpression, there was a significant reduction in the levels of HIF1α associated with BAT. Of note, mitochondrial genes, including UCP1 and PGC1α were markedly up-regulated as well (Figure 3F). Collectively, these data suggest that local VEGF-A overexpression in BAT while the mice are subjected to HFD increased vascularization of BAT, leading to reduced hypoxia, thereby ameliorating the consequences of obesity. Even though we did not examine inflammatory markers in adipose tissue directly, we suspect that these values would also be reduced in accordance with the improved metabolic profile.
3.7. Local overexpression of VEGF-A in BAT leads to an increase in energy expenditure under high-fat diet
To further evaluate the metabolic benefits brought about by local overexpression of VEGF-A, we monitored the energy expenditure of the VEGF-A transgenic mice in metabolic chambers 6 weeks post initiation of HFD feeding. The oxygen consumption (VO2) was increased in the dark cycle (Figure 4A, 4174 vs. 4323 ml/hr/kg; *p < 0.05), reflecting an increase in energy expenditure in the VEGF-A transgenic mice. VEGF-A transgenic mice had no significant differences in food intake and in the respiratory exchange rates (RER) (Figure 4B and data not shown). Notably, VEGF-A transgenic mice generated slightly more heat during the dark cycle (Figure 4C, 19.86 vs. 20.53 kcal/hr/kg; *p < 0.05), further highlighting the potential for enhanced energy expenditure in these mice. Local overexpression of VEGF-A in BAT, therefore, increases energy expenditure on a high-fat diet.
Figure 4.
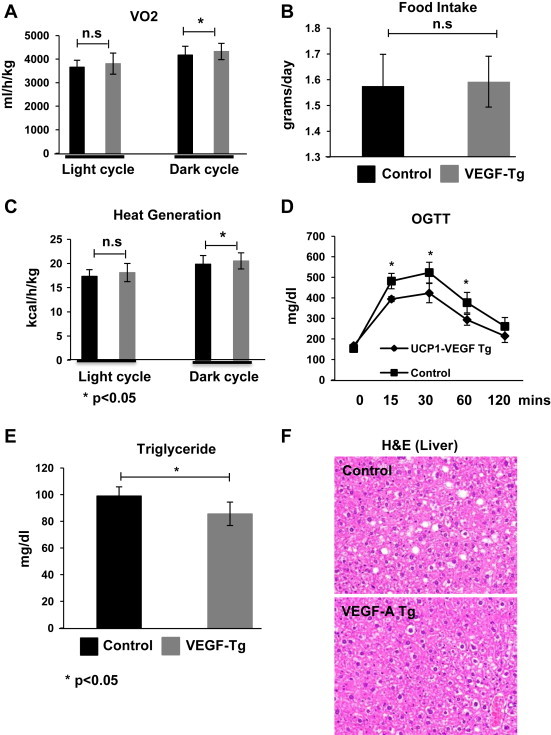
BAT-specific overexpression of VEGF-A increases energy expenditure and improves glucose and lipid metabolism upon HFD exposure. (A) Oxygen consumption; (B) Food intake; and (C) Heat generation in VEGF-A transgenic and their littermate controls under HFD challenging for 6 weeks measured by a TSA system (n = 6 per group, Student's t-test, *p < 0.05 vs. controls). (D) An OGTT in VEGF-A transgenic and their littermate control mice after HFD feeding for 5 weeks (n = 5 per group, Student's t-test, *p < 0.05 vs. controls). (E) Circulating triglyceride levels in VEGF-A transgenic and their littermate control mice (n = 5 per group, Student's t-test, *p < 0.05). (F) Representative of H&E staining of liver tissue in VEGF-A transgenic and their littermate control mice (n = 2 per group).
3.8. BAT-specific VEGF-A transgenic mice exhibit improved glucose and lipid metabolism and a decrease in HFD-induced hepatic steatosis
To investigate whether the changes above trigger any metabolic consequences due to VEGF-A stimulated BAT activation, we performed oral glucose tolerance tests (OGTTs) at the fifth week after HFD plus Dox induction. Of note, at that stage, there were not yet any significant body weight differences between the groups. The OGTTs demonstrate that the response to an oral glucose challenge is significantly improved in the VEGF-A transgenic mice (Figure 4D). The circulating levels of triglyceride in the VEGF-A transgenic mice were significantly lower (Figure 4E). H&E staining of histological slides indicates fewer and smaller lipid droplets in hepatocytes from VEGF-A transgenic mice (Figure 4F). Activation of BAT by local VEGF overexpression, therefore, results in significant systemic improvements in both glucose and lipid metabolism.
4. Discussion
It is well established that BAT is the predominant organ for non-shivering thermogenesis in rodents [13]. The thermogenic capacity of BAT is heavily dependent on a high rate of perfusion via its high density of blood vessels. Angiogenesis is rapidly induced in BAT when the animals are exposed to cold [12,25–27]. Many elegant previous reports have demonstrated that VEGF-A expression in BAT is up-regulated upon cold exposure, and it plays a critical role for the cold-induced angiogenesis in the tissue [12,26,28,29]. Recent findings further suggest potential roles for VEGF-A in BAT in brown preadipocyte proliferation, mitochondrial development, and energy expenditure and also suggest an anti-inflammatory role [7,9,30], highlighting its relevance as an important metabolic factor. All of these observations warranted further experimentation regarding the physiological significance of this growth factor in BAT. Here, we are using a genetic model to achieve a BAT-specific inducible expression selective to the brown adipocyte. Using this model, we demonstrate that VEGF-A can trigger local angiogenesis in BAT. This is associated with an activation of brown adipogenesis, along with an up-regulation of PGC-1α and UCP1, stimulating mitochondrial function in BAT and promoting enhanced thermogenesis in the transgenic mice. In a high-fat diet-exposed mouse model, we find that VEGF-A mediated angiogenesis also facilitates the functional expansion of BAT. As a result, the mice retain a higher level of metabolic fitness, with improved glucose tolerance, lipid clearance, and energy expenditure. These findings highlight a central role of VEGF-A for BAT physiology.
Angiogenesis is an integral component of adipose tissue remodeling [4], even under conditions when weight is stable. While many angiogenic factors were identified in the past decades, VEGF-A is a key factor governing angiogenesis in adipose tissue in both WAT and BAT [3,4,31,32]. Recent studies of VEGF-A were mainly focused on its functions in WAT. The conclusions of all these studies were that VEGF triggers angiogenesis, as reflected by enhanced vascular density in adipose tissue. Phenotypically, enhancing vascular density triggers improvements in systemic metabolism [2,5,7]. In contrast to WAT, BAT does not store triglycerides to the same extend, instead fuels the process of energy consumption to produce heat when necessary [13]. Thus, BAT is the major site for adaptive non-shivering thermogenesis in mammals during environmental challenges, such as reduced temperatures, to maintain body temperature, and protect the organism from hypothermia [33]. The process of thermogenesis takes place in the mitochondria. PGC1α is a transcriptional master regulator for mitochondrial biogenesis and oxidative metabolism in this process in brown adipocytes [34–36]. As a transcriptional co-activator, PGC1α tightly regulates multiple genes that control many aspects of oxidative metabolism, including mitochondrial biogenesis and respiration [34]. A main target of PGC1α is UCP1, one of the key thermogenic proteins in BAT [13,34]. Indeed, the phenomenon of adaptive adrenergic non-shivering thermogenesis in the intact animal is fully dependent on the presence of UCP1. This notion is supported by previous studies indicating that in obese rats and mice that lack UCP1, diet-induced thermogenesis by BAT is impaired [37]. In this study, we show that upon a very short-term VEGF-A induction (5 days), both PGC1α and UCP1 were markedly up-regulated in BAT of transgenic mice. Furthermore, in BAT of diet-induced obese mice, these genes were also induced to a higher level in the transgenic group, further suggesting their role in energy expenditure to combat diet-induced obesity. The underlying mechanisms leading to the significant up-regulation of these mitochondrial genes in the VEGF-A transgenic models are currently under investigation. What is clear is that the VEGF-A-induced expansion of BAT and the VEGF-A-induced beigeing of WAT are acting on a unique population of precursor cells. We have previously demonstrated that cold-induced as well as β3-adrenergic agonist-induced beigeing of subcutaneous WAT involves de novo adipogenesis [38]. We will still have to formally demonstrate that the same de novo differentiation processes takes place with VEGF-A-induced beigeing. If this is so, it suggests a unique role for VEGFR2, the key receptor for VEGF-A, on the precursor cell pool that gives rise to beige and classical brown adipocytes. Indeed, VEGFR2 increases during differentiation of brown adipocytes, and anti-VEGFR2 antibodies increase brown adipocyte apoptosis [30].
Intrigued by the up-regulation of the mitochondrial genes, we further investigated the functional consequences of these effects. We only found a trend towards improved cold tolerance in the transgenic group in short-term cold exposure assays. However, after a longer-term cold acclimation, the VEGF-A transgenic mice showed significantly elevated baseline body temperatures and improved cold tolerance upon further challenge in the absence of food. In further support of this observation, mitochondria isolated from the chronically cold-exposed VEGF-A transgenic mice exhibited dramatically higher oxygen consumption rates (OCRs) in vitro. During the acute cold exposure, the mice can defend the body temperature by shivering or exercise [13]. After a longer-term cold acclimation, non-shivering thermogenesis becomes the predominant way to generate heat and only UCP1 can mediate this adaptive thermogenic response under these conditions [13,39]. Furthermore, during the chronic phase in the cold environment, our UCP1 promoter driven inducible overexpression of VEGF-A may also work in the brite/beige cells of subcutaneous WAT. These locally overexpressed VEGF-A molecules lead to enhanced “brown fat-like” phenotypes in the subcutaneous fat pads by local up-regulation of PGC1α and UCP1 proteins [2]. This in turn further contributes to the thermogenic effects in the transgenic state [2,7,9].
Aside from its potent influence on thermogenesis and energy expenditure with its profound effects on metabolism, BAT also secretes adipokines and hormones, such as leptin, adiponectin, resistin, and triiodothyronine that play potent endocrine effects in metabolism [13]. All these properties make BAT a very important contributor to improvements during metabolic challenges. Supporting this further are manipulations that trigger activation of BAT that also reduce adiposity and protect mice from diet-induced obesity [40,41]. On the other hand, the inhibition of BAT activity leads to pathological expansion. For example, obese BAT suffers from hypoxic conditions that trigger HIF1α induction. HIF1α further initiates multiple transcriptional programs that lead to metabolic dysfunction [4,20,21,42]. In our high-fat diet-induced obese model, BAT in the VEGF-A transgenic group is more functional than in the control group, as indicated by reduced HIF1α levels. Improved functionality is also apparent by higher levels of UCP1 and PGC1α gene expression compared with the controls. Histologically, brown adipocytes in the control mice display a “whitening” phenotype characterized by larger and more often unilocular lipid droplets after HFD exposure. In contrast, brown fat cells in the VEGF-A transgenic group contained smaller and more multilocular lipid droplets. All these phenomena demonstrate that BAT in VEGF-A transgenic mice is an indicative that BAT undergoes a “healthier” expansion under HFD.
As a “leaner” phenotype of BAT prevails, VEGF-A transgenic mice gained slightly less overall body weight, likely due to the smaller size of WAT. Intriguingly, in contrast to WAT, BAT in these mice was larger than the control group. This finding is in agreement with a recent report that VEGF-A increases brown fat cell survival and proliferation in vitro [30]. The effect is mainly through activation of VEGF receptor 2 that is uniquely overexpressed in brown adipocytes upon VEGF-A stimulation [30].
Diet-induced obesity leads to systemic insulin resistance [4,42]. The consequence of the increased free fatty acid release is an elevated lipotoxicity in other tissues, particularly in liver and muscle [43,44]. Indeed, we found impaired glucose tolerance, increased circulating triglyceride, and large lipid droplets in the liver tissue after HFD challenging. However, the VEGF-A transgenic mice showed normal glucose tolerance, lower lipotoxicity, and normal liver histology. Since our BAT-specific VEGF-A overexpression has no angiogenic effects in the liver, we believe that these metabolically beneficial effects are direct effects of activation of BAT by local VEGF-A.
In conclusion, our results demonstrate a direct role for VEGF signaling in brown adipose tissue physiology. A greater understanding of the molecular mechanism by which VEGF-A manipulates BAT activity will further help to identify new strategies to activate BAT for the goal of reducing obesity and its associated metabolic dysfunction. While the important role of BAT in rodents is well established and there is growing appreciation for the dynamic nature of browning/beigeing in human WAT as well, the potential contributions of the phenomenon toward overall energy homeostasis in humans still remain to be better understood in quantitative terms.
Acknowledgments
We thank Dr. Robert A. Koza at the Pennington Biomedical Research Center for the UCP1 promoter fragment and Dr. Jeffrey Pollard from the MRC Centre for Reproductive Health at the University of Edinburgh for the TRE-VEGF mice. We also thank Angelica Sifuentes, Amy Song, and Steven Connell in the Scherer laboratory for technical help; John Sheldon from the pathology core facility at the University of Texas (UT) Southwestern for help with histology; Dr. Bob Hammer and the transgenic core facility at UT Southwestern for generating the UCP1-rtTA transgenic mice; the Metabolic Core Unit at UT Southwestern for phenotyping efforts. This work was supported by National Institutes of Health Grants R01-DK55758, R01-DK099110 and P01DK088761 (to P.E.S.). Q.A.W. is supported by a postdoctoral fellowship from the ADA (7-11-MN-47). C.M.K. is supported by was supported by a fellowship from the Juvenile Diabetes Research Foundation International (JDRF 3-2008-130). W.L.H. is supported by National Institutes of Health Grant (R00-DK094973) and AHA Beginning Grant in Aid 12BGI-A8910006.
Conflict of interest
None declared.
References
- 1.Cao Y. Adipose tissue angiogenesis as a therapeutic target for obesity and metabolic diseases. Nature Reviews Drug Discovery. 2010;9:107–115. doi: 10.1038/nrd3055. [DOI] [PubMed] [Google Scholar]
- 2.Sun K., Wernstedt-Asterholm I., Kusminski C.M., Bueno A.C., Wang Z.V., Pollard J.W. Dichotomous effects of VEGF-A on adipose tissue dysfunction. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:5874–5879. doi: 10.1073/pnas.1200447109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cao Y. Angiogenesis modulates adipogenesis and obesity. The Journal of Clinical Investigation. 2007;117:2362–2368. doi: 10.1172/JCI32239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun K., Kusminski C.M., Scherer P.E. Adipose tissue remodeling and obesity. The Journal of Clinical Investigation. 2011;121:2094–2101. doi: 10.1172/JCI45887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sung H.K., Doh K.O., Son J.E., Park J.G., Bae Y., Choi S. Adipose vascular endothelial growth factor regulates metabolic homeostasis through angiogenesis. Cell Metabolism. 2013;17:61–72. doi: 10.1016/j.cmet.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 6.Neufeld G., Cohen T., Gengrinovitch S., Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. Federation of American Societies for Experimental Biology Journal. 1999;13:9–22. [PubMed] [Google Scholar]
- 7.Elias I., Franckhauser S., Ferre T., Vila L., Tafuro S., Munoz S. Adipose tissue overexpression of vascular endothelial growth factor protects against diet-induced obesity and insulin resistance. Diabetes. 2012;61:1801–1813. doi: 10.2337/db11-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yilmaz M., Hotamisligil G.S. Damned if you do, damned if you don't: the conundrum of adipose tissue vascularization. Cell Metabolism. 2013;17:7–9. doi: 10.1016/j.cmet.2012.12.014. [DOI] [PubMed] [Google Scholar]
- 9.Elias I., Franckhauser S., Bosch F. New insights into adipose tissue VEGF-A actions in the control of obesity and insulin resistance. Adipocyte. 2013;2:109–112. doi: 10.4161/adip.22880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nedergaard J., Cannon B. UCP1 mRNA does not produce heat. Biochimica et Biophysica Acta. 2013;1831:943–949. doi: 10.1016/j.bbalip.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 11.Rosen E.D., Spiegelman B.M. What we talk about when we talk about fat. Cell. 2014;156:20–44. doi: 10.1016/j.cell.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xue Y., Petrovic N., Cao R., Larsson O., Lim S., Chen S. Hypoxia-independent angiogenesis in adipose tissues during cold acclimation. Cell Metabolism. 2009;9:99–109. doi: 10.1016/j.cmet.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 13.Cannon B., Nedergaard J. Brown adipose tissue: function and physiological significance. Physiological Reviews. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 14.Bartelt A., Bruns O.T., Reimer R., Hohenberg H., Ittrich H., Peldschus K. Brown adipose tissue activity controls triglyceride clearance. Nature Medicine. 2011;17:200–205. doi: 10.1038/nm.2297. [DOI] [PubMed] [Google Scholar]
- 15.Nedergaard J., Bengtsson T., Cannon B. Unexpected evidence for active brown adipose tissue in adult humans. American Journal of Physiology. Endocrinology Metabolism. 2007;293:E444–E452. doi: 10.1152/ajpendo.00691.2006. [DOI] [PubMed] [Google Scholar]
- 16.Muzzin P. The uncoupling proteins. Annales d'endocrinologie (Paris) 2002;63:106–110. [PubMed] [Google Scholar]
- 17.Golozoubova V., Cannon B., Nedergaard J. UCP1 is essential for adaptive adrenergic nonshivering thermogenesis. American Journal of Physiology. Endocrinology and Metabolism. 2006;291:E350–E357. doi: 10.1152/ajpendo.00387.2005. [DOI] [PubMed] [Google Scholar]
- 18.Nedergaard J., Golozoubova V., Matthias A., Asadi A., Jacobsson A., Cannon B. UCP1: the only protein able to mediate adaptive non-shivering thermogenesis and metabolic inefficiency. Biochimica et Biophysica Acta. 2001;1504:82–106. doi: 10.1016/s0005-2728(00)00247-4. [DOI] [PubMed] [Google Scholar]
- 19.Rim J.S., Kozak L.P. Regulatory motifs for CREB-binding protein and Nfe2l2 transcription factors in the upstream enhancer of the mitochondrial uncoupling protein 1 gene. Journal of Biological Chemistry. 2002;277:34589–34600. doi: 10.1074/jbc.M108866200. [DOI] [PubMed] [Google Scholar]
- 20.Halberg N., Khan T., Trujillo M.E., Wernstedt-Asterholm I., Attie A.D., Sherwani S. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Molecular and Cellular Biology. 2009;29:4467–4483. doi: 10.1128/MCB.00192-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun K., Halberg N., Khan M., Magalang U.J., Scherer P.E. Selective inhibition of hypoxia-inducible factor 1alpha ameliorates adipose tissue dysfunction. Molecular and Cellular Biology. 2013;33:904–917. doi: 10.1128/MCB.00951-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin E.Y., Li J.F., Bricard G., Wang W., Deng Y., Sellers R. Vascular endothelial growth factor restores delayed tumor progression in tumors depleted of macrophages. Molecular Oncology. 2007;1:288–302. doi: 10.1016/j.molonc.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kusminski C.M., Holland W.L., Sun K., Park J., Spurgin S.B., Lin Y. MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nature Medicine. 2012;18:1539–1549. doi: 10.1038/nm.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rogers G.W., Brand M.D., Petrosyan S., Ashok D., Elorza A.A., Ferrick D.A. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS One. 2011;6:e21746. doi: 10.1371/journal.pone.0021746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tonello C., Giordano A., Cozzi V., Cinti S., Stock M.J., Carruba M.O. Role of sympathetic activity in controlling the expression of vascular endothelial growth factor in brown fat cells of lean and genetically obese rats. FEBS Letters. 1999;442:167–172. doi: 10.1016/s0014-5793(98)01627-5. [DOI] [PubMed] [Google Scholar]
- 26.Asano A., Kimura K., Saito M. Cold-induced mRNA expression of angiogenic factors in rat brown adipose tissue. The Journal of Veterinary Medicine Science. 1999;61:403–409. doi: 10.1292/jvms.61.403. [DOI] [PubMed] [Google Scholar]
- 27.Asano A., Morimatsu M., Nikami H., Yoshida T., Saito M. Adrenergic activation of vascular endothelial growth factor mRNA expression in rat brown adipose tissue: implication in cold-induced angiogenesis. The Biochemical Journal. 1997;328(Pt 1):179–183. doi: 10.1042/bj3280179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fredriksson J.M., Lindquist J.M., Bronnikov G.E., Nedergaard J. Norepinephrine induces vascular endothelial growth factor gene expression in brown adipocytes through a beta-adrenoreceptor/cAMP/protein kinase A pathway involving Src but independently of Erk1/2. Journal of Biological Chemistry. 2000;275:13802–13811. doi: 10.1074/jbc.275.18.13802. [DOI] [PubMed] [Google Scholar]
- 29.Fredriksson J.M., Nikami H., Nedergaard J. Cold-induced expression of the VEGF gene in brown adipose tissue is independent of thermogenic oxygen consumption. FEBS Letters. 2005;579:5680–5684. doi: 10.1016/j.febslet.2005.09.044. [DOI] [PubMed] [Google Scholar]
- 30.Bagchi M., Kim L.A., Boucher J., Walshe T.E., Kahn C.R., D'Amore P.A. Vascular endothelial growth factor is important for brown adipose tissue development and maintenance. Federation of American Societies for Experimental Biology Journal. 2013;27:3257–3271. doi: 10.1096/fj.12-221812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Q.X., Magovern C.J., Mack C.A., Budenbender K.T., Ko W., Rosengart T.K. Vascular endothelial growth factor is the major angiogenic factor in omentum: mechanism of the omentum-mediated angiogenesis. The Journal of Surgical Research. 1997;67:147–154. doi: 10.1006/jsre.1996.4983. [DOI] [PubMed] [Google Scholar]
- 32.Fukumura D., Ushiyama A., Duda D.G., Xu L., Tam J., Krishna V. Paracrine regulation of angiogenesis and adipocyte differentiation during in vivo adipogenesis. Circulation Research. 2003;93:e88–e97. doi: 10.1161/01.RES.0000099243.20096.FA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith R.E. Thermoregulatory and adaptive behavior of brown adipose tissue. Science. 1964;146:1686–1689. doi: 10.1126/science.146.3652.1686. [DOI] [PubMed] [Google Scholar]
- 34.Spiegelman B.M. Transcriptional control of energy homeostasis through the PGC1 coactivators. Novartis Foundation Symposium. 2007;286:3–6. [discussion: 6–12, 162–163, 196–203] [PubMed] [Google Scholar]
- 35.Puigserver P., Wu Z., Park C.W., Graves R., Wright M., Spiegelman B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 36.Puigserver P., Spiegelman B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocrine Reviews. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- 37.Kozak L.P. Brown fat and the myth of diet-induced thermogenesis. Cell Metabolism. 2010;11:263–267. doi: 10.1016/j.cmet.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Q.A., Tao C., Gupta R.K., Scherer P.E. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nature Medicine. 2013;19:1338–1344. doi: 10.1038/nm.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Golozoubova V., Hohtola E., Matthias A., Jacobsson A., Cannon B., Nedergaard J. Only UCP1 can mediate adaptive nonshivering thermogenesis in the cold. Federation of American Societies for Experimental Biology Journal. 2001;15:2048–2050. doi: 10.1096/fj.00-0536fje. [DOI] [PubMed] [Google Scholar]
- 40.Guerra C., Koza R.A., Yamashita H., Walsh K., Kozak L.P. Emergence of brown adipocytes in white fat in mice is under genetic control. Effects on body weight and adiposity. The Journal of Clinical Investigation. 1998;102:412–420. doi: 10.1172/JCI3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ghorbani M., Claus T.H., Himms-Hagen J. Hypertrophy of brown adipocytes in brown and white adipose tissues and reversal of diet-induced obesity in rats treated with a beta3-adrenoceptor agonist. Biochemical Pharmacology. 1997;54:121–131. doi: 10.1016/s0006-2952(97)00162-7. [DOI] [PubMed] [Google Scholar]
- 42.Sun K., Scherer P.E. Springer-Verlag; Berlin Heidelberg: 2010. Adipose tissue dysfunction: a multistep process. [Google Scholar]
- 43.Unger R.H., Clark G.O., Scherer P.E., Orci L. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochimica et Biophysica Acta. 2010;1801:209–214. doi: 10.1016/j.bbalip.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 44.Unger R.H., Scherer P.E. Gluttony, sloth and the metabolic syndrome: a roadmap to lipotoxicity. Trends in Endocrinology and Metabolism. 2010;21:345–352. doi: 10.1016/j.tem.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]