

Abstract
Brain mitochondrial activity is centrally involved in the central control of energy balance. When studying mitochondrial functions in the brain, however, discrepant results might be obtained, depending on the experimental approaches. For instance, immunostaining experiments and biochemical isolation of organelles expose investigators to risks of false positive and/or false negative results. As an example, the functional presence of cannabinoid type 1 (CB1) receptors on brain mitochondrial membranes (mtCB1) was recently reported and rapidly challenged, claiming that the original observation was likely due to artifact results. Here, we addressed this issue by directly comparing the procedures used in the two studies. Our results show that the use of appropriate controls and quantifications allows detecting mtCB1 receptor with CB1 receptor antibodies, and that, if mitochondrial fractions are enriched and purified, CB1 receptor agonists reliably decrease respiration in brain mitochondria. These data further underline the importance of adapted experimental procedures to study brain mitochondrial functions.
Keywords: Brain bioenergetics, CB1 receptor, mitochondria, electron microscopy, antibodies, organelle purification
Abbreviations: CB1, cannabinoid type 1 receptor; BSA, bovine serum albumin; DAB–Ni, Ni-intensified 3,3ʹ-diaminobenzidine–4HCl; DMSO, dimethyl sulfoxide; WT, wild-type; KO, knock-out; Slp2, stomatin-like protein 2; WIN, WIN55,212-2; SDHA, succinate dehydrogenase a; LDHa, lactate dehydrogenase a
1. Introduction
Mitochondrial oxidative phosphorylation (OXPHOS) converts most of the energy contained in nutrients into ATP, required for cellular endergonic reactions. These key organelles also regulate several other important physiological processes, including calcium homeostasis, oxidative stress, apoptosis, and steroidogenesis. Mitochondrial structure and functions are constantly adjusted to maintain cellular metabolic homeostasis. Defects in mitochondrial functions have been associated to different pathologies, including obesity, type-2 diabetes, and neurodegenerative diseases [1–4]. In peripheral tissues, insulin-resistance is associated with mitochondrial dysfunctions in myocytes, hepatocytes, adipocytes, and islets β-cells [2,5]. Mitochondria within the central nervous system are also important for coordinating energy intake and expenditure. For example, mitochondrial dynamics within specific neurons of the arcuate nucleus of the hypothalamus, a brain region involved in whole-body energy homeostasis, regulate diet-induced obesity [3,4].
Therefore, the study of brain mitochondrial activity and regulation became an important step to fully understand brain functioning in general and the central control of the body energy balance in particular [6,7]. However, these types of studies in the brain present intrinsic specific difficulties that should be taken into account. Ex vivo bioenergetics studies are traditionally conducted on relatively homogeneous tissues and organs (e.g. heart, liver, skeletal muscle, or adipose tissue) that are formed by few types of cells. Conversely, the brain is a highly heterogeneous organ formed by extremely diverse cell types, which are themselves more complex than any other cellular element in the body. For instance, neurons and glial cells are continuously classified into new subgroups by the identification of specific markers, morphological features, and functions. This complexity is also reflected in the mitochondrial functions in the brain, rendering bioenergetics experiments particularly delicate [8]. Thus, neuroanatomical and biochemical features of brain mitochondria need to be carefully evaluated to draw firm conclusions on the mechanisms regulating brain bioenergetics. Such a complexity exposes investigators to the potential risk of false positive and/or negative results, and contrasting conclusions among different experimental approaches are commonly observed. Recently, the case of the functional presence of the seven transmembrane G protein coupled type-1 cannabinoid receptor (CB1) at mitochondrial membrane [9] emerged as a prototypical example of these potential controversies [10].
CB1 receptors were identified as the molecular targets of exogenous and endogenous cannabinoids and they are present in different tissues, including brain, liver, skeletal muscle, pancreas, and fat, where their activation contributes to cellular and organism metabolic functions [11,12]. For instance, genetic deficiency or pharmacological blockade of CB1 receptors has been shown beneficial against metabolic alterations, such as obesity and metabolic syndrome [12–15].
The impact of exogenous cannabinoids on mitochondrial processes has been proposed since the Seventies [16], long before the discovery of cannabinoid receptors [17]. With the identification of cannabinoid receptors as typical plasma membrane receptors [17,18], the mitochondrial effects of cannabinoids were interpreted either as indirect consequences of plasma membrane CB1 receptors activation, or as unspecific alterations of mitochondrial membranes by these lipid compounds [19,20]. Our team recently challenged this concept by demonstrating that CB1 receptors are also present in brain mitochondria (called thereafter as mtCB1 receptors) [9]. The specific localization of CB1 receptors within CA1 hippocampal mitochondrial membranes was first established by using immunogold electron microscopy. Approximately 30% of wild-type hippocampal mitochondria were observed to contain CB1 receptors labeling, whereas only 3% of mitochondria showed non-specific immunoreactivity in CB1-KO mice [9]. Moreover, different exogenous agonists of CB1 receptors [Δ9-tetrahydrocannabinol (THC), WIN55,212-2, HU210, or JZL195 (an inhibitor of the two enzymes responsible for the degradation of endocannabinoids)] dose-dependently decreased respiration rates of purified brain mitochondria from wild-type mice, but not from CB1-KO mice [9]. These data suggested a direct link between (endo)cannabinoids, CB1 receptors, and brain cellular bioenergetics, which could constitute a new target for the treatment of metabolic disorders.
However, using different organelle isolation and immunohistochemistry techniques, a recent study challenged these results by showing that (i) CB1 receptor immunoreactivity was still present in CB1-KO mitochondria and (ii) WIN55,212-2 did not affect respiration rates of “mitochondria enriched fractions” [10]. These discrepant results illustrate the difficulty of identifying and characterizing brain mitochondrial proteins. For instance, antibodies can detect off-target epitopes, and biochemical purification procedures may lack sufficient power to reliably isolate the organelles of interest.
In this study, we hypothesized that the discrepant results obtained in different laboratories might be caused by different experimental approaches, further underlying the specific cautions that should be taken when addressing brain bioenergetics processes. Thus, we performed a series of experiments to directly compare the different procedures used in the two studies [9,10]. Three main aspects were considered: (i) electron microscopic immunodetection of CB1 receptors and quantification of staining in wild-type and CB1-KO mice adopting the methods used in [9] and [10], followed by different quantification procedures; (ii) Western immunoblotting and immunoprecipitation experiments to further test the presence of the CB1 receptor in mitochondria; (iii) comparison of purity, respiration, and cannabinoid effects on mitochondrial preparations obtained using the methods described by [9] and by [10], respectively.
2. Material and methods
2.1. Mice
Experiments were approved by the Committee on Animal Health and Care of INSERM and the French Ministry of Agriculture and Forestry (authorization number 3306369) and by the Committee of Ethics for Animal Welfare of the University of the Basque Country (CEBA/93/2010/GRANDESMORENO). Mice were housed under standard conditions (food and water ad libitum; 12h/12h light/dark cycle, light on 7 a.m.; experiments were performed between 9 a.m. and 12 a.m.). Wild-type (CB1-WT) and CB1-KO littermate female mice (2–4 months old) were obtained, bred, and genotyped as described [21].
2.2. Electron microscopy assays
Mice were deeply anesthetized by intraperitoneal injection of ketamine/xylazine (80/10 mg/kg body weight) and were transcardially perfused at room temperature (RT, 20–25 °C) with phosphate buffered saline (0.1 M PBS, pH 7.4) for 20 s, followed by the fixative solution made up of 4% formaldehyde (freshly depolymerized from paraformaldehyde), 0.2% picric acid, and 0.1% glutaraldehyde in phosphate buffer (0.1 M PB, pH 7.4) for 10–15 min. Then, brains were removed from the skull and post-fixed in the fixative solution for approximately 1 week at 4 °C. Afterwards, brains were stored at 4 °C in 1:10 diluted fixative solution until used.
2.2.1. Immunogold electron microscopy
Coronal hippocampal vibrosections were cut at 50 μm and collected in 0.1 M PB (pH 7.4) at RT. Sections were preincubated in a blocking solution of 10% bovine serum albumin (BSA), 0.1% sodium azide, and 0.02% saponin prepared in Tris–HCl buffered saline (TBS 1×, pH 7.4) for 30 min at RT. A pre-embedding silver-intensified immunogold method was used for the localization of CB1 protein. Hippocampal sections were incubated with a primary goat anti-CB1 antibody binding to a 31 amino acids sequence of the C-terminus (CB1 C-ter31; 2 μg/ml; Cat. N.: CB1-Go-Af450-1; Frontier Institute; Japan) in 10% BSA/TBS containing 0.1% sodium azide and 0.004% saponin on a shaker for 1 day at RT.
After several washes in 1% BSA/TBS, tissue sections were incubated in a secondary 1.4 nm gold-labeled rabbit anti-goat IgG (Fab' fragment, 1:100, Nanoprobes Inc., Yaphank, NY, USA) in 1% BSA/TBS with 0.004% saponin on a shaker for 4 h at RT. Thereafter, the tissue was washed in 1% BSA/TBS overnight at 4 °C and post-fixed in 1% glutaraldehyde in TBS for 10 min at RT. Following washes in double-distilled water, gold particles were silver-intensified with a HQ Silver kit (Nanoprobes Inc., Yaphank, NY, USA) for about 12 min in the dark and then washed in 0.1 M PB (pH 7.4). Stained sections were osmicated (1% osmium tetroxide, OsO4, in 0.1 M PB pH 7.4, 20 min), dehydrated in graded alcohols to propylene oxide, and plastic-embedded flat in Epon 812. Ultrathin sections of 80 nm were collected on mesh nickel grids, stained with 2.5% lead citrate for 20 min, and examined in a Philips EM208S electron microscope. Tissue preparations were photographed by using a digital camera coupled to the electron microscope. Figure compositions were made at 300 dots per inch (dpi). Labeling and minor adjustments in contrast and brightness were made using Adobe Photoshop (CS, Adobe Systems, San Jose, CA, USA).
2.2.2. Semi-quantification of mtCB1 receptor immunostaining using immunogold
Hippocampal CA1 sections from each animal genotype (n = 3 each) showing good and reproducible silver-intensified gold particles were cut at 80 nm. Electron micrographs (18,000×) were taken from grids with similar labeling intensity indicating that selected areas were at the same depth. To avoid false negatives, only ultrathin sections in the first 1.5 μm from the surface were examined. To determine the proportion of CB1-positive mitochondria, the number of labeled mitochondria was normalized to the total number of mitochondria in the images. The percentages of CB1 immunopositive mitochondria in CB1-WT and CB1-KO mice were calculated taking into account only particles (at least one) on mitochondrial membrane segments far away from other membranes (distance ≥80 nm). Image-J (version 1.36) was used to measure this distance. Graphs and statistical analyses were performed using GraphPad software (version 5.0). Results were expressed as means of independent data points ± S.E.M. Statistical significance between groups was tested using unpaired Student's t test (two-sided); p < 0.05.
2.2.3. Pre-embedding DAB–Ni immunoperoxidase method for electron microscopy
Coronal hippocampal vibrosections were cut at 40 μm and collected in 0.1 M PB at RT. Sections were preincubated in a blocking solution of 10% BSA, 0.1% sodium azide, and 0.02% saponin in TBS for 30 min at RT. Then, they were incubated with the same primary goat anti-CB1 antibody described above (CB1 C-ter31; 2 μg/ml; Cat. N.: CB1-Go-Af450-1; Frontier Institute; Japan) prepared in the blocking solution with 0.004% saponin, on a shaker for 2 days at 4 °C. After several washes in 1% BSA/TBS, tissue sections were incubated in a secondary biotinylated horse anti-goat IgG (1:200, Vector Laboratories, Burlingame, CA, USA) prepared in the solution of the primary antibody for 1 h on a shaker at RT. After washing, sections were processed by a conventional avidin–biotin peroxidase complex method (ABC, Elite, Vector Laboratories, Burlingame, CA, USA). Tissue was incubated in the avidin–biotin complex (1:50) prepared in the washing solution for 1 h at RT. Then, sections were washed and incubated with 0.05% diaminobenzidine in 0.1 M PB containing 0.01% hydrogen peroxide and a few drops of nickel solution (Vector Laboratories, Burlingame, CA, USA) for 5 min at RT. After washing in 0.1 M PB, tissue was osmicated (1% OsO4 in 0.1 M PB, 20 min), dehydrated in graded alcohols to propylene oxide and plastic-embedded flat in Epon 812. As described above, 80-nm ultrathin sections were collected on mesh nickel grids, stained with lead citrate, and examined in a Philips EM208S electron microscope. Tissue preparations were photographed by using a digital camera coupled to the electron microscope and figure compositions were made at 300 dots per inch (dpi). Labeling and minor adjustments in contrast and brightness were made using Adobe Photoshop (CS, Adobe Systems, San Jose, CA, USA).
2.2.4. Semi-quantification of mtCB1 receptor immunostaining using DAB–Ni immunoperoxidase
Hippocampal CA1 sections showing good and reproducible DAB–Ni immunoperoxidase staining were cut at 80 nm. Electron micrographs (18,000×) were taken from grids containing DAB–Ni immunoperoxidase staining. As above, all of them showed a similar staining intensity and only ultrathin sections in the first 1.5 μm from the surface of the tissue block were examined. Positive labeling was considered when DAB–Ni immunoprecipitation was observed inside or surrounding the membrane of mitochondria. Graphs and statistical analyses were performed using GraphPad software (version 5.0). Results were expressed as means of independent data points ± S.E.M. Statistical significance between groups was tested using chi-square test; p < 0.05.
2.3. Isolation of mitochondrial fractions
For isolation of purified brain mitochondria, adult wild-type mice were killed by cervical dislocation, and the brains were immediately dissected on ice. Mitochondria were then isolated and purified using discontinuous Ficoll gradient as previously described [8,9,22,23]. In detail, mice brain were extracted in ice-cold isolation buffer (sucrose 250 mM, Tris 10 mM, EDTA 1 mM, NaF 2 mM, and pH 7.6) and homogenized using a Teflon potter. Homogenates were centrifuged at 1,500 g for 5 min (4° C). The supernatant was then centrifuged at 12,500 g (4 °C). The pellet was collected and the cycle of centrifugation was repeated. To purify mitochondria, the final pellet was resuspended in 400 μL of isolation buffer, layered on top of a discontinuous Ficoll gradient (10% and 7% fractions) and centrifuged at 100,000 g for 1 h (4 °C). The pellet was finally resuspended in 100 μL of isolation buffer.
In parallel, the procedure used by [10] was repeated to prepare mitochondrial-enriched fractions from brain samples obtained on the same days of experiments. After death, brains were extracted, homogenized in ice-cold isolation buffer (mannitol 225 mM, sucrose 75 mM, HEPES 5 mM, EGTA 1 mM, bovine serum albumin 1 mg/ml, protease type VIII 0.3125 mg/ml, and pH 7.4) and centrifuged at 2,000 g during 5 min. The pellet, including the synaptosomal layer, was resuspended in isolation buffer containing 0.02% digitonin and centrifuged at 12,000 g for 10 min. The pellet, without the synaptosomal layer, was resuspended in isolation buffer and centrifuged at 12,000 g during 10 min. This last pellet was finally resuspended in 100 μL of resuspension buffer (mannitol 225 mM, sucrose 75 mM, HEPES 5 mM, and pH 7.4). All experiments using freshly isolated brain mitochondria were performed within 3 h following purification.
2.4. Measurements of protein content
Samples' protein content was measured using the Pierce BCA protein assay kit (Thermo Scientific, France).
2.5. Immunoprecipitation
Freshly purified brain mitochondria were resuspended in PBS (5 mg/ml) supplemented with a protease inhibitor cocktail (Roche, France) and 2 mM NaF, and solubilized with 1% lauryl maltoside for 30 min (4 °C). Proteins were incubated with (i) primary rabbit anti-CB1 antibody binding to a 12 amino acids sequence of the C-terminus (CB1 C-ter12; 1:10; Cat. N.: 10006590; Cayman, USA), (ii) primary rabbit anti-CB1 antibody binding to a 14 amino acids sequence of the N-terminus (CB1 N-ter14; 1:10; Cat. N.: AB5636P; Millipore; USA), and (iii) primary goat anti-CB1 antibody binding to a 31 amino acids sequence of the C-terminus (CB1 C-ter31; 1:10; Cat. N.: CB1-Go-Af450-1; Frontier Institute; Japan) overnight (4 °C). Protein A/G PLUS-Agarose beads (Santa Cruz, USA) were then added and incubation continued for 4 h (4 °C). Elution was carried out using glycine buffer (0.2 M glycine, 0.05% lauryl maltoside, pH 2.5) and samples were processed for Western immunoblotting.
2.6. Western immunoblotting
Proteins were separated on Tris–glycine 7% or 10% acrylamide gels and transferred to PVDF membranes. Membranes were blocked in 5% milk powder in TBS–Tween 20 (0.05% Tween 20). Proteins were immunodetected using antibodies against SDHA (Abcam, 1:20,000, 1 h, RT), synaptophysin (Millipore, 1:20,000, 1 h, RT), LDHa (Santa Cruz, 1:500, 1 h, RT), Tom20 (Santa Cruz, 1:2,000, 1 h, RT), Slp2 (Santa Cruz, 1:1,000, 2 h, RT), and β-actin (Sigma–Aldrich, 1:50,000, 1 h, RT). Antisera directed against the C-terminus of the CB1 receptor (CB1 C-ter12; Cayman, 1:200, overnight, 4 °C and CB1 C-ter31; Frontier Institute, 1:200, overnight, 4 °C) and one antiserum directed against the N-terminus of the CB1 receptor (CB1 N-ter14; Millipore, 1:200, overnight, 4 °C) were applied on membranes for immunoprecipitation assays. Then, membranes were washed and incubated with appropriate secondary HRP-coupled antibodies (1 h, RT). For immunoprecipitation assays, an HRP-coupled light-chain specific anti-rabbit IgG (for CB1 C-ter12 and CB1 N-ter14) was used. Finally, HRP signal was revealed using the ECL plus reagent (Amersham, France) and detected by the Bio-Rad Quantity One system (Bio-Rad, France).
2.7. Oxygen consumption measurements
Respiration rates of mitochondrial preparations were monitored at 37 °C in a glass chamber equipped with a Clark oxygen electrode (Hansatech, UK). Purified mitochondria or mitochondria-enriched fractions (75–100 μg) were suspended in 500 μL of respiration buffer (75 mM mannitol, 25 mM sucrose, 10 mM KCl, 10 mM Tris–HCl, pH 7.4, and 50 mM EDTA) in the chamber. A stock solution of 500 μM WIN55,212-2 in dimethyl sulfoxyde (DMSO) was stored at −20 °C. WIN55,212-2 or DMSO was then diluted in respiration buffer and directly added into the chamber (1:10,000 for WIN55,212-2 50 nM and 1:5,000 for WIN55,212-2 100 nM). Pyruvate (5 mM), malate (2 mM), and ADP (5 mM) were successively directly added into the chamber to follow mitochondrial respiration. Graphs and statistical analyses were performed using GraphPad software (version 5.0). Results were expressed as means of independent data points ± S.E.M. Respiration rates were analyzed by unpaired T test. Two groups were considered statistically different when p < 0.05.
3. Results
3.1. Detection of CB1 receptors in brain mitochondrial membranes by different approaches
3.1.1. Electron microscopy
Specific localization of CB1 receptors within hippocampal mitochondrial membranes was first established using semi-quantitative immunogold electron microscopy [9]. In that study, CB1 receptor immunolabeling was quantified by considering all mitochondrial gold particles. Hence, approximately 30% of wild-type hippocampal mitochondrial sections displayed CB1 receptors labeling, whereas only 3% of mitochondrial sections showed non-specific immunoreactivity in CB1-KO mice [9]. However, several mitochondrial sections are in close vicinity of linear densities, such as plasma and endoplasmic reticulum membranes, making it possible that gold particles present on these structures were wrongly identified as mitochondrial labeling. Thus, to avoid misidentification of mitochondrial CB1 receptor labeling, we performed a more strict quantification, now considering only mitochondrial gold particles located at more than 80 nm (the average diameter of silver-intensified gold particles was ≈30 nm) from these linear densities (Figure 1A–G). This approach revealed that 21.6 ± 1.4% of CB1-WT mitochondrial sections were CB1 receptor immunopositive, whereas only 2.9 ± 0.6% of mitochondrial sections displayed CB1 receptor labeling in CB1-KO mice (Figure 1I).
Figure 1.
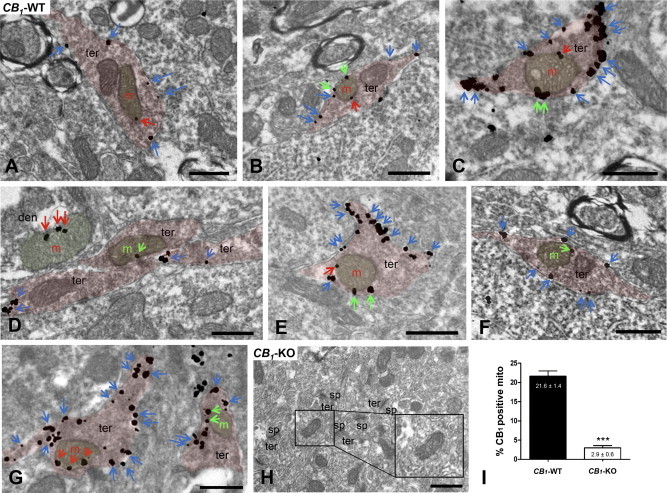
Immunoelectron detection of CB1 receptors in neuronal CA1 hippocampal mitochondria by a goat anti-CB1 C-ter31 antibody combined with a pre-embedding silver-intensified immunogold method. In CB1-WT (A–G), CB1 immunoparticles are on presynaptic terminal (ter) membranes (blue arrows) and on mitochondrial (m) membrane segments close to (distance ≤80 nm; green arrows) or far away from (distance ≥80 nm; red arrows) other membranes. Note in (D), red arrows pointing to silver metals on a dendritic (den) mitochondria. (H) The CB1 pattern is abolished in CB1-KO. Scale bars: 0.5 μm. sp: dendritic spines. (I) Semi-quantitative analyses of the proportion of CB1 immunolabeled mitochondria in CA1 hippocampi from CB1-WT and CB1-KO mice. Mitochondria with particles distant from other membranes (≥80 nm) were only considered. Data are expressed as mean ± S.E.M. ***p < 0.001 as compared to WT.
Using a more sensitive pre-embedding immunoperoxidase (ABC) method with Ni-intensified 3,3ʹ-diaminobenzidine–4HCl (DAB–Ni) as a chromogen, Morozov et al. [10] showed that the CB1 receptor mitochondrial immunoreactivity was also present in CB1-KO brain tissue. However, these results are rather difficult to interpret, since no quantification was provided in order to compare the CB1 receptors labeling between wild-type and CB1-KO mice [10]. Therefore, we repeated the ABC method to further determine the specificity of CB1 receptors labeling on mitochondria. Similar to our previous results obtained with immunogold electron microscopy [9], the ABC method revealed that 27.8% ± 2.2% of wild-type mitochondrial sections showed CB1 receptors immunoreactivity (Figure 2A–C and H), whereas 9.5% ± 1.0% of CB1-KO mitochondrial sections displayed unspecific labeling (Figure 2D, E, and H). These results confirm that low levels of unspecific mitochondrial CB1 receptor immunoreactivity are still present in CB1-KO brain tissue, although the diffuse nature of DAB–Ni staining impedes higher resolution discrimination between different cellular membrane elements. In addition, using the DAB–Ni method, the background staining levels were higher than those obtained using immunogold procedures: approximately 3% with immunogold [9] and approximately 9% with the ABC method in the present study, respectively (Figure 2H). The unspecific CB1 immunoreactivity was also evaluated in CB1-KO tissues without primary antibody: 6.3% ± 1.0% of CB1-KO mitochondria were still labeled using this procedure (Figure 2F–H). Despite the fact that semi-quantification analyses confirmed that approximately 20–25% of wild-type brain mitochondrial sections contained mtCB1 receptors (Figures 1 and 2I), these results indicate that, due to its diffuse staining and its higher levels of background staining, the DAB–Ni method is less suitable to evaluate the detection of the low levels of mtCB1 receptors as compared with the immunogold technique.
Figure 2.
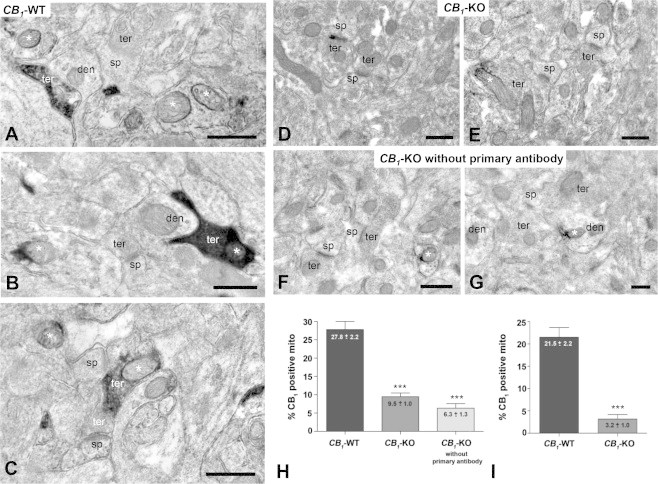
Ultrastructural localization of CB1 receptors in mitochondrial sections with the immunoperoxidase DAB–Ni method using a goat anti-CB1 C-ter31 antibody. (A–C) In CB1-WT mouse, CB1 immunoreactivity was observed in mitochondrial sections (asterisks), some of them were contained in CB1-positive synaptic terminals (black immunoreaction product). (D and E) Absence of CB1 immunodeposits in mitochondrial sections and synaptic terminals of CB1-KO mouse. Only some scattered mitochondrial sections display DAB–Ni precipitates (asterisk in E). (F and G) Unspecific immunodeposits on mitochondrial sections (asterisks) and dendritic profiles of CB1-KO mouse when the primary antibody was omitted. (H) Semi-quantitative analyses of the proportion of CB1 immunopositive mitochondrial sections in CB1-WT and CB1-KO mice, and in CB1-KO mice without primary antibody. (I) Semi-quantitative analyses of the proportion of CB1 immunopositive mitochondrial section in CB1-WT and CB1-KO mice after subtraction of the percentage value obtained in CB1-KO mice without primary antibody. Data are expressed as mean ± S.E.M. ***p < 0.0001 according to chi-square test. ter: presynaptic terminals; den: dendrites; sp: dendritic spines. Scale bars: 0.5 μm.
3.1.2. Immunoprecipitation and Western immunoblotting
Most antibodies display certain levels of unspecific labeling of off-target antigens. Therefore, anti-CB1 antisera could unspecifically react with other mitochondrial proteins. However, our previous and present data showed that it is possible to specifically detect the presence of the CB1 receptors in mitochondria when appropriate control experiments are performed. Figure 3A shows an immunoblotting experiment, in which several unspecific bands were detected. Whereas the number of these bands is variable amongst experiments, we expressly chose to show an experiment, in which several bands were detected to illustrate the need of proper negative controls. Given their presence in CB1-KO tissues (Figure 3A), the large majority of these bands cannot be considered as specific CB1 protein. However, one band at the apparent molecular weight of approximately 50–55 kDa was observed in wild-type, but not in CB1-KO tissue (Figure 3A). This band can be considered as the CB1 receptor protein.
Figure 3.
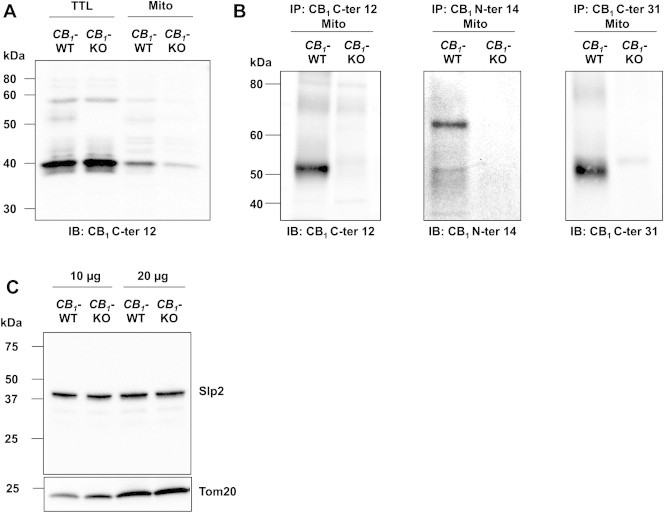
Specific identification of CB1 receptors in purified brain mitochondria by Western blot analyses. (A) Representative immunoblotting of CB1 receptors from CB1-WT and CB1-KO brain lysates, using rabbit anti-CB1 C-ter12 antibody. (B) Representative immunoblotting (three independent experiments) showing that CB1 receptors are specifically immunoprecipitated from wild-type purified brain mitochondria with different CB1 receptor antibodies. CB1 C-ter12: primary rabbit anti-CB1 antibody binding to a 12 amino acids sequence of the C-terminus; CB1 N-ter14: primary rabbit anti-CB1 antibody binding to a 14 amino acids sequence of the N-terminus; CB1 C-ter31: primary goat anti-CB1 antibody binding to a 31 amino acids sequence of the C-terminus. (C) Immunoblot of Slp2 from CB1-WT and CB1-KO brain lysates (10 and 20 μg), Tom20 used as mitochondrial loading control.
To improve the quality of immunoblotting by decreasing the number of unspecific bands, and further to test for the presence of CB1 receptors in brain mitochondria, CB1 proteins were immunoprecipitated from purified brain mitochondria using three different CB1 receptor antisera: one specific for a 12 amino acids sequence within its C-terminus (CB1 C-ter12), one specific for a 14 amino acid sequence at its N-terminus (CB1 N-ter12), and one specific for a 31 amino acid sequence within its C-terminus (CB1 C-ter31), followed by immunoblotting with the same antisera, respectively. Immunoblots showed that all CB1 receptor antisera tested were able to specifically immunoprecipitate mtCB1 receptors from brain purified mitochondria. Both antisera against the CB1 receptor C-terminus (CB1 C-ter12 and CB1 C-ter31) revealed a specific band at approximately ≈52 kDa, corresponding to the known molecular weight of native CB1 receptor [24–26], whereas no band was detected in the immunoprecipitates from CB1-KO mitochondria (Figure 3B). When the antiserum against CB1 receptor N-terminus (CB1 N-ter14) was used, both a band at ≈52 kDa and a band at ≈64 kDa clearly appeared in wild-type immunoprecipitates whereas no band was detected in CB1-KO samples (Figure 3B). The 64 and 52 kDa bands observed with different antibodies could likely represent different forms of CB1 receptors with different post-translational modifications, e.g. glycosylated and non-glycosylated, respectively [26,27]. Altogether, our electron microscopy, immunoprecipitation, and Western immunoblotting results confirmed the specific presence of CB1 receptor in brain mitochondria. They also confirmed that CB1 receptor antibodies, as most antibodies, may react unspecifically and generate some background staining as shown in Ref. [10], illustrating the necessity of using appropriate controls and quantifications to obtain reliable results.
The use of knock-out mice is a powerful tool to control the specificity of antisera: the lack (or significantly lower levels after quantification) of staining in immunohistochemical experiments or the absence of bands in Western immunoblotting approaches are clear indications that the antisera used can detect the specific target protein. However, there is also the possibility that the target protein might be somehow linked to the expression of off-target epitopes of the antisera. In other words, the deletion of the target protein could lead to decreased expression levels of the unspecific target. This is of course a remote possibility that cannot be tested for all possible proteins. However, in some cases, the putative identity of unspecific targets of antibodies is known. For instance, antisera raised against the CB1 receptor have been proposed to recognize the mitochondrial stomatin-like protein 2 (Slp2) [10]. Therefore, we decided to control if CB1-KO brain tissues display a reduction or even an ablation of Slp2 protein. Western immunoblotting experiments using an antiserum against Slp2 revealed no difference in the amount of the protein in brain extracts of wild-type and CB1-KO littermates (Figure 3C), clearly indicating that the deletion of the CB1 gene does not alter the expression levels of a potential off-target of anti-CB1 antisera.
3.2. The effect of the synthetic cannabinoid WIN55,212-2 in different mitochondrial preparations from mouse brain
It is well established that mitochondria are difficult to isolate and purify from brain tissues [8,28]. Simple differential centrifugation protocols usually largely remove nuclei, undisrupted cells, and cytosolic material, resulting in mitochondrial-enriched fractions. However, a further purification step removing synaptosomes and myelin is required to obtain purified mitochondria from brain samples [8,28]. As CB1 receptor is highly expressed at plasma membranes of synaptosomes, a minimal elimination of contamination seems essential to study mitochondrial CB1 receptor. Such purification of brain mitochondria can be done by performing a differential centrifugation protocol followed by a Ficoll density gradient ultracentrifugation step [8,28], as performed in Ref. [9]. This allows obtaining highly purified mitochondria almost totally free of synaptosomes and other non-mitochondrial components [8]. In contrast, Morozov et al. prepared mitochondrial fractions using a different protocol without any purification step [10]. Therefore, we replicated both protocols to assess the purity, the respiration rates, and the effect of WIN55,212-2 on respiration rates in two different brain mitochondrial preparations: (i) purified brain mitochondria [as in Ref. 9] and (ii) mitochondria-enriched brain fractions [as in Ref. 10].
Immunoblots of different cellular components revealed important differences in the levels of purity of mitochondrial fractions obtained through the two different protocols (Figure 4A). Purified brain mitochondrial fractions presented a clear enrichment of SDHA, the subunit A of the component of the electron transport system succinate dehydrogenase. These preparations were almost completely devoid of synaptophysin (a component of synaptic vesicles enriched in synaptosomes) and of the cytosolic proteins LDHa (a subunit of lactate dehydrogenase), and actin (cytoskeleton component) (Figure 4A). In contrast, mitochondria-enriched brain fractions produced preparations poorly enriched in SDHA and highly contaminated with other non-mitochondrial material (Figure 4A). Notably, these results are in agreement with electron micrographic analysis of mitochondria-enriched brain fractions, which also revealed the presence of non-mitochondrial components [10]. These different levels of purity were also demonstrated by the disparity of mitochondrial respiration rates between the two preparations (Figure 4B). Indeed, ADP-stimulated respiration states of purified brain mitochondria were of 696.8 ± 49.4 nmol O2 min−1 mg−1, whereas mitochondria-enriched brain fractions, obtained by strictly following the procedure used in Ref. [10], had respiration rates of 145.2 ± 11.7 nmol O2 min−1 mg−1 (Figure 4B).
Figure 4.
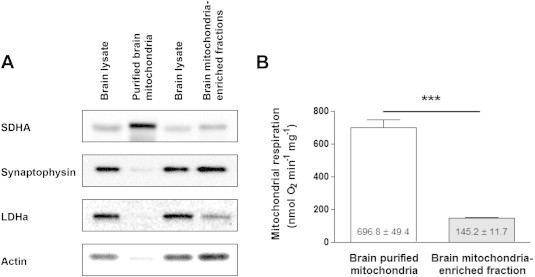
Characterization of two types of brain mitochondrial preparations: purified mitochondria [9] and mitochondria-enriched fractions [10]. (A) Representative immunoblotting showing mitochondrial content (SDHA: succinate dehydrogenase a), synaptosomal content (synaptophysin), cytosolic content (LDHa: lactate dehydrogenase a; actin) in total brain lysate, and mitochondrial fractions (three independent experiments). (B) Basal mitochondrial ADP-stimulated respiration rates in brain purified mitochondria and mitochondria-enriched fraction (n = 6). Data are expressed as mean ± S.E.M. ***p < 0.001 according to unpaired T test.
The effect of the synthetic CB1 agonist WIN55,212-2 on ADP-stimulated mitochondrial respiration was then measured in both mitochondrial preparations. WIN55,212-2 dose-dependently decreased mitochondrial respiration of purified brain mitochondria: significant decreases of 6.6% ± 1.9% at 50 nM and of 19.9% ± 3.9% at 100 nM were observed (Figure 5A). In contrast, WIN55,212-2 did not exert any dose-dependent effect on the respiration rates of mitochondrial-enriched fractions (Figure 5B). These results suggest that the purity of mitochondrial fractions is a key factor for studying the role of mtCB1 receptors on mitochondrial functions.
Figure 5.
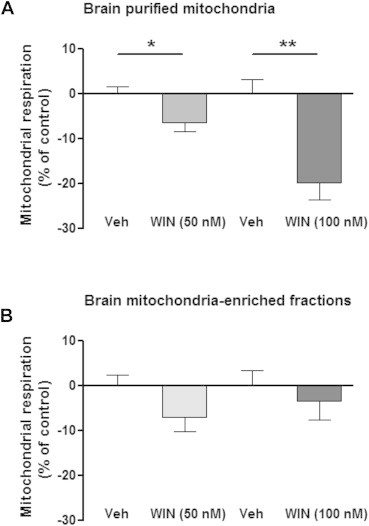
Effect of WIN55,212-2 on mitochondrial ADP-stimulated respiration in brain purified mitochondria and mitochondria-enriched fractions. (A) WIN55,212-2 dose-dependently decreases mitochondrial respiration in purified brain mitochondria, whereas (B) brain mitochondria-enriched fractions are insensitive to WIN. Data are expressed as mean ± S.E.M. *p < 0.05, p < 0.01, according to unpaired T test.
4. Discussion
Mitochondrial activity bears a growing importance in general brain functions and, particularly, for the regulation of energy balance in the body [3,4,7,29]. However, due to its intrinsic complexity, the brain requires specific methodological approaches for bioenergetics studies. Indeed, the use of different methods can lead to discrepant results, with the risk to draw conclusions on false positive results on one hand, or to consider interesting results as mere artifacts on the other hand. In this study, we addressed one example of such discrepancies in the literature, due to the use of different methodological approaches: the case of mitochondrial CB1 receptors [9,10]. By directly comparing different approaches, our results indicate that CB1 receptors are indeed functionally present on brain mitochondria and that this can be revealed using adapted anatomical and functional methods.
Antibodies against CB1 receptors have enabled great advances in revealing the broad impact of the endocannabinoid system in brain physiology and pathophysiology. However, the epitope of most antibodies, including CB1 receptor antisera, are usually covering small portion(s) of the total amino acid sequence of a protein, leaving the possibility of unspecific binding to other proteins. To avoid misinterpretations, the use of tissues from knock-out animals for the protein of interest is an essential step to evaluate the specificity of an antibody. Then, quantifications are necessary to verify if the signal observed in wild-type is higher or equivalent to the background signal observed in knock-out tissues. This is particularly necessary when low levels of specific staining are expected, as it is the case for mtCB1 receptors (approximately 15% of total brain CB1 protein [9]). Indeed, in these cases, simple subjective visual inspection might lead to erroneous interpretations of the results, which are avoided by objective quantifications.
Recently, mitochondrial CB1 immunoreactivity was also observed in CB1-KO mice [10]. The authors suggested that this observation might have been missed in the study of Benard et al. [9]. However, quantifications of mitochondrial CB1 receptor labeling in CB1-KO mice were provided in Ref. [9]: approximately 30% of wild-type CA1 hippocampal mitochondria displayed mtCB1 receptor labeling, whereas only 3% of CA1 mitochondria showed non-specific immunoreactivity in CB1-KO mice [9]. In electron microscopy experiments, Morozov et al. [10] mainly used the pre-embedding immunoperoxidase (ABC) method with Ni-intensified 3,3ʹ-diaminobenzidine–4HCl (DAB–Ni) as a chromogen. In some experiments, they also used a pre-embedding immunogold technique, similar to the one used in Ref. [9], but this technique was not tested in CB1-KO tissues [10]. The authors rightly claimed that the ABC method is more sensitive than the pre-embedding immunogold technique. However, the ABC method is also more prone to unspecific staining, due to the high chance of precipitation of chromophores [30]. Indeed, the electron microscopic images obtained through this technique displayed a fuzzy black immunostaining, suggesting unspecific precipitates of the DAB–Ni in both CB1-WT and CB1-KO tissue (see Ref. [10] and Figure 2). Nevertheless, our present data show that still approximately 20–25% of wild-type mitochondrial sections display specific CB1 receptor immunoreactivity using the DAB–Ni technique. The background staining levels observed in these conditions, however, were higher than using immunogold (approximately 9% with DAB–Ni versus approximately 3% with immunogold, see Ref. [9]), which could be due, for instance, to the fact that mitochondria contain endogenously biotinylated proteins [31]. Therefore, it is not surprising that the DAB–Ni staining produce some level of labeling on mitochondria, even without primary antibodies. These results clearly show the specificity of the CB1 receptor antiserum used in our laboratory for electron microscopic immunostaining experiments, confirm the need for careful negative controls and quantifications, and corroborate that background labeling depends on the technique used. In particular, the diffuse staining obtained by the DAB–Ni technique and the higher levels of background staining obtained with this method as compared with immunogold, clearly indicate that the latter method should be used to precisely evaluate the presence of mitochondrial proteins, especially when their expression levels are particularly low.
Another important consideration when evaluating immunogold staining of outer membrane mitochondrial proteins is the possibility that gold particles might be wrongly identified on mitochondrial membranes, when they are instead located on other close membranes. Despite the high resolution of immunogold detection, silver-intensified gold particles have minimal dimensions (20–40 nm) that could actually overlap between closely located membranes. Thus, CB1 immunogold staining could be actually on plasma or endoplasmic reticulum membranes and just “seem” to be located on proximal mitochondria. To further control for this potential caveat, we adopted a more stringent way to quantify mtCB1 expression by counting exclusively gold particles that appear on mitochondria, but that are also at least 80 nm from any other recognizable membrane. As expected, using this more stringent evaluation, the percentage of CB1-positive mitochondria in the CA1 hippocampal region of both wild-type and CB1-KO mice decreased to approximately 20% and 2%, respectively, maintaining, however, a highly significant difference between genotypes. Thus, we can conclude that, depending on the stringency of quantification, a range of 20–30% of mitochondria in the CA1 hippocampal region contain mtCB1 receptor protein.
In immunoblotting experiments, Morozov et al. [10] detected a 40 kDa band, identified as stomatin-like protein 2 (Slp2). Our immunoprecipitation and Western immunoblotting experiments, performed with tissues from CB1-KO mice as negative controls, confirmed the specific detection of bands at approximately 52 and 64 kDa, absent in CB1-KO tissues. However, as already previously observed in Ref. [9], additional bands were detected in the present experiments. Given the presence of these bands in both wild-type and CB1-KO tissues, they have to be considered as unspecific staining. The detection of additional unspecific bands is a common experience in many immunoblotting experiments, and particularly when using anti-CB1 antisera (common experience from several researchers in this field). Therefore, particular attention should be given to negative control conditions: only bands that are not present in CB1-KO tissues should be considered specific. Moreover, when potential off-targets are proposed, such as Slp2 for anti-CB1 antisera, it is good practice to control that the deletion of the target protein in knock-out mice does not impair expression of the putative off-target one. In this study, we performed this control, and we found that Slp2 levels (correctly identified as a 40 kDa band in Western immunoblotting experiments) are not changed in the brain of CB1-KO mice, further confirming the specificity of the anti-CB1 antisera used to detect mtCB1 expression.
Morozov et al. [10] stated “that the previously reported effect of synthetic cannabinoid WIN55,212-2 on mitochondrial complex III respiration [which should have been termed mitochondrial respiration, see 32] is not detectable in purified mitochondrial preparations”. However, the mitochondrial isolation protocol they used generated fractions that were poorly enriched in mitochondria, heavily contaminated with non-mitochondrial components and not sensitive to WIN55,212-2. Notably, the occurrence of abundant synaptosomes revealed by our Western immunoblots and by electron micrography [10] suggests that important levels of plasma membrane CB1 receptor might be present in these preparations. Since mtCB1 receptors represent only an estimate of 15% of the total pool of CB1 receptors [9], it is not surprising that WIN55,212-2 was not sufficient to activate mtCB1 receptors in mitochondrial preparations highly contaminated by synaptosomes, which likely contain much more abundant levels of the receptor. These data illustrate that precautions have to be taken when studying brain-isolated mitochondria. For instance, digitonin should be avoided as much as possible when working with these preparations, because it disrupts membranes through its binding with cholesterol and can lead to alterations of mitochondrial (especially the outer) membranes [8]. Therefore, digitonin should be avoided when components of mitochondrial outer membrane, including mtCB1 receptor [9], are studied.
In conclusion, our previous [9] and present studies provide converging anatomical and biochemical evidence that functional CB1 receptors are located at brain mitochondria, though at low levels. These data allow concluding with a reasonable degree of confidence that a direct relationship between CB1 receptor and mitochondrial functions in the brain is highly likely and can be detected if experimental procedures including accurate controls and quantifications are applied. Additional techniques, such as binding studies will also provide valuable information on the subject. In general, these data underline the particular importance of choosing specific adapted methodological approaches to decipher bioenergetics processes in a complex organ such as the brain.
Acknowledgements
We thank Delphine Gonzales, Nathalie Aubailly and all the personnel of the Animal Facility of the NeuroCentre Magendie for mouse care and genotyping. We thank all the members of Marsicano's lab for useful discussions. Supported by INSERM (G.M.), EU-Fp7 (REPROBESITY, HEALTH-F2-2008-223713, G.M.), European Research Council (ENDOFOOD, ERC-2010-StG-260515, G.M.), Fondation pour la Recherche Medicale (G.M.), Region Aquitaine (G.M.), BRAIN ANR-10-LABX-0043 (G.M.), Ministerio de Economía y Competitividad (BFU2012-33334 P.G.), The Basque Government (IT764-13, P.G.), Red de Trastornos Adictivos, RETICS, Instituto de Salud Carlos III (RD12/0028/0004, P.G.), The Basque Country University (UFI11/41, P.G.), AFM trampoline grant RAE13001GGA (GB). L. Reguero held a Postdoctoral Specialization Contract from the University of the Basque Country UPV/EHU. We are very grateful to Prof. Tamas Horvath (Yale School of Medicine) for critically reading the manuscript and valuable suggestions.
Conflict of interest
None declared.
References
- 1.Bournat J.C., Brown C.W. Mitochondrial dysfunction in obesity. Current Opinion in Endocrinology Diabetes and Obesity. 2010;17:446–452. doi: 10.1097/MED.0b013e32833c3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hojlund K., Mogensen M., Sahlin K., Beck-Nielsen H. Mitochondrial dysfunction in type 2 diabetes and obesity. Endocrinology and Metabolism Clinics of North America. 2008;37:713–731. doi: 10.1016/j.ecl.2008.06.006. x. [DOI] [PubMed] [Google Scholar]
- 3.Schneeberger M., Dietrich M.O., Sebastian D., Imbernon M., Castano C., Garcia A. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell. 2013;155:172–187. doi: 10.1016/j.cell.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dietrich M.O., Liu Z.W., Horvath T.L. Mitochondrial dynamics controlled by mitofusins regulate Agrp neuronal activity and diet-induced obesity. Cell. 2013;155:188–199. doi: 10.1016/j.cell.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sivitz W.I., Yorek M.A. Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxidants & Redox Signaling. 2010;12:537–577. doi: 10.1089/ars.2009.2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diano S., Horvath T.L. Mitochondrial uncoupling protein 2 (UCP2) in glucose and lipid metabolism. Trends in Molecular Medicine. 2012;18:52–58. doi: 10.1016/j.molmed.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 7.Abizaid A., Horvath T.L. Brain circuits regulating energy homeostasis. Regulatory Peptides. 2008;149:3–10. doi: 10.1016/j.regpep.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sims N.R., Anderson M.F. Isolation of mitochondria from rat brain using Percoll density gradient centrifugation. Nature Protocols. 2008;3:1228–1239. doi: 10.1038/nprot.2008.105. [DOI] [PubMed] [Google Scholar]
- 9.Benard G., Massa F., Puente N., Lourenco J., Bellocchio L., Soria-Gomez E. Mitochondrial CB(1) receptors regulate neuronal energy metabolism. Nature Neuroscience. 2012;15:558–564. doi: 10.1038/nn.3053. [DOI] [PubMed] [Google Scholar]
- 10.Morozov Y.M., Dominguez M.H., Varela L., Shanabrough M., Koch M., Horvath T.L. Antibodies to cannabinoid type 1 receptor co-react with stomatin-like protein 2 in mouse brain mitochondria. The European Journal of Neuroscience. 2013;38:2341–2348. doi: 10.1111/ejn.12237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piomelli D. The molecular logic of endocannabinoid signalling. Nature Reviews Neuroscience. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- 12.Silvestri C., Di Marzo V. The endocannabinoid system in energy homeostasis and the etiopathology of metabolic disorders. Cell Metabolism. 2013;17:475–490. doi: 10.1016/j.cmet.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 13.Nam D.H., Lee M.H., Kim J.E., Song H.K., Kang Y.S., Lee J.E. Blockade of cannabinoid receptor 1 improves insulin resistance, lipid metabolism, and diabetic nephropathy in db/db mice. Endocrinology. 2012;153:1387–1396. doi: 10.1210/en.2011-1423. [DOI] [PubMed] [Google Scholar]
- 14.Vickers S.P., Webster L.J., Wyatt A., Dourish C.T., Kennett G.A. Preferential effects of the cannabinoid CB1 receptor antagonist, SR 141716, on food intake and body weight gain of obese (fa/fa) compared to lean Zucker rats. Psychopharmacology. 2003;167:103–111. doi: 10.1007/s00213-002-1384-8. [DOI] [PubMed] [Google Scholar]
- 15.Pagotto U., Marsicano G., Cota D., Lutz B., Pasquali R. The emerging role of the endocannabinoid system in endocrine regulation and energy balance. Endocrine Reviews. 2006;27:73–100. doi: 10.1210/er.2005-0009. [DOI] [PubMed] [Google Scholar]
- 16.Bartova A., Birmingham M.K. Effect of delta9-tetrahydrocannabinol on mitochondrial NADH-oxidase activity. The Journal of Biological Chemistry. 1976;251:5002–5006. [PubMed] [Google Scholar]
- 17.Matsuda L.A., Lolait S.J., Brownstein M.J., Young A.C., Bonner T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 18.Munro S., Thomas K.L., Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 19.Athanasiou A., Clarke A.B., Turner A.E., Kumaran N.M., Vakilpour S., Smith P.A. Cannabinoid receptor agonists are mitochondrial inhibitors: a unified hypothesis of how cannabinoids modulate mitochondrial function and induce cell death. Biochemical and Biophysical Research Communications. 2007;364:131–137. doi: 10.1016/j.bbrc.2007.09.107. [DOI] [PubMed] [Google Scholar]
- 20.Howlett A.C., Barth F., Bonner T.I., Cabral G., Casellas P., Devane W.A. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacological Reviews. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- 21.Marsicano G., Wotjak C.T., Azad S.C., Bisogno T., Rammes G., Cascio M.G. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- 22.Benard G., Faustin B., Passerieux E., Galinier A., Rocher C., Bellance N. Physiological diversity of mitochondrial oxidative phosphorylation. American Journal of Physiology. Cell Physiology. 2006;291:C1172–C1182. doi: 10.1152/ajpcell.00195.2006. [DOI] [PubMed] [Google Scholar]
- 23.Clark J.B., Nicklas W.J. The metabolism of rat brain mitochondria. Preparation and characterization. The Journal of Biological Chemistry. 1970;245:4724–4731. [PubMed] [Google Scholar]
- 24.Howlett A.C., Song C., Berglund B.A., Wilken G.H., Pigg J.J. Characterization of CB1 cannabinoid receptors using receptor peptide fragments and site-directed antibodies. Molecular Pharmacology. 1998;53:504–510. doi: 10.1124/mol.53.3.504. [DOI] [PubMed] [Google Scholar]
- 25.McIntosh H.H., Song C., Howlett A.C. CB1 cannabinoid receptor: cellular regulation and distribution in N18TG2 neuroblastoma cells. Brain Research Molecular Brain Research. 1998;53:163–173. doi: 10.1016/s0169-328x(97)00294-5. [DOI] [PubMed] [Google Scholar]
- 26.Fukudome Y., Ohno-Shosaku T., Matsui M., Omori Y., Fukaya M., Tsubokawa H. Two distinct classes of muscarinic action on hippocampal inhibitory synapses: M2-mediated direct suppression and M1/M3-mediated indirect suppression through endocannabinoid signalling. The European Journal of Neuroscience. 2004;19:2682–2692. doi: 10.1111/j.0953-816X.2004.03384.x. [DOI] [PubMed] [Google Scholar]
- 27.Song C., Howlett A.C. Rat brain cannabinoid receptors are N-linked glycosylated proteins. Life Sciences. 1995;56:1983–1989. doi: 10.1016/0024-3205(95)00179-a. [DOI] [PubMed] [Google Scholar]
- 28.Kristian T. Isolation of mitochondria from the CNS. Current Protocols in Neuroscience. 2010:22. doi: 10.1002/0471142301.ns0722s52. Chapter 7, Unit 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horvath T.L., Diano S., Tschop M. Brain circuits regulating energy homeostasis. Neuroscientist. 2004;10:235–246. doi: 10.1177/1073858403262151. [DOI] [PubMed] [Google Scholar]
- 30.Maunsbach A.B., Afzelius B.A. Academic Press; San Diego: 1999. Biomedical electron microscopy. Illustrated methods and interpretations. [Google Scholar]
- 31.Hollinshead M., Sanderson J., Vaux D.J. Anti-biotin antibodies offer superior organelle-specific labeling of mitochondria over avidin or streptavidin. The Journal of Histochemistry and Cytochemistry. 1997;45:1053–1057. doi: 10.1177/002215549704500803. [DOI] [PubMed] [Google Scholar]
- 32.Chance B., Williams G.R. A method for the localization of sites for oxidative phosphorylation. Nature. 1955;176:250–254. doi: 10.1038/176250a0. [DOI] [PubMed] [Google Scholar]