

Abstract
Ca2+/calmodulin-dependent protein kinase II (CaMKII) functions both in regulation of insulin secretion and neurotransmitter release through common downstream mediators. Therefore, we hypothesized that pancreatic ß-cells acquire and store the information contained in calcium pulses as a form of “metabolic memory”, just as neurons store cognitive information. To test this hypothesis, we developed a novel paradigm of pulsed exposure of ß-cells to intervals of high glucose, followed by a 24-h consolidation period to eliminate any acute metabolic effects. Strikingly, ß-cells exposed to this high-glucose pulse paradigm exhibited significantly stronger insulin secretion. This metabolic memory was entirely dependent on CaMKII. Metabolic memory was reflected on the protein level by increased expression of proteins involved in glucose sensing and Ca2+-dependent vesicle secretion, and by elevated levels of the key ß-cell transcription factor MAFA. In summary, like neurons, human and mouse ß-cells are able to acquire and retrieve information.
Keywords: Insulin secretion, Metabolic memory, CaMKII
1. Introduction
Ionic calcium (Ca2+) controls multiple cellular signaling processes in all eukaryotic cells, including proliferation, gene expression, and neurotransmitter release. Ca2+-binding proteins such as calmodulin play a pivotal role in Ca2+ signal transmission and amplification. Increases in the concentration of intracellular Ca2+ activate specific protein targets, among which the Ca2+/calmodulin-dependent protein kinase II (CaMKII) is a critical signal mediator [1].
In response to an increase in intracellular Ca2+, Thr-286 of CaMKII becomes exposed and phosphorylated in an intraholoenzyme reaction [2]. The coupling of Ca2+-calmodulin to one of the CaMKII subunits allows for the phosphorylation of an adjacent subunit at Thr286. This process, known as “calmodulin trapping”, confers Ca2+-calmodulin-independent kinase activity to the complex and thus prolongs the Ca2+ signal. Thus, calmodulin trapping represents a molecular mechanism of memory [3,4], which is defined as the capacity to acquire, store (consolidate), and retrieve (evocate) information [5].
CaMKII is a major synaptic protein that is activated during the induction of long-term potentiation (LTP) by Ca2+ influx through N-methyl-d-aspartate (NMDA) receptors. Calmodulin trapping allows CaMKII to remain activated long after the initial Ca2+ signal has dissipated, suggesting that CaMKII is a memory molecule crucial for LTP [4,5]. Consistent with this notion, CaMKII-null mice present with impaired memory formation, and CaMKII is essential for genesis and maintenance of LTP in postsynaptic neurons [2]. Following presynaptic stimulation, CaMKII is activated in postsynaptic neurons, which creates a physiological imprint of the initial Ca2+ signal, and increases translocation of NMDA receptors to the plasma membrane [6]. Because of its capacity to remain activated long after the initial pulse of Ca2+ signaling, CaMKII perpetuates Ca2+ effects and modulates gene expression and the epigenetic profile of postsynaptic neurons [7].
CaMKII also participates in glucose-stimulated insulin secretion (GSIS), as multiple insulin secretagogues increase CaMKII activity [8]. In the perfused rat pancreas, the dynamics of CaMKII activation correlate with the amplitude of GSIS, and CaMKII activation is temporally associated with insulin secretion [8]. CaMKII is essential for appropriate GSIS and is involved in several steps of this process, including the synthesis of insulin granules, the modulation of cytoplasmic content of ATP, and the activation of Synapsin [8]. Importantly, CaMKII also regulates transcription factors central for ß-cell function such as CREB [9] and MafA [10,11].
Here, we investigated whether pancreatic ß-cells, like neurons, acquire and store the information contained in calcium pulses as a form of “metabolic memory”. Indeed, we find that ß-cells retain memory of prior activation, and describe the molecular mechanism that contributes to this memory.
2. Materials and methods
2.1. The glucose pulse paradigm
MIN6 insulinoma cells or mouse or human pancreatic islets were distributed into three groups: control, pulse, and pulse + KN93 (10 μM). Initially, all groups were maintained in the same medium with 5.6 mM glucose for 24 h. After this acclimation period, the control group was exposed to 3-mM glucose for 24 h, while the pulse and pulse + KN93 (10 μM) groups were exposed to 30 mM of glucose for four 1-h periods intercalated with 7-hour periods of 3 mM glucose. KN93 (a CaMKII inhibitor) at 10 μM was only present during the 30 mM glucose pulses. All groups were then maintained at 3 mM glucose for a 24-h consolidation period.
2.2. Insulin secretion assays
Insulin secretion of MIN6 cells [12] and mouse pancreatic islets [13] was conducted following standard procedures, and insulin was measured by RIA.
2.3. Human islet perfusion
One hundred and fifty islets were handpicked under a light microscope and pre-incubated for 1 h in KRBB (Krebs bicarbonate buffer) containing 0.3 g/l BSA and 2.8 mM glucose. The medium was discarded and the islets incubated for an additional hour in 500-μl KRBB containing 2.8 or 16.8 mM glucose. Subsequently, the supernatant was collected to evaluate insulin secretion by RIA. For perifusion assays, 150 treated human islets were placed into a perifusion chamber (Millipore, Billerica, MA, USA). A computer-controlled fast-performance HPLC system allowed programmable rates of flow and concentrations of the appropriate solutions held in a 37 °C water bath. Islets were perfused for 80 min with KRBB in the absence of glucose, followed by KRBB with increasing concentrations of glucose (glucose was ramped from 0 to 30 mm at 0.75 mM/min). At the end of each experiment, islets were tested for maximal insulin secretion by adding 30 mM KCl to the perifusate [14,15]. Samples were collected at 2 ml/min and insulin content was determined by using RIA.
2.4. Statistics
Data are expressed as means ± SEM. Statistical analyses were performed using Student's t-test or a two-way ANOVA with a Bonferroni's posttest, as required. Statistical significance was set at p < 0.05.
3. Results and discussion
3.1. Mouse ß-cells exhibit long-lasting metabolic memory
In order to investigate metabolic memory of insulin-producing ß-cells, we first had to develop a paradigm in which such memory was evident. As detailed below, we established that exposure of cultured MIN6 ß-cells to four 1-h pulses of high glucose, interspersed with 7-h intervals of low glucose and thus mimicking postprandial glucose spikes in vivo, were able to elicit robust metabolic memory. The detailed experimental paradigm is outlined in Figure 1A. As shown in Figure 1B, MIN6 ß-cells exposed to the glucose pulse regimen showed higher insulin secretion both when exposed to low (2.8 mM) or high (16.8 mM) glucose than ß-cells, which had been cultured continuously at low glucose. This increase in insulin secretion was accompanied by a significant rise in the levels of phosphorylated CaMKII, consistent with the notion that CaMKII might be a molecular mediator of this metabolic memory (Figure 1C and D). Importantly, both the increases in basal and stimulated insulin secretion in the glucose pulse group were abolished by KN93 (Figure 1B), a specific CaMKII inhibitor.
Figure 1.
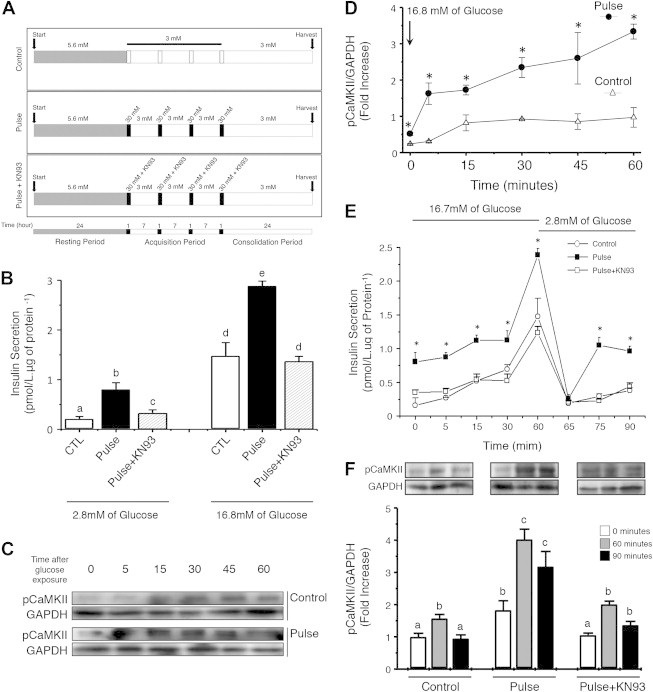
MIN6 insulinoma cells acquire, store, and recall metabolic information. (A) Experimental paradigm to elicit metabolic memory. ß-cells were exposed to four pulses of high (30 mM) glucose, interspersed with three intervals of low (3 mM) glucose, followed by a 24-h consolidation period before analysis of insulin secretion. Parallel dishes were cultured in low glucose continuously, or exposed to the CaMKII inhibitor KN93 only during the glucose pulses. (B) Acute insulin secretion of MIN6 ß-cells 24 h after the last glucose pulse at 2.8 or 16.8 mM glucose for 1 h. Letters indicate significant differences at p < 0.05 between groups. a – CTL 2.8 vs all groups; b – Pulse 2.8 vs all groups; c – Pulse + KNP3 vs all groups; d – CTL 16.7/Pulse + KN93 16.7 vs CTL 2.8/Pulse 2.8/Pulse + KN93 2.8/Pulse 16.7; and e – Pulse 16.7 vs all groups. Data represent six independent experiments. (C and D) MIN6 ß-cells of the control (white triangle) or glucose pulse group (black circle) were acutely exposed to 16.7 mM of glucose. Phosphorylation of CaMKII (pCaMKII/GAPDH) was assessed at 0, 5, 15, 30, 45, and 60 min of exposure. Values are presented as fold-increase over time zero. *p < 0.05. (E) Insulin secretion during the acute stimulus of 16.7 mM glucose over time (0, 5, 15, 30, and 60 min), and after the removal of the stimulus (65, 75, and 90 min). *p < 0.05. (F) CaMKII phosphorylation of the three experimental groups in basal conditions, after 60 min of acute glucose stimulus, and 30 min after the removal of glucose (“90 minutes”). Letters denote significant differences between the groups at p < 0.05: a – CTL 0′/CTL 90′/Pulse+KN93 0′ vs all groups; b – CTL 60′/Pulse 0′/Pulse+KN93 60′/Pulse+KN93 90′ vs all groups; c – Pulse 60′/Pulse 90′ vs all groups. Data are means ± SEM of four independent experiments.
Next, we evaluated the kinetics of the ß-cell response to acute glucose exposure, and observed that the pulse group secreted more insulin at basal (time zero) and stimulatory concentrations of glucose (16.7 mM) between 5 and 60 min following stimulation (Figure 1E). This effect persisted even after glucose concentrations were returned to 2.8 mM. In parallel, the phosphorylation of CaMKII was higher in the glucose pulse group at both glucose concentrations, and remained elevated even after the glucose concentration was reduced to 2.8 mM (Figure 1F). Thus, MIN6 ß-cells had acquired metabolic memory of prior exposure to glucose pulses, which they retained through a 24-h consolidation period.
3.2. Metabolic memory extends to human ß-cells
It was important to determine if metabolic memory is confined to MIN6 insulinoma cells, or if it is also a property of primary human ß-cells. Therefore, we cultured human islets from multiple non-diabetic deceased organ donors (see Table 1 for donor information) and exposed them to pulses of 30 mM glucose in the same paradigm as described above. Pulse treatment increased insulin secretion at basal (2.8 mM) and stimulatory (16.8 mM) glucose concentrations (Figure 2A). As was the case in MIN6 ß-cells, the pulse group showed increased CaMKII phosphorylation at both basal and stimulatory glucose concentrations (Figure 2B). Next, we determined the human islet response to a glucose challenge in the islet perifusion assay. Islets from the glucose pulse group showed both increased first and second phase insulin secretion compared with islets from the control group (Figure 2C and D). As before, these effects were abolished by the CaMKII inhibitor KN93 (Figure 2A–D). We also tested metabolic memory of ß-cells using isolated mouse pancreatic islets, with similar outcome (Extended Data Figure 1). We also found, using live cell calcium imaging of mouse islets, that the increases in cytoplasmic calcium levels following acute glucose stimulation, representing the penultimate step in insulin secretion, are significantly higher in the pulse group compared with controls (Extended Data Figure 1C).
Table 1.
Parameters of islet donors.
| Gender | Age | Height (m) | Weight (kg) | BMI | Diabetes |
|---|---|---|---|---|---|
| M | 27 | 1.77 | 64 | 20.2 | No |
| M | 44 | 1.70 | 75 | 25.8 | No |
| F | 36 | 1.62 | 92 | 34.8 | No |
| M | 43 | 1.95 | 117 | 30.6 | No |
Figure 2.
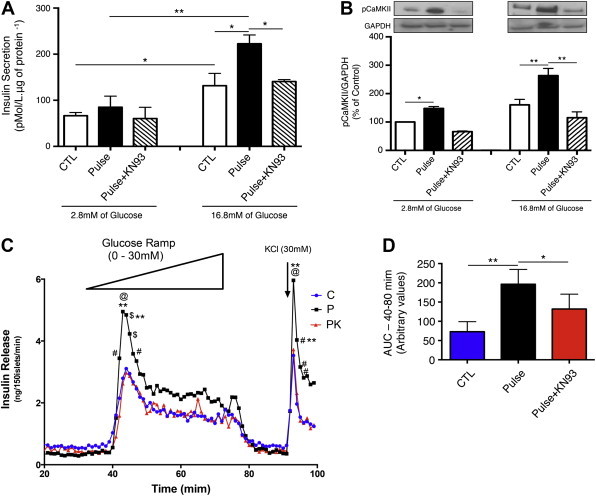
Human pancreatic islets display metabolic memory of prior glucose exposure. Twenty-four hours after the last glucose pulse we determined (A) insulin secretion and (B) phosphorylation of CaMKII in human islets from non-diabetic deceased organ donors exposed to KRBB with 2.8 or 16.8 mM glucose for 1 h. (C) Representative insulin secretion from perfused islets. After the consolidation period, 150 human islet per group were placed into a perifusion chamber and exposed to KRBB without glucose for 30 min, followed by KRBB with increasing concentrations of glucose from 0 to 30 mM, increasing at 0.75 mM/min. Samples were collected at 2 ml/min and insulin levels determined by RIA. (D) The area under the curve (AUC) summarizing insulin secretion during acute glucose exposure. In panel c: #p < 0.05 vs Control, @p < 0.001 vs Control, **p < 0.05 vs Pulse + KN93, and $p < 0.01 vs Control. In panels A, B, D, and E *p < 0.05, **p < 0.01. Data are means ± SEM of 3–5 independent experiments.
3.3. Molecular mediators of metabolic memory
To investigate the molecular mechanisms by which the glucose pulse paradigm induces memory in ß-cells, we evaluated the expression of essential mediators of the insulin secretory response. The glucose pulse paradigm induced expression of glucokinase (GCK), the main glucose sensor of ß-cells (Figure 3A), the voltage-gated Ca2+ channel (Cav1.2) (Figure 3B), SNAP25, an essential component of the exocytotic machinery (Figure 3C), and MafA, a key transcription factor of the insulin gene and the mature ß-cell phenotype in general (Figure 3D). All the effects of the pulsed glucose paradigm on protein levels were reduced or abolished by treatment with KN93 during the high glucose exposure only.
Figure 3.
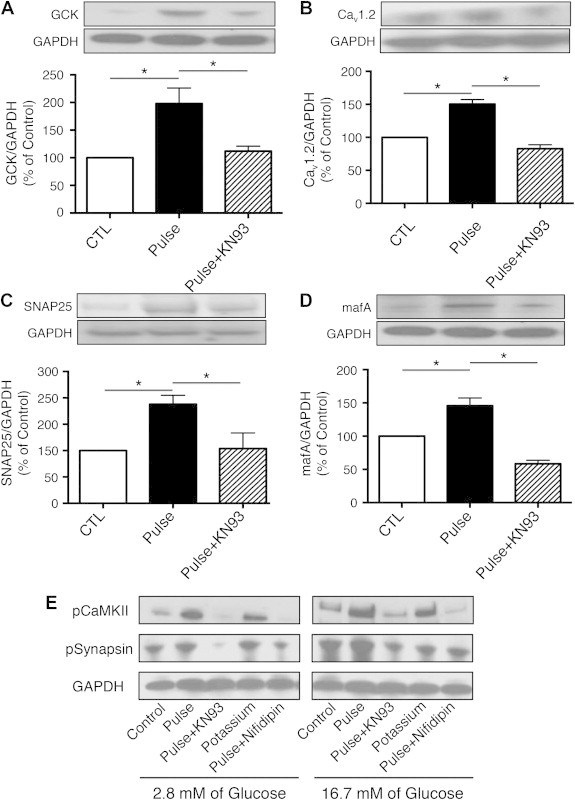
Metabolic memory is reflected in expression changes of key players in insulin secretion. Twenty-four hours after the last pulse of glucose, human islets were harvested and protein extracted to quantify expression of the following proteins: (A) glucokinase (GCK, expression normalized to GAPDH), (B) voltage-dependent Ca2+ channel (Cav1.2/GAPDH), (C) synaptosomal-associated protein 25 (SNAP-25/GAPDH), and (D) the transcriptional factor MAFA (MafA/GAPDH). Data are means ± SEM of 3–5 independent experiments. *p < 0.05. (E) Expression of pCaMKII, pSynapsin, and GAPDH of human islet from the groups Control, Pulse, Pulse + KN93, potassium chloride, and Pulse + Nifidipin exposed for 1 h to KRBB with 2.8 or 16.8 mM of glucose.
Finally, we asked if metabolic memory of ß-cells is dependent on glucose metabolism itself, or if this memory can be produced by membrane depolarization alone. To this end, we treated human islets with pulses of 30 mM KCl, which causes membrane depolarization, opening of voltage-gated Ca2+ channels, and insulin secretion without a prior increase in intracellular ATP levels. As shown in Figure 3E, depolarization by potassium chloride during the acquisition period produced similar, if somewhat lower, increases in pCaMKII and pSynapsin levels 24 h after the last KCl pulse. Thus, increases in intracellular calcium elicited by membrane depolarization are sufficient to activate CaMKII and produce metabolic memory. Total CaMKII protein levels were not affected by any of the treatments used (data not shown).
Glucose-stimulated insulin secretion is a complex process that translates glycolytic flux and elevated ATP production to increased cytoplasmic Ca2+ levels and finally fusion of insulin granules with the plasma membrane. This process is accompanied by increased phosphorylation of CaMKII (Extended Data Figure 2), a process now shown to be part of the establishment of metabolic memory in ß-cells. Cerasi and colleagues had previously reported time-dependent potentiation of insulin secretion, in that exposure of rat islets to high glucose (27.7 mM) up to 60 min prior to the test stimulation with glucose increased subsequent insulin secretion [16,17]. However, these studies focused on analyzing acute effects of high glucose pre-treatment, and did not yet investigate the molecular mechanism underlying the phenomenon. Here, we demonstrate that human and mouse pancreatic ß-cells are able to acquire, consolidate, and retrieve information, induced by high glucose exposure, and determined that this ability is dependent on CaMKII.
In conclusion, we have shown here that, like neurons, human and mouse ß-cells are able to acquire, store, and retrieve information in a process similar to neuronal LTP. This process, presented schematically in Figure 4, is dependent on the activation of CaMKII, as is the case for neuronal memory. These findings provide further evidence that the similarity of neurons and ß-cells on the transcriptome and epigenome level is not accidental, but an important aspect of the biology of these embryologically distinct cell types. Metabolic memory of ß-cells likely represents a useful evolutionary adaptation to variation in food availability. As periods of food abundance in nature can vary dramatically in length, metabolic memory of repeated carbohydrate loads in ß-cells ensures higher insulin secretion and thus more efficient uptake of excess glucose into skeletal muscle and adipose tissue for long-term storage, contributing to the adaptation to periods of starvation. Given the pivotal role of altered insulin secretion in metabolic disorders, future work will need to establish to what extent metabolic memory of pancreatic ß-cells contributes to the pathophysiology of these diseases.
Figure 4.
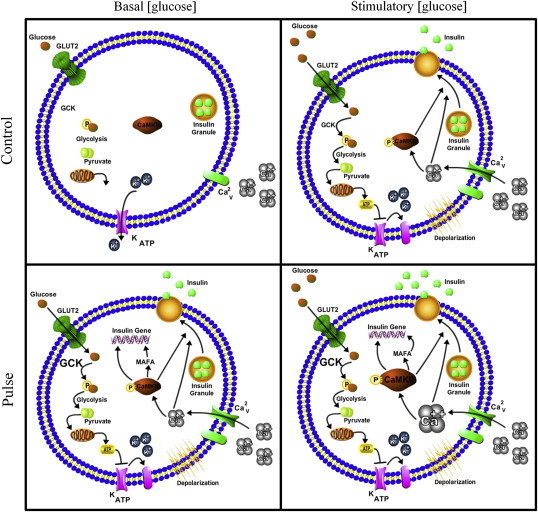
Model of the proposed mechanism of CaMKII-dependent memory formation in pancreatic ß-cells. In low glucose concentrations (2.8 mM), control islets exhibit basal insulin secretion. Islets from the glucose pulse group show, even 24 h after the last high glucose pre-treatment, an increase in acute glucose-induced insulin secretion. This is due to the fact that ß-cells in the pulse group have increased expression of GCK, calcium channels, and phosphorylation of CaMKII. When exposed to high glucose, islets from the group control increase insulin secretion because of the increased glycolytic flux through GCK and elevated ATP production. ß-cells previously exposed to high glucose pulses have higher levels of GCK, calcium channel, Calcium influx, CaMKII phosphorylation, and therefore respond with higher insulin secretion to acute glucose.
Acknowledgments
We thank Drs. Le Lay and Blendy for critical comments on the manuscript, CNPq, FAPESP (BEPE 2011/09012-6, CEPID 2013/07607-8 and Temático 2011/09012-6 to AC Boschero) and NIH (NIDDK R01-DK-055342 and R01-DK-088383 to K.H. Kaestner) for financial support, and Marise MC Brunelli (Biologist, IB/Unicamp), Priscila de Oliveira Marques (IB/Unicamp), Wei Qin (IDOM/Upenn), and Heather Collins (TCL/Upenn) for technical assistance. Santos G.J. belongs to the Obesity and Comorbities Research Center/Sao Paulo Research Foundation.
Contributor Information
Klaus H. Kaestner, Email: kaestner@mail.med.upenn.edu.
Antonio Carlos Boschero, Email: boschero@unicamp.br.
Conflict of interest
None declared.
Appendix A. Supplementary data
The following are the supplementary data related to this article:
Supplementary Figure 1.
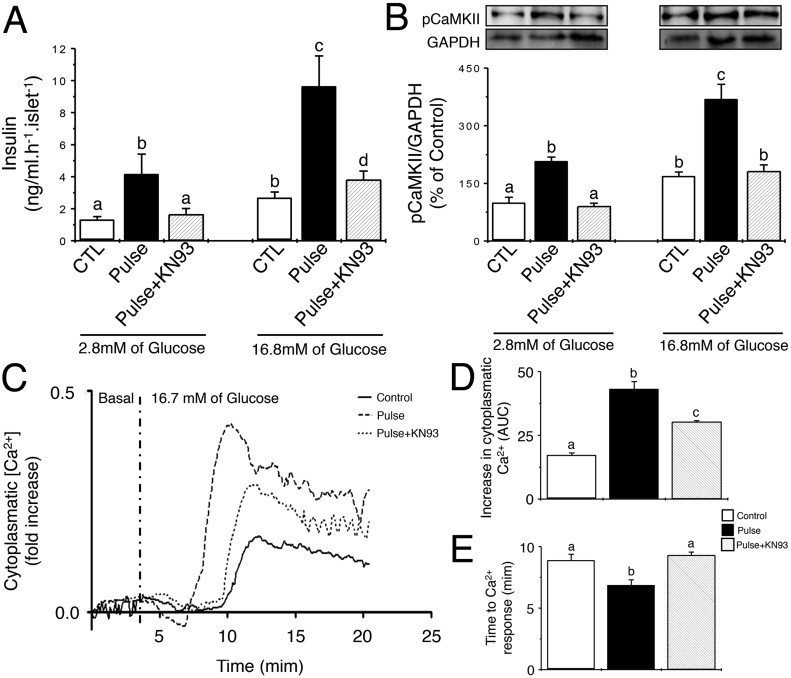
Mouse pancreatic islets acquire, store, and evoke information. Following the glucose paradigm and the consolidation period, pancreatic islet from Swiss mice were exposed to KRBB with 5.6 mM of glucose for 1 h, and then exposed to KRBB with 2.8 or 16.8 mM of glucose for additional 1 h. After this period, we determined (A) insulin secretion and (B) phosphorylation of CaMKII (pCaMKII/GAPDH). (C) Representative curves of intracellular Ca2+ concentrations in response to 16.7 mM of glucose for mouse islets pre-treated with the various glucose exposure paradigms. Values are the ratios of F340/F380 of FURA2-AM fluorescence are presented as fold-increase over the initial ratio. (D) The total rise in intracellular calcium was determined as ‘area under the curve’ (AUC) was during the stimulatory period. (E) The time delay from glucose exposure to initial rise in intracellular Ca2+ depends on prior glucose exposure. Values are represented in minutes. Different letters means significant differences at p < 0.05. Data are means ± SEM of 3–4 independent experiments.
Supplementary Figure 2.
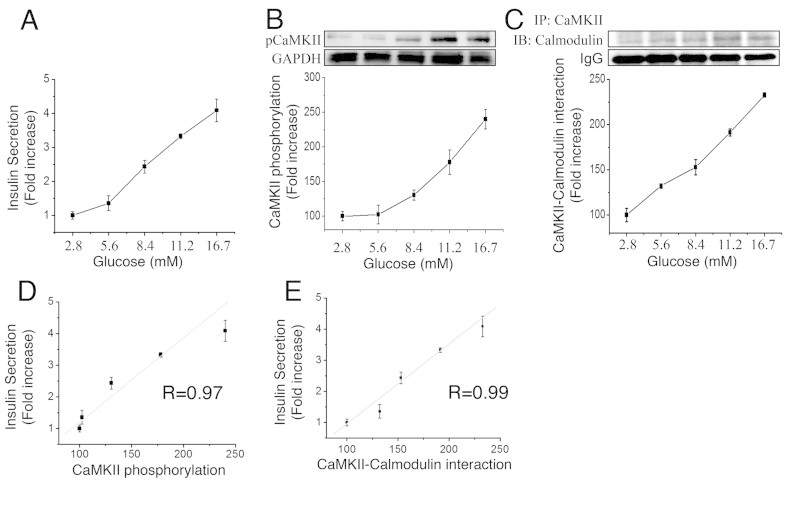
Insulin secretion in MIN6 correlates with acute CaMKII phosphorylation. MIN6 cells were maintained in RPMI medium without glucose for 1 h, and then exposed to KRBB with increasing glucose concentrations for 1 h. At the end of the 1-h exposure, we assessed insulin secretion, P-CaMKII, and the Calmodulin/P-CaMKII interaction. (A) Glucose-induced insulin secretion (GSIS) and (B) CaMKII phosphorylation (P-CaMKII/GAPDH) following acute glucose stimulation. (C) Calmodulin associated with pCaMKII. Protein extract were immunoprecipitated with a pCaMKII antibody followed by immunoblot for Calmodulin. (D) Correlation of insulin secretion and P-CaMKII following acute glucose exposure. (E) Correlation of insulin secretion and Calmodulin/P-CaMKII association following acute glucose exposure. Values are expressed as fold-increase over basal. Data are means ± SEM of four independent experiments.
References
- 1.Clapham D. Calcium signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 2.Yamauchi T. Neuronal Ca2+/calmodulin-dependent protein kinase II–discovery, progress in a quarter of a century, and perspective: implication for learning and memory. Biological and Pharmaceutical Bulletin. 2005;28:1342–1354. doi: 10.1248/bpb.28.1342. [DOI] [PubMed] [Google Scholar]
- 3.Waxham M., Tsai A., Putkey J. A mechanism for calmodulin (CaM) trapping by CaM-kinase II defined by a family of CaM-binding peptides. Journal of Biological Chemistry. 1998;273:17579–17584. doi: 10.1074/jbc.273.28.17579. [DOI] [PubMed] [Google Scholar]
- 4.Rongo C. A fresh look at the role of CaMKII in hippocampal synaptic plasticity and memory. BioEssays. 2002;24:223–233. doi: 10.1002/bies.10057. [DOI] [PubMed] [Google Scholar]
- 5.Fukunaga K., Miyamoto E. A working model of CaM kinase II activity in hippocampal long-term potentiation and memory. Journal of Neuroscience Research. 2000;38:3–17. doi: 10.1016/s0168-0102(00)00139-5. [DOI] [PubMed] [Google Scholar]
- 6.Hudmon A., Schulman H. Neuronal CA2+/calmodulin-dependent protein kinase II: the role of structure and autoregulation in cellular function. Annual Review of Biochemistry. 2002;71:473–510. doi: 10.1146/annurev.biochem.71.110601.135410. [DOI] [PubMed] [Google Scholar]
- 7.Lipsky R.H. Epigenetic mechanisms regulating learning and long-term memory. International Journal of Developmental Neuroscience. 2012 doi: 10.1016/j.ijdevneu.2012.10.110. [DOI] [PubMed] [Google Scholar]
- 8.Easom R. CaM kinase II: a protein kinase with extraordinary talents germane to insulin exocytosis. Diabetes. 1999;48:675–684. doi: 10.2337/diabetes.48.4.675. [DOI] [PubMed] [Google Scholar]
- 9.Suefuji M., Furukawa N., Matsumoto K., Oiso H., Shimoda S., Yoshinaga T. The impact of Ca2+/calmodulin-dependent protein kinase II on insulin gene expression in MIN6 cells. Biochemical and Biophysical Research Communication. 2012;421:801–807. doi: 10.1016/j.bbrc.2012.04.091. [DOI] [PubMed] [Google Scholar]
- 10.Hang Y., Stein R. MafA and MafB activity in pancreatic β cells. Trends in Endocrinology and Metabolism. 2011;22:364–373. doi: 10.1016/j.tem.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang H., Brun T., Kataoka K., Sharma A.J., Wollheim C.B. MAFA controls genes implicated in insulin biosynthesis and secretion. Diabetologia. 2007;50:348–358. doi: 10.1007/s00125-006-0490-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watson M.L., Macrae K., Marley A.E., Hundal H.S. Chronic effects of palmitate overload on nutrient-induced insulin secretion and autocrine signalling in pancreatic MIN6 beta cells. PLoS One. 2011;6:e25975. doi: 10.1371/journal.pone.0025975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rezende L.F., Santos G.J., Santos-Silva J.C., Carneiro E.M., Boschero A.C. Ciliary neurotrophic factor (CNTF) protects non-obese Swiss mice against type 2 diabetes by increasing beta cell mass and reducing insulin clearance. Diabetologia. 2012;55:1495–1504. doi: 10.1007/s00125-012-2493-5. [DOI] [PubMed] [Google Scholar]
- 14.Doliba N.M., Qin W., Vatamaniuk M.Z., Li C., Zelent D., Najafi H. Restitution of defective glucose-stimulated insulin release of sulfonylurea type 1 receptor knockout mice by acetylcholine. American Journal of Physiology. Endocrinology and Metabolism. 2004;286:E834–E843. doi: 10.1152/ajpendo.00292.2003. [DOI] [PubMed] [Google Scholar]
- 15.Deng S., Vatamaniuk M., Huang X., Doliba N., Lian M.M., Frank A. Structural and functional abnormalities in the islets isolated from type 2 diabetic subjects. Diabetes. 2004;53:624–632. doi: 10.2337/diabetes.53.3.624. [DOI] [PubMed] [Google Scholar]
- 16.Efendić S., Cerasi E., Luft R., Gladnikoff G. Potentiation of glucose-induced insulin release by glucose in the isolated pancreas of fed and fasted rats. Diabetes. 1976;25:949–954. doi: 10.2337/diab.25.10.949. [DOI] [PubMed] [Google Scholar]
- 17.Grill V., Adamson U., Cerasi E. Immediate and time-dependent effects of glucose on insulin release from rat pancreatic tissue. Evidence for different mechanisms of action. Journal of Clinical Investigation. 1978;61:1034–1043. doi: 10.1172/JCI109002. [DOI] [PMC free article] [PubMed] [Google Scholar]